
Increasing the Efficacy of Seproxetine as an Antidepressant Using Charge–Transfer Complexes

 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
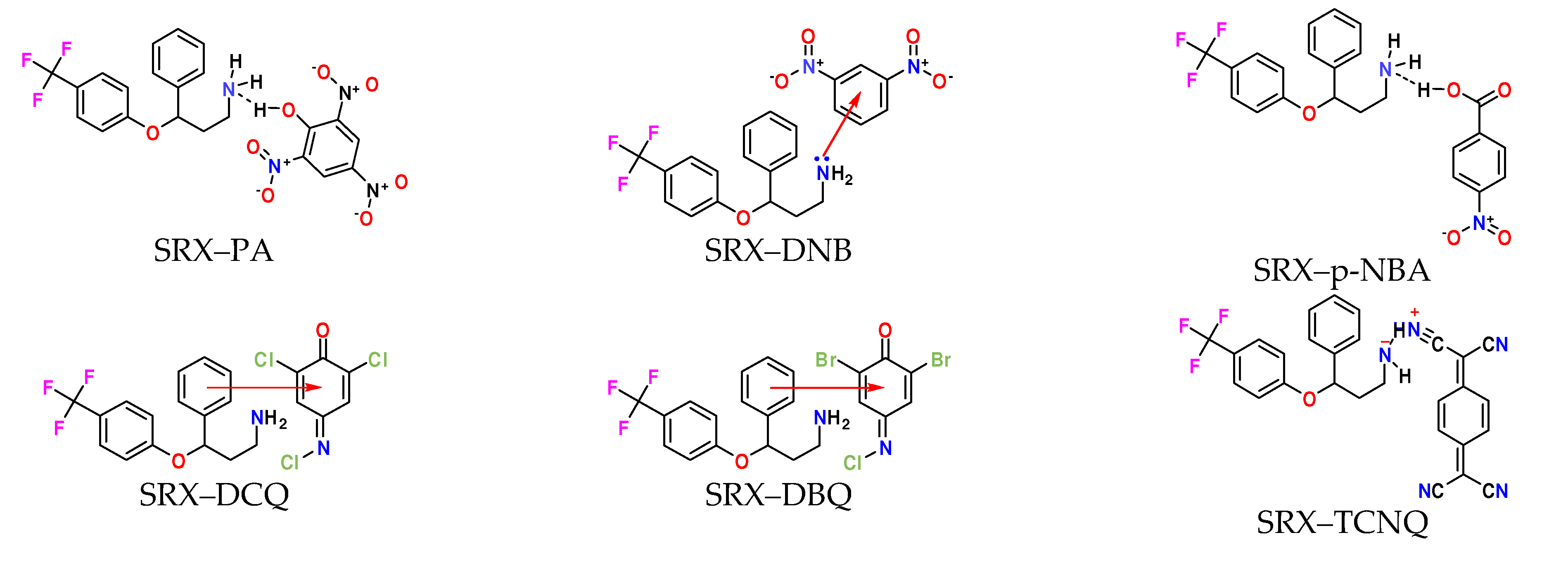
2.1. Synthesis of [(SRX)(π-Acceptor)] Charge–Transfer Complexes
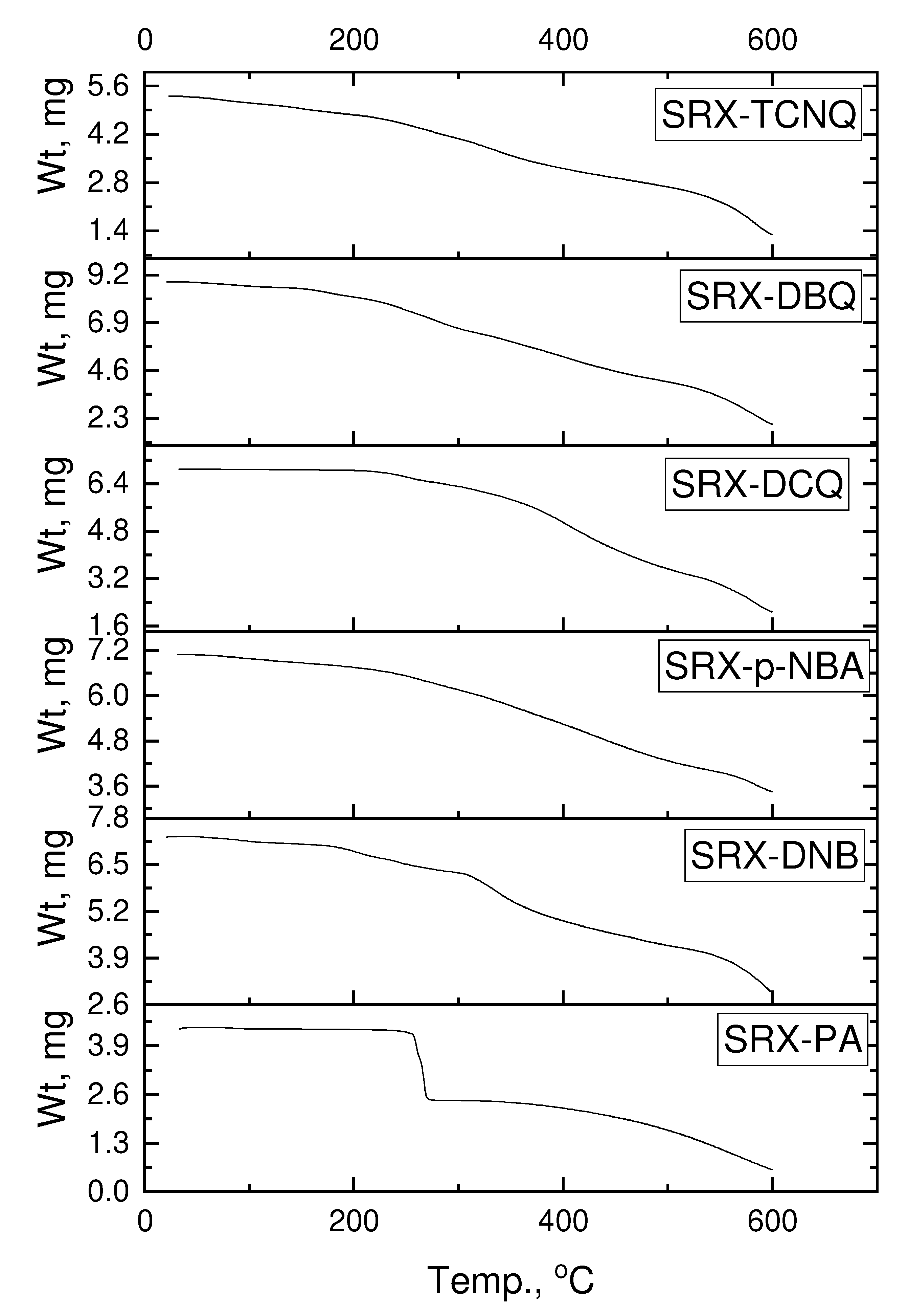
2.2. Instruments and Measurements
2.3. Molecular Docking
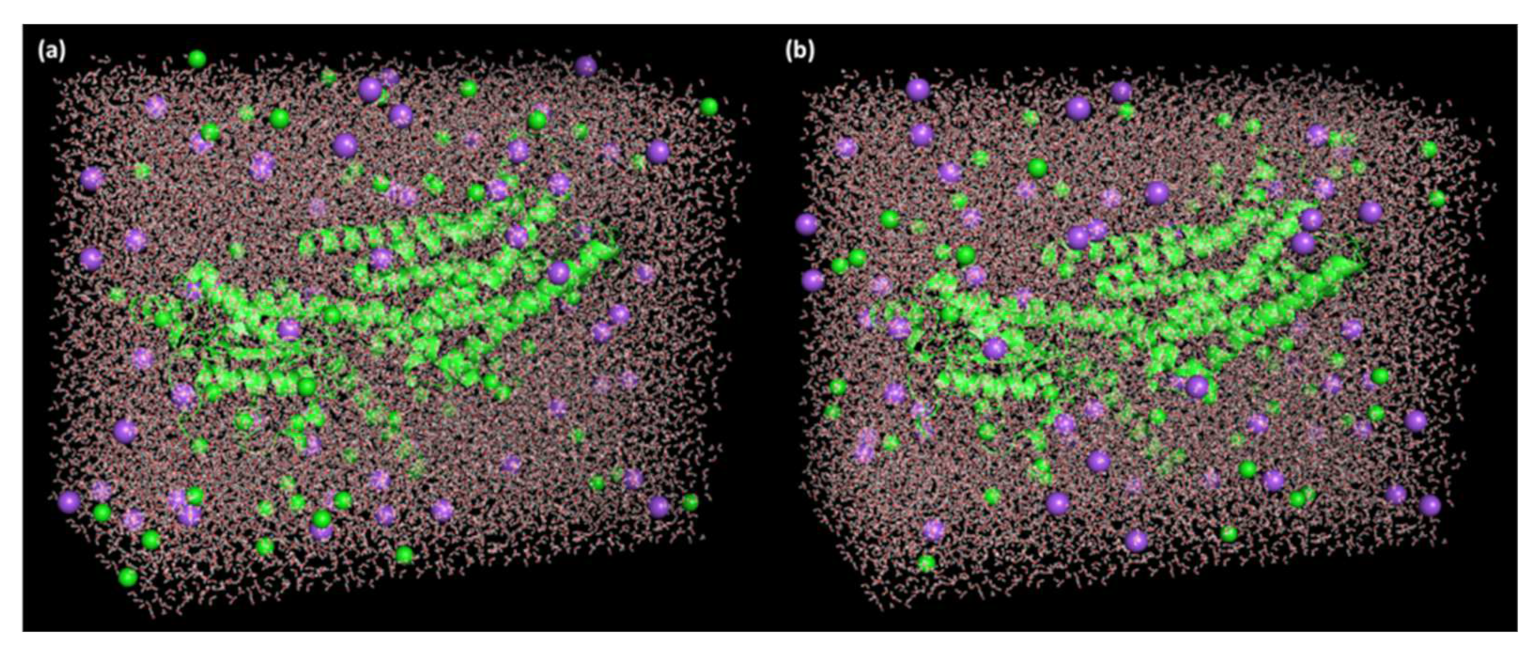
2.4. Molecular Dynamics (MD) Simulation
2.5. Computational Structural Analysis
3. Results and Discussion
3.1. Preapprehension
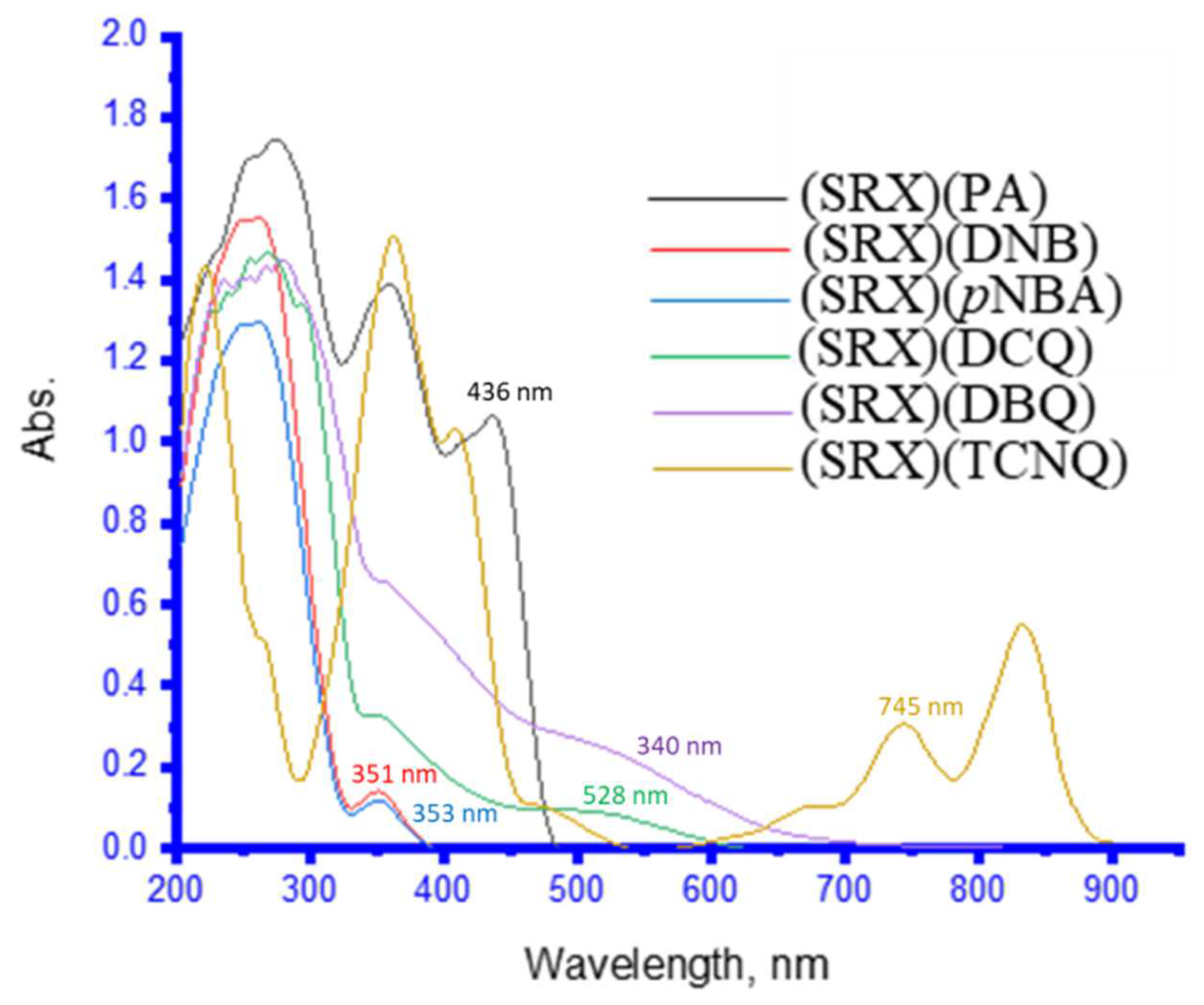
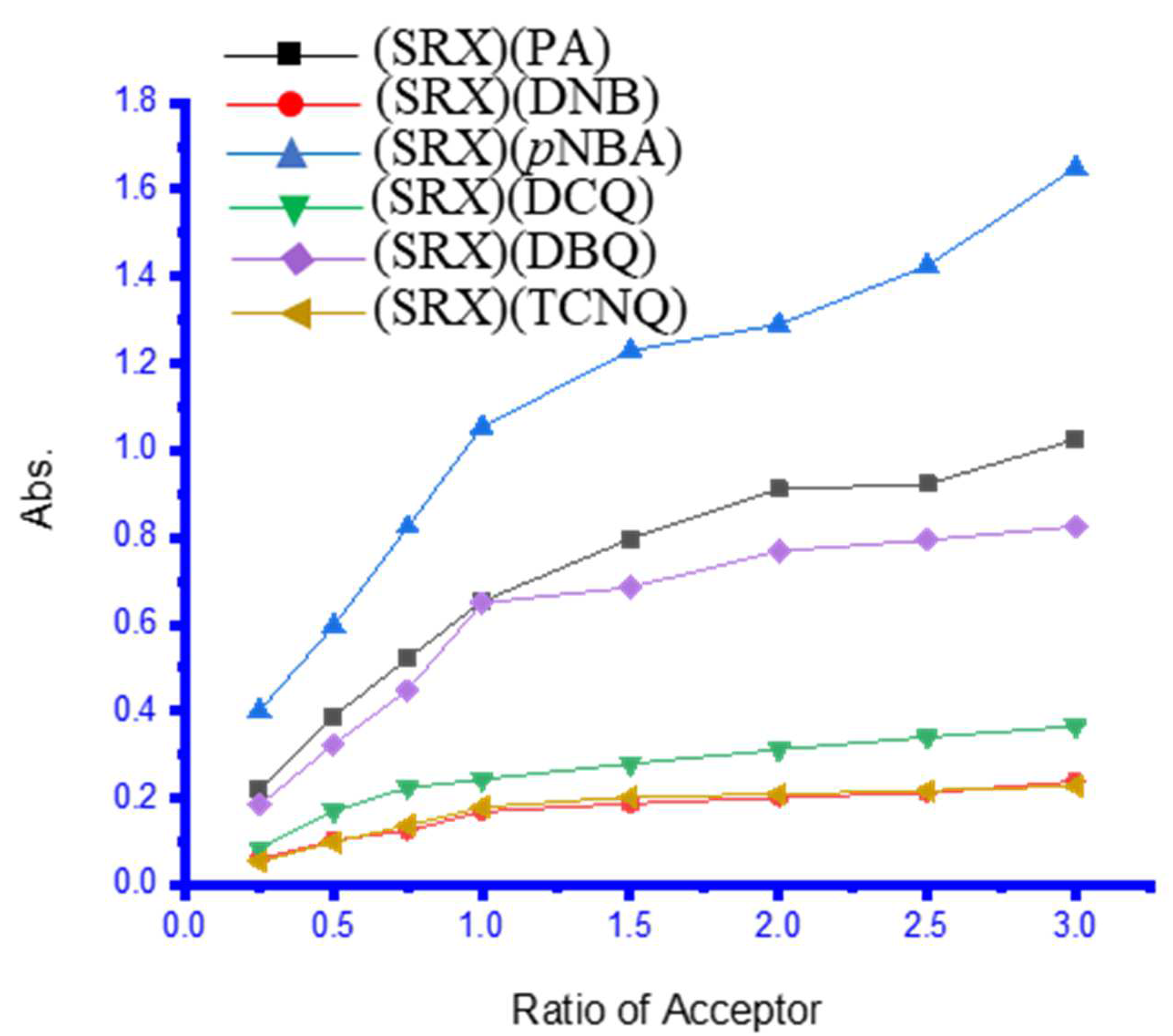
3.2. UV–Vis Spectra and Photometric Titration
3.3. 1H-NMR Spectra
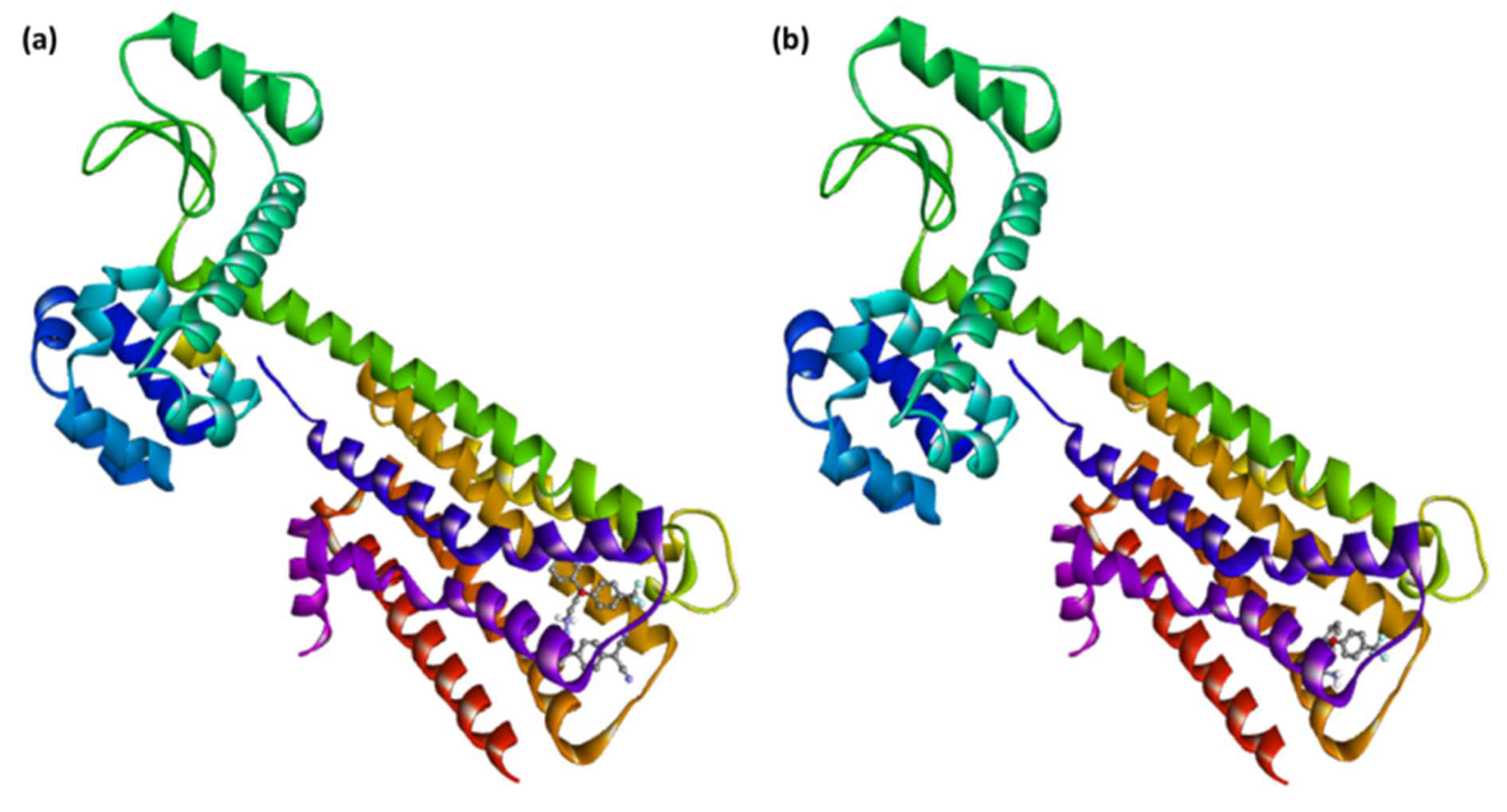
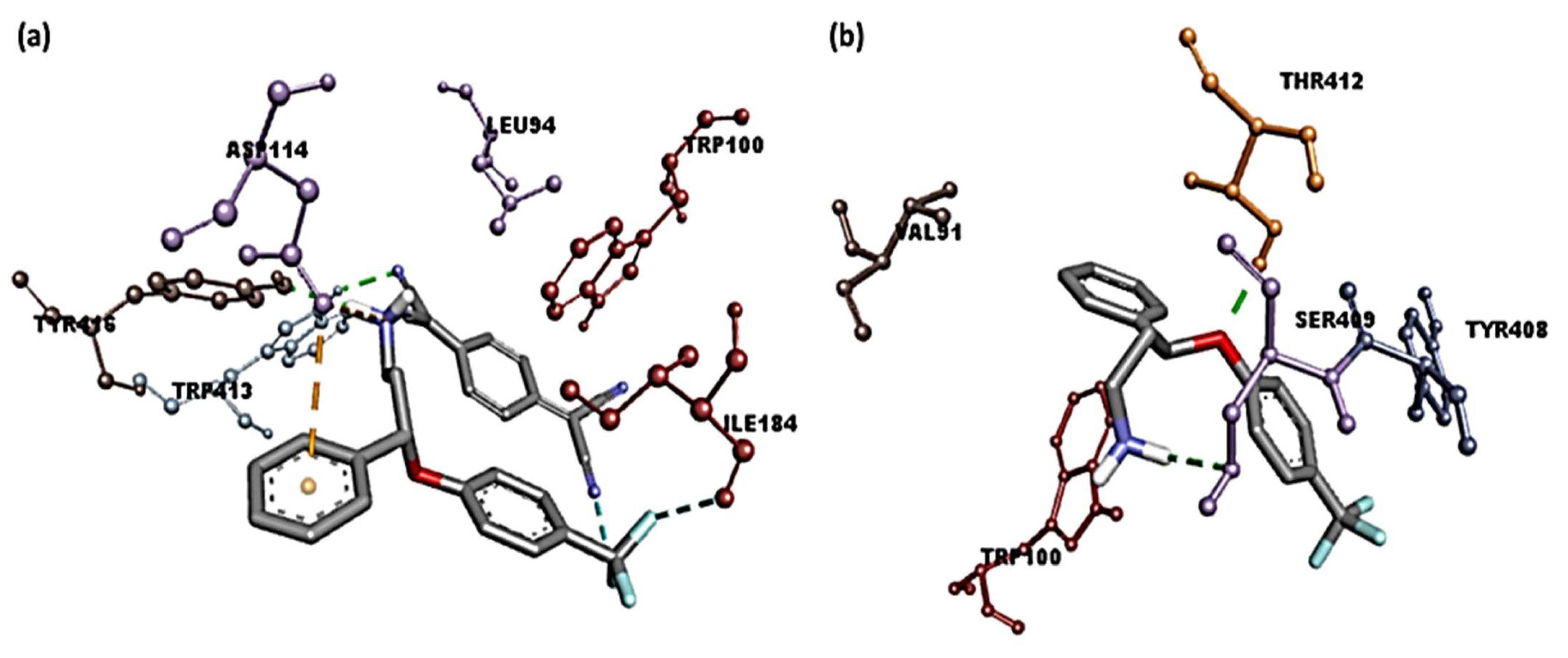
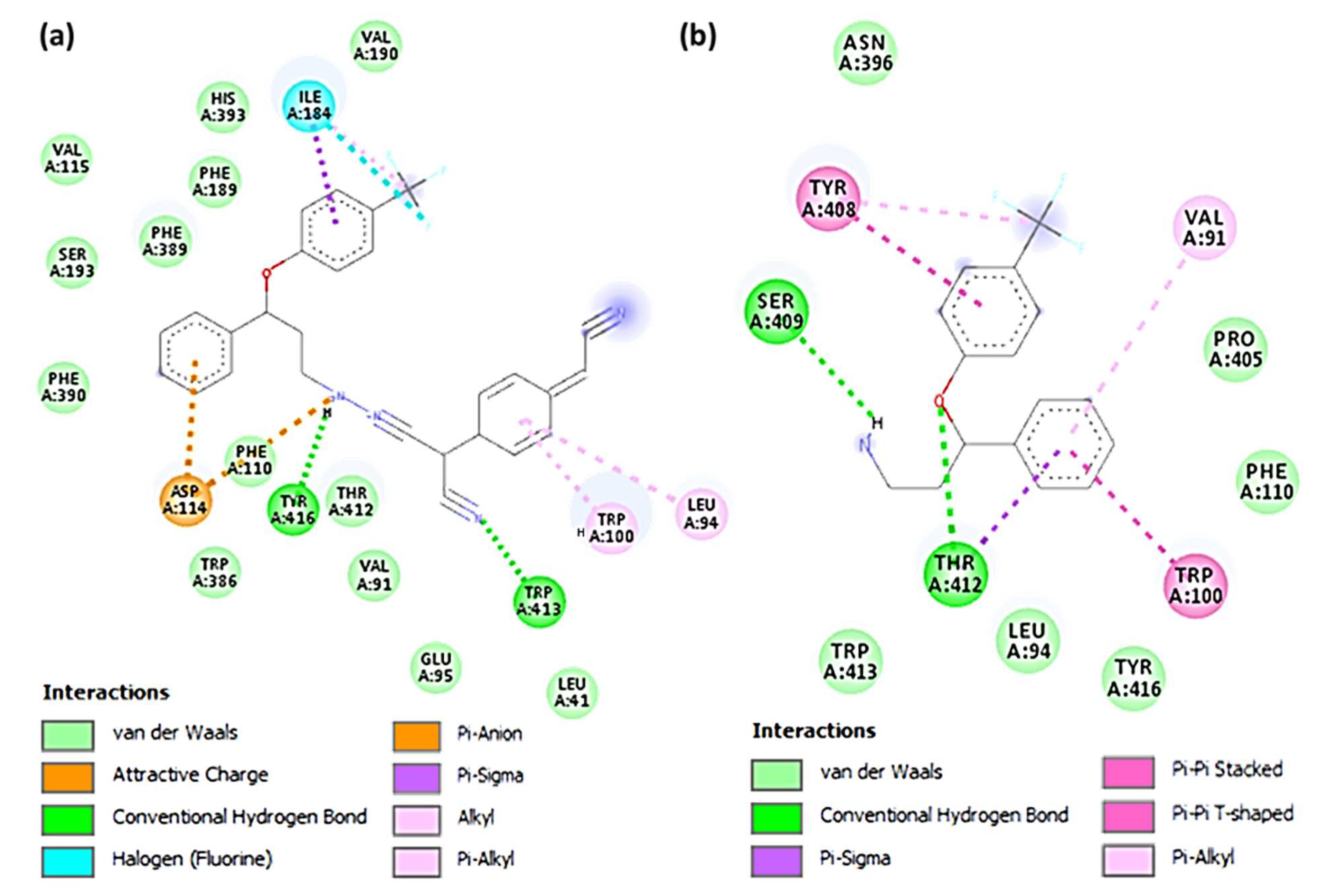
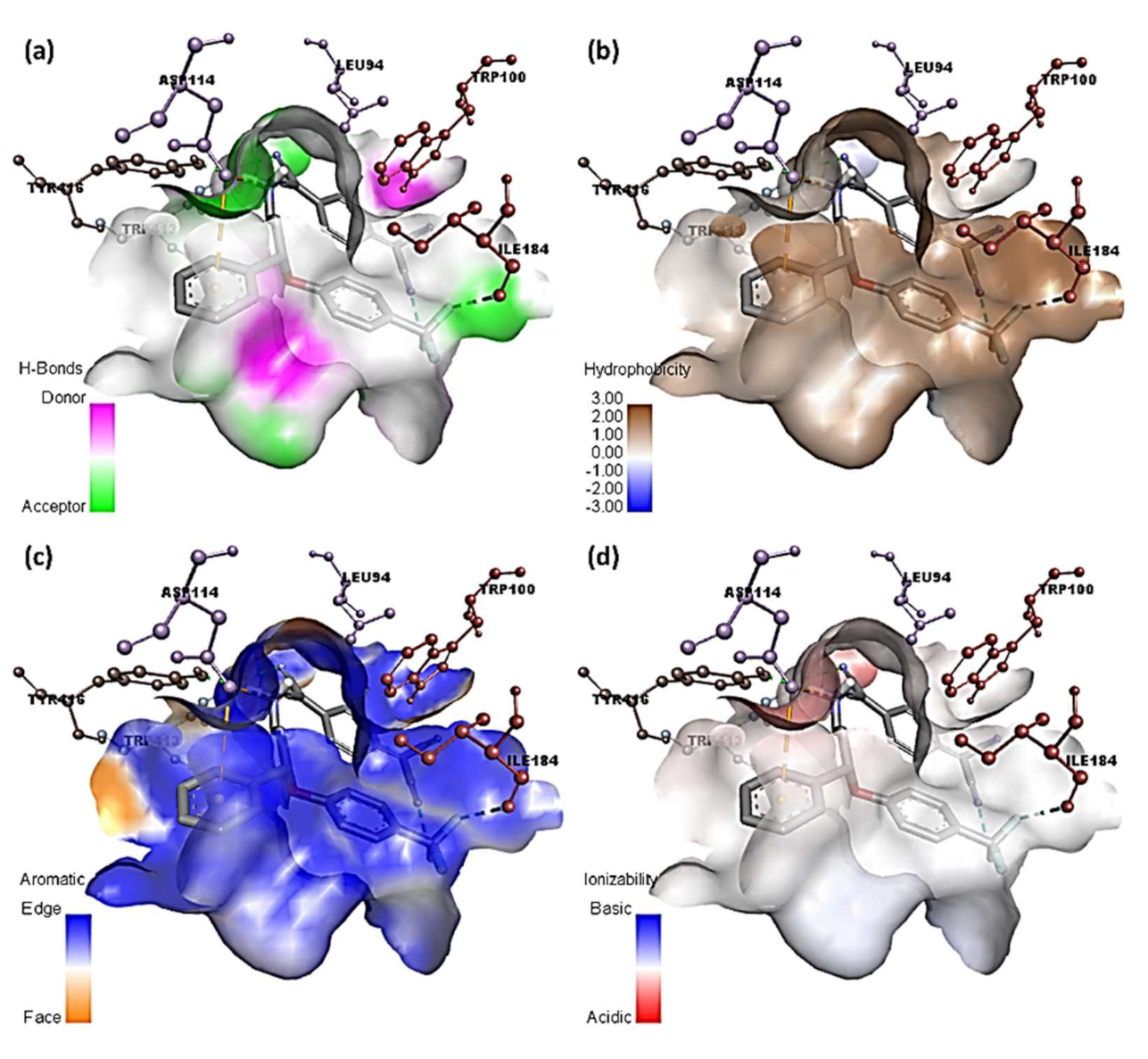
3.4. Molecular Docking Studies
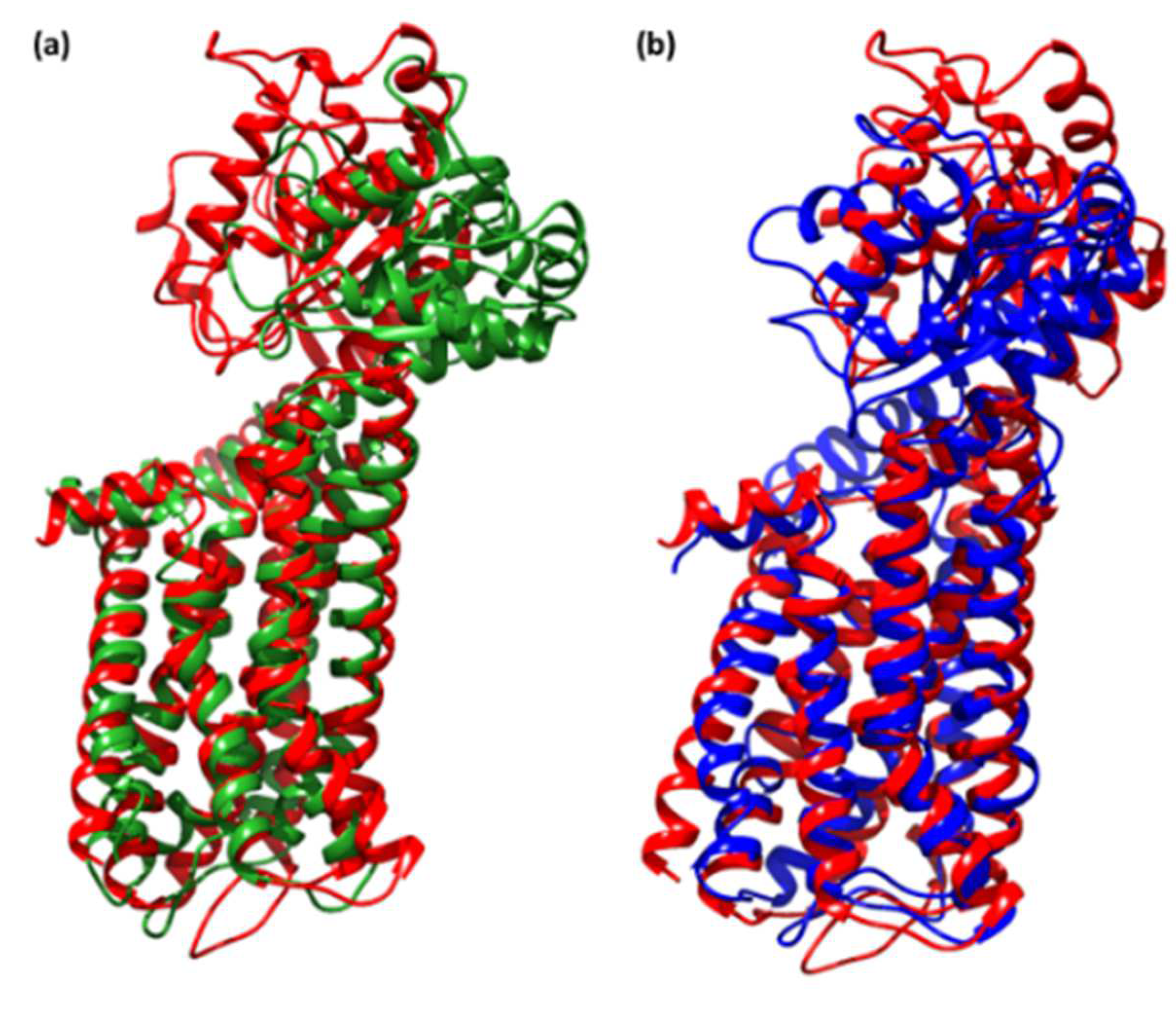
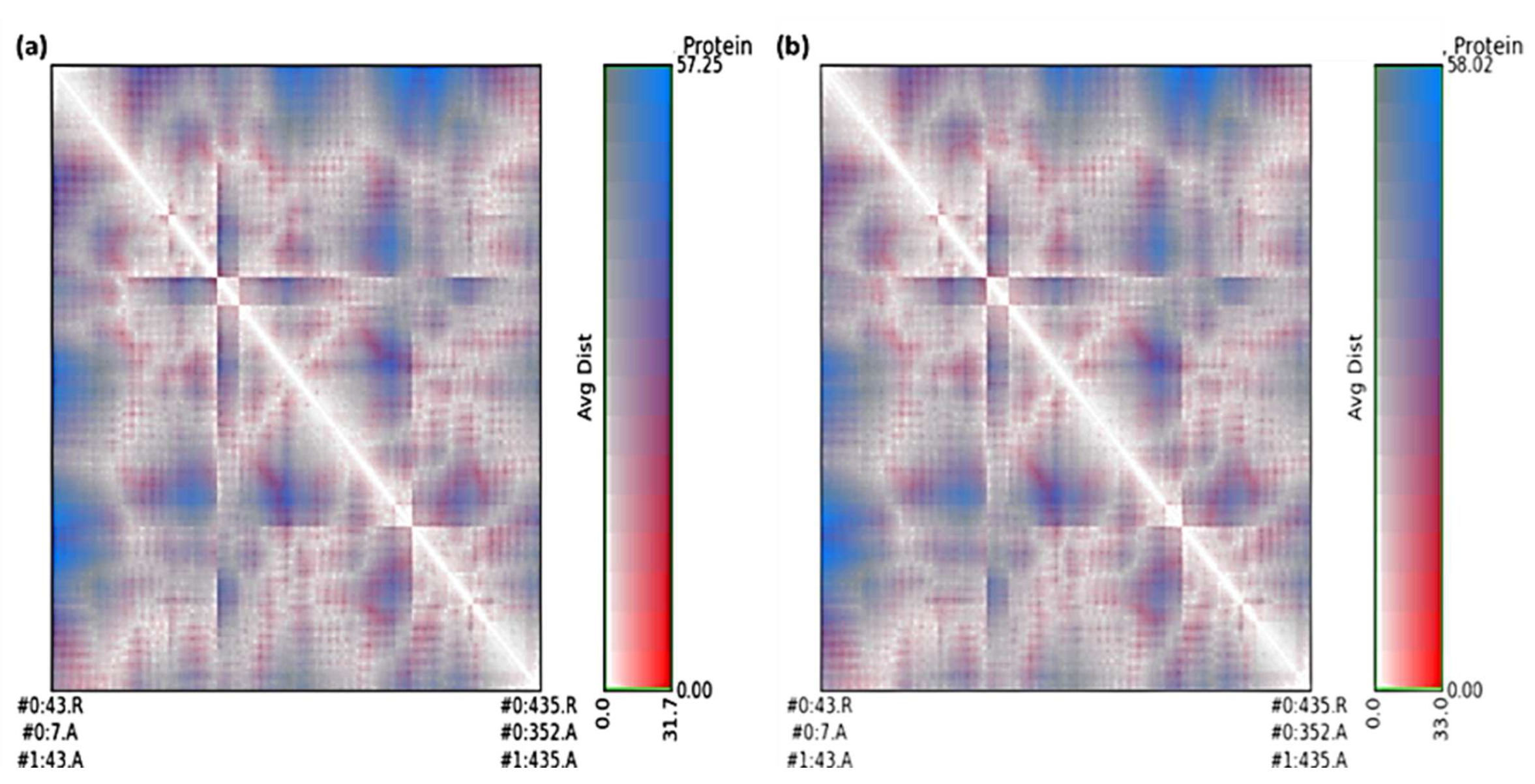
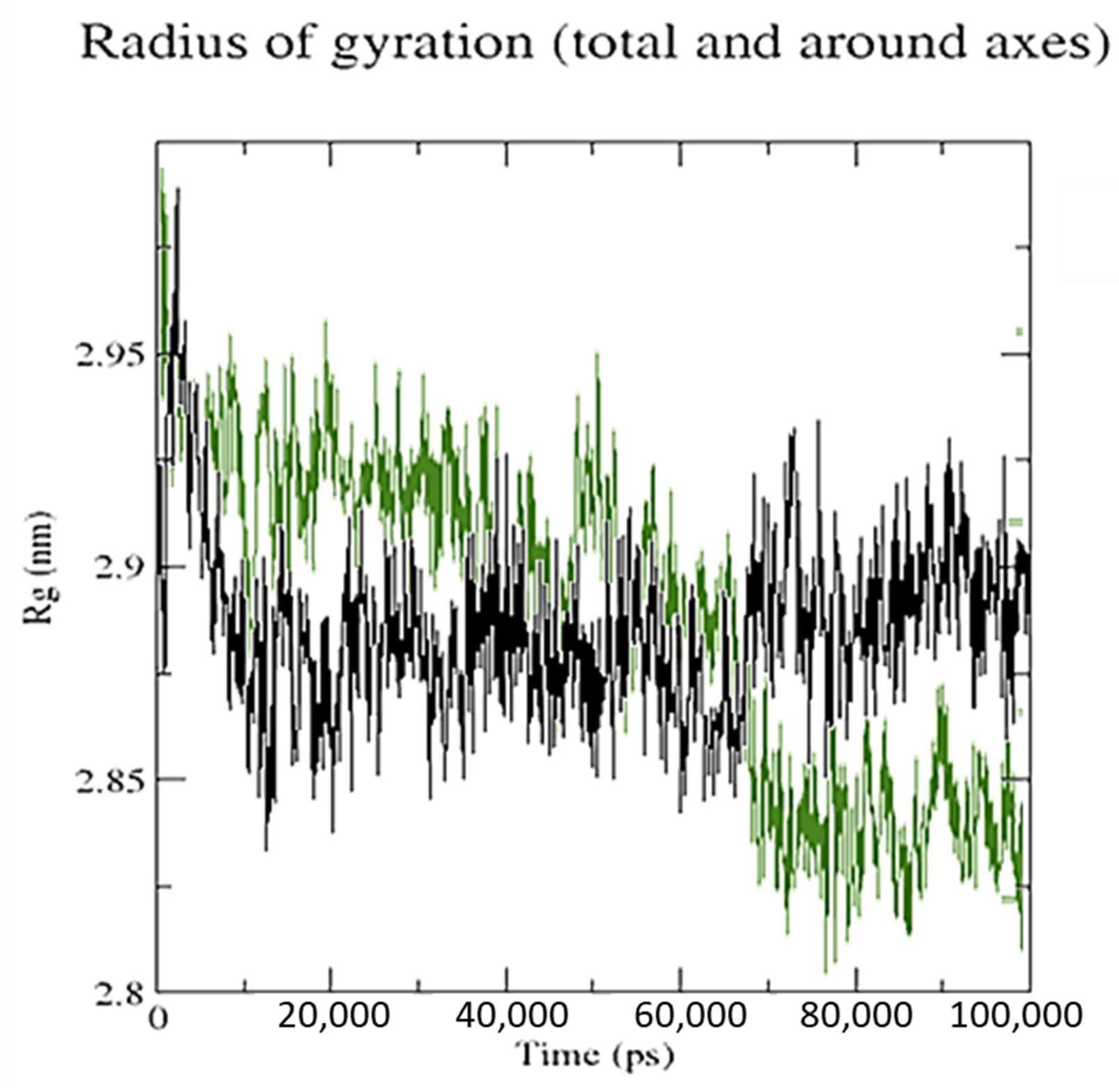
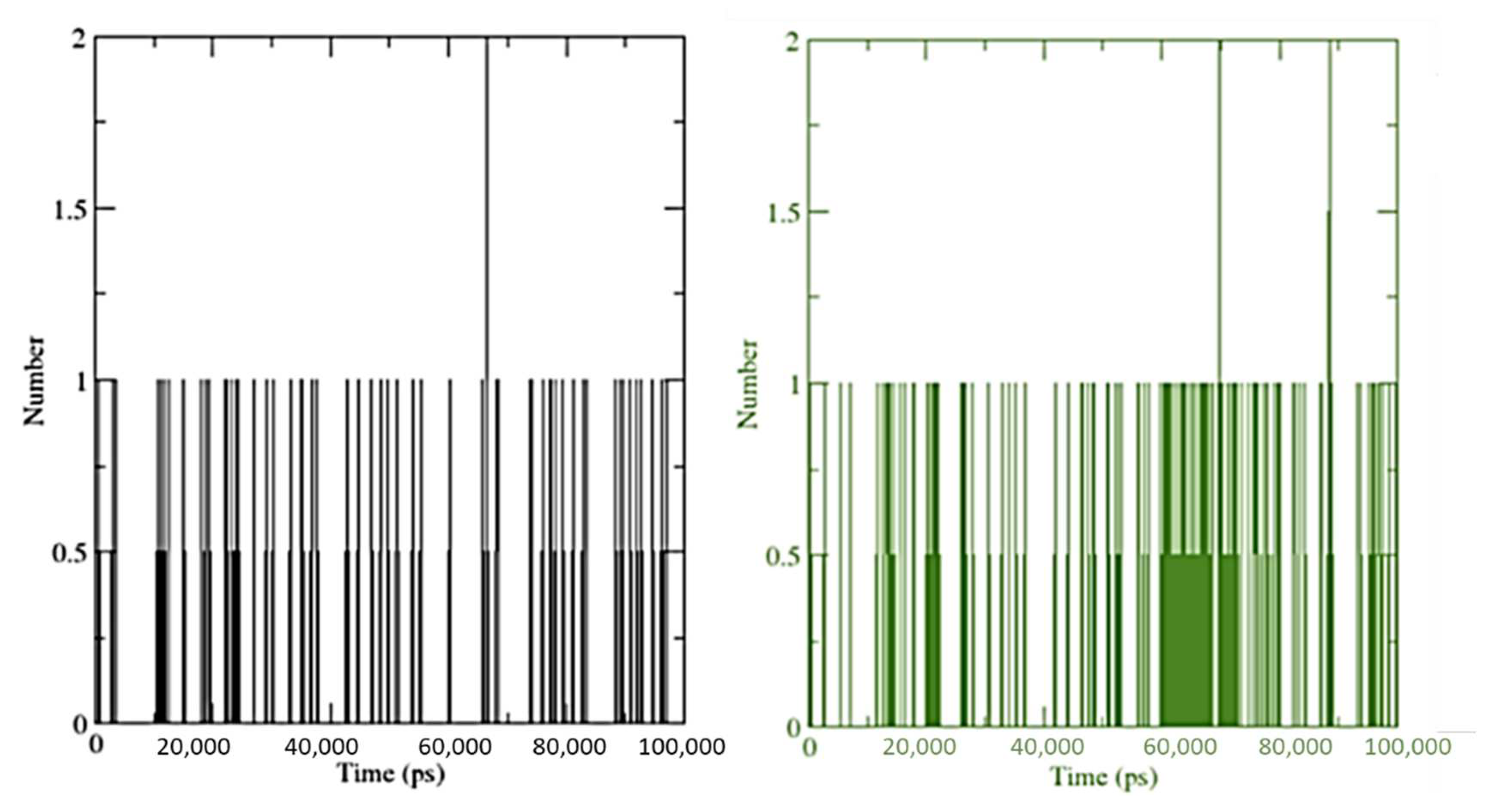
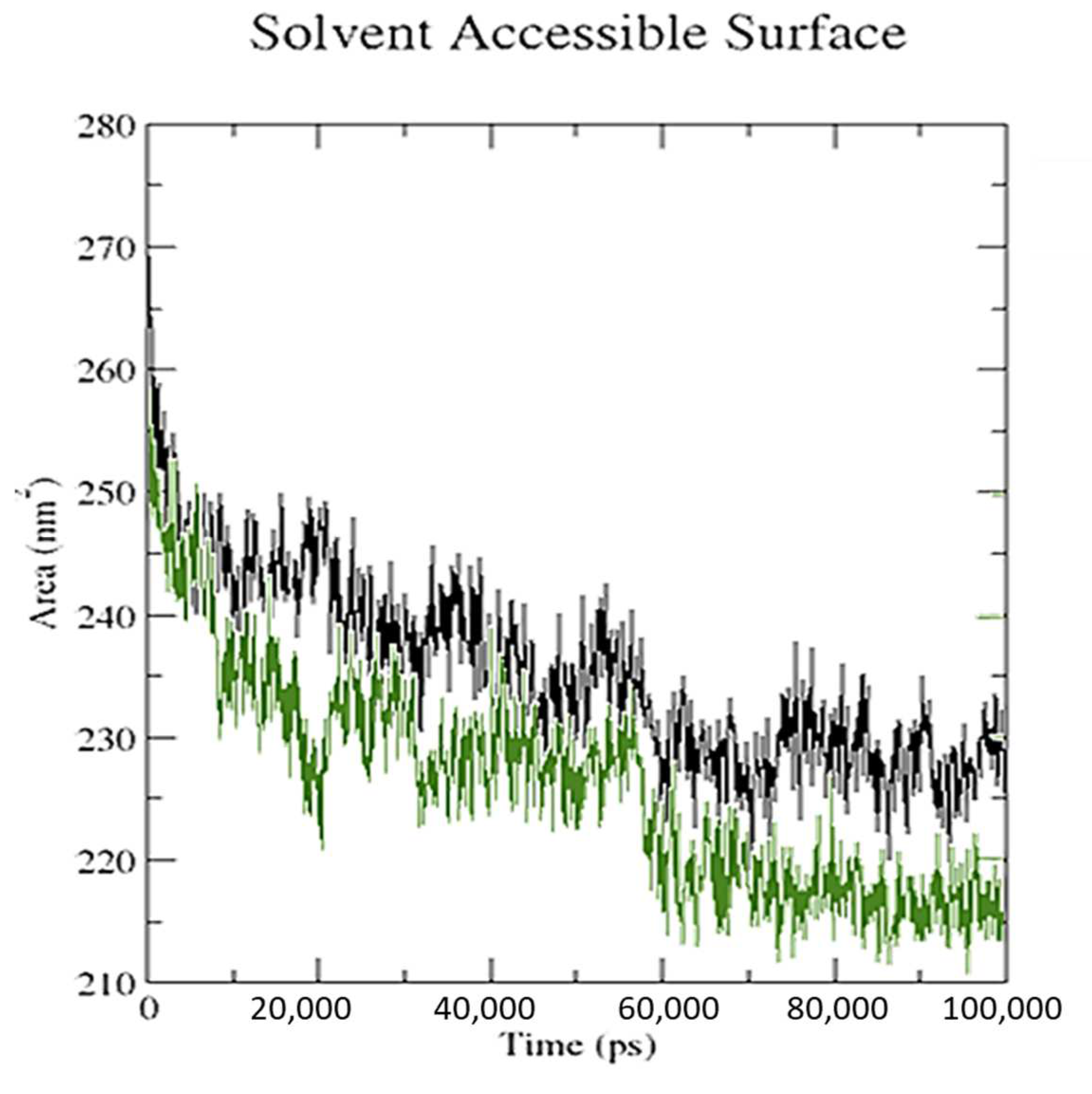
3.5. Molecular Dynamics Simulation
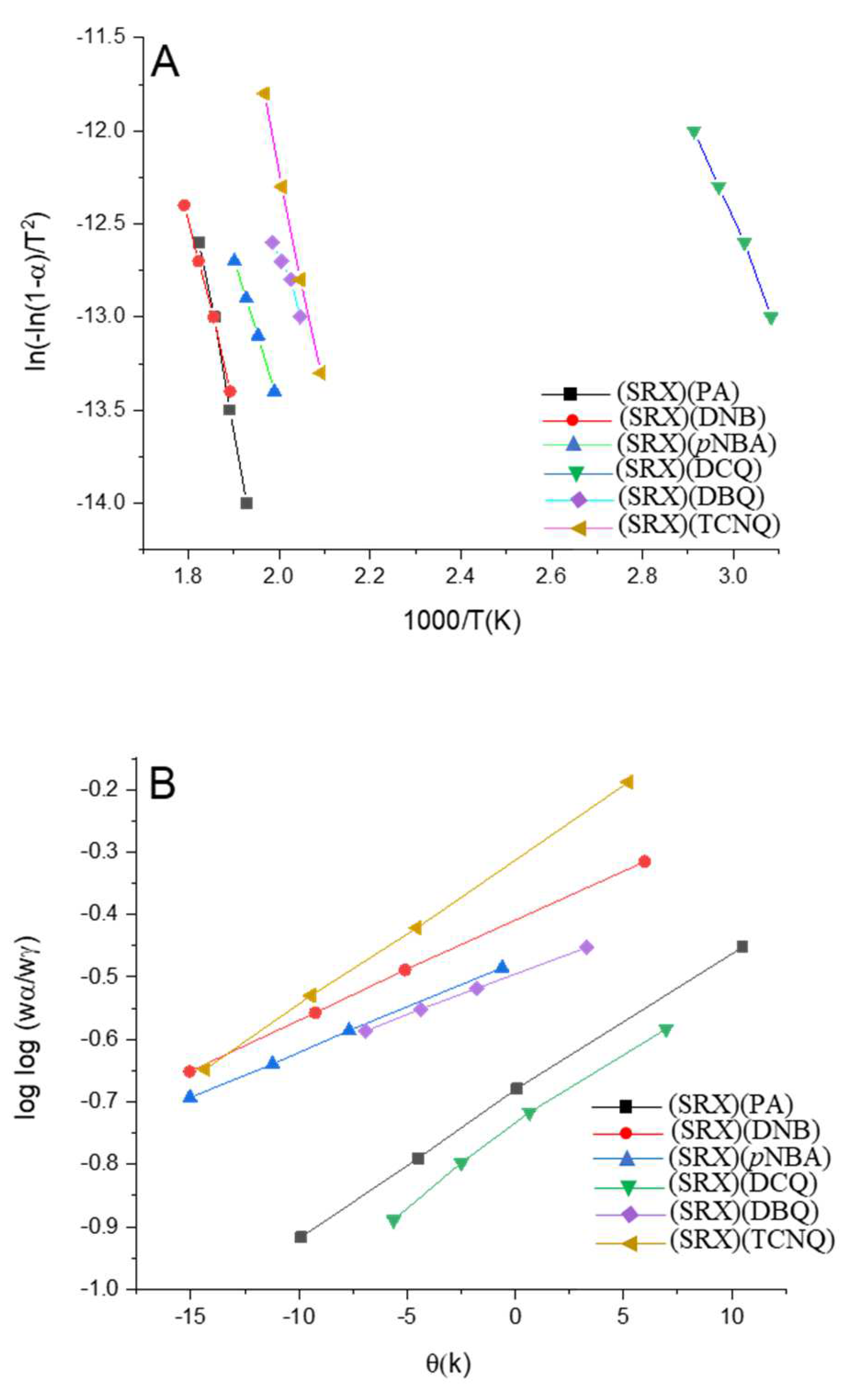
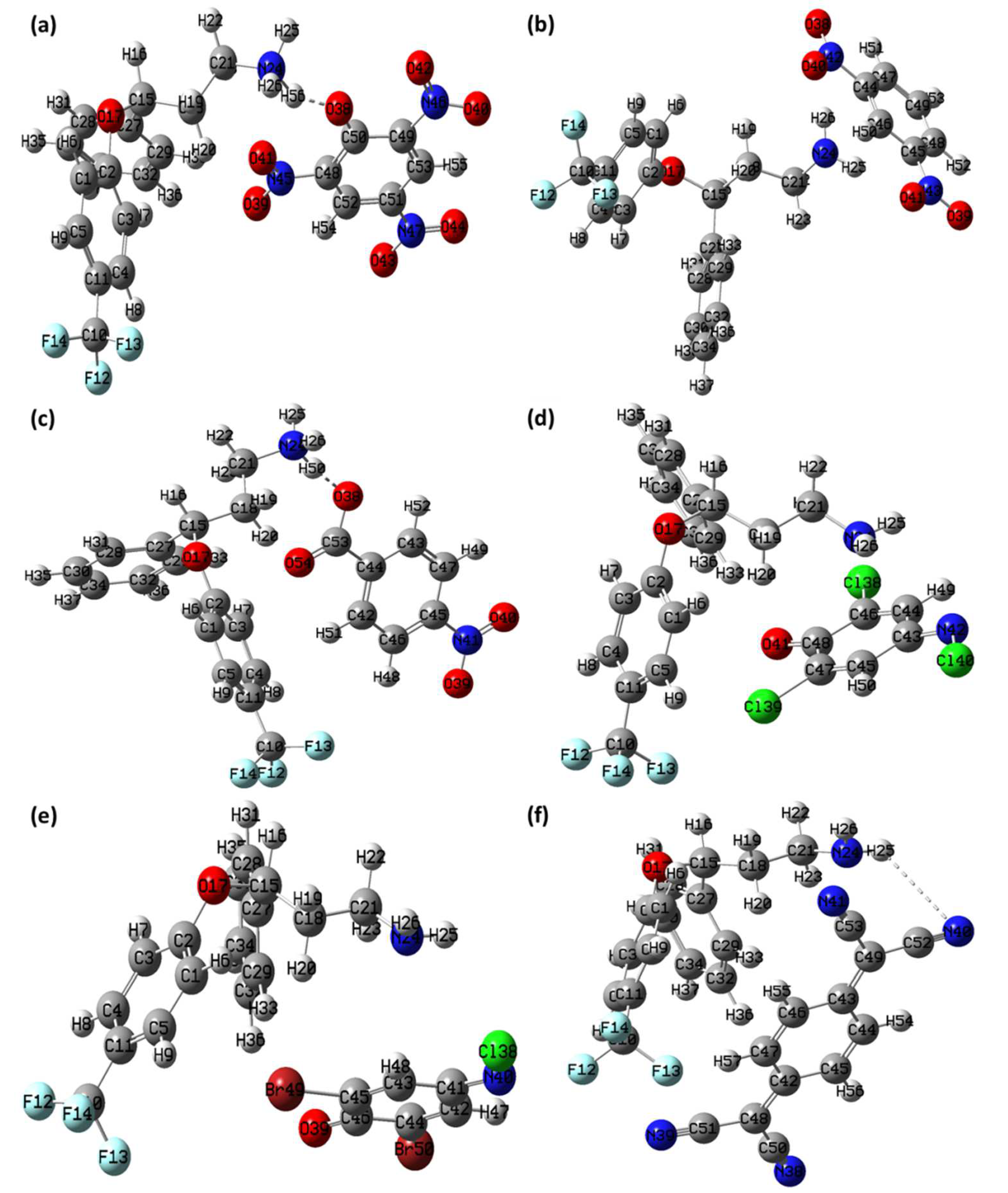
3.6. Theoretical Structural Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Friedrich, M.J. Depression Is the Leading Cause of Disability Around the World. JAMA 2017, 317, 1517. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.S. Depression: The disorder and the burden. Indian J. Psychol. Med. 2010, 32, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yohn, C.N.; Gergues, M.M.; Samuels, B.A. The role of 5-HT receptors in depression. Mol. Brain 2017, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Al-Humaidi, J.Y.; Refat, M.S. Solution, and solid investigations on the charge–transfer complexation between seproxetine as a selective serotonin reuptake inhibitor drug with six kinds of π–electron acceptors. J. Mol. Liq. 2021, 332, 115831. [Google Scholar] [CrossRef]
- Hyttel, J. Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs). Int. Clin. Psychopharmacol. 1994, 9 (Suppl. S1), 19–26. [Google Scholar] [CrossRef]
- Van Oekelen, D.; Luyten, W.H.; Leysen, J.E. 5-HT2A and 5-HT2C receptors and their atypical regulation properties. Life Sci. 2003, 72, 2429–2449. [Google Scholar] [CrossRef]
- Horton, J.R.; Liu, X.; Wu, L.; Zhang, K.; Shanks, J.; Zhang, X.; Rai, G.; Mott, B.T.; Jansen, D.J.; Kales, S.C.; et al. Insights into the Action of Inhibitor Enantiomers against Histone Lysine Demethylase 5A. J. Med. Chem. 2018, 61, 3193–3208. [Google Scholar] [CrossRef]
- Rajamani, S.; Eckhardt, L.L.; Valdivia, C.R.; Klemens, C.A.; Gillman, B.M.; Anderson, C.L.; Holzem, K.M.; Delisle, B.P.; Anson, B.D.; Makielski, J.C.; et al. Drug-induced long QT syndrome: HERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br. J. Pharmacol. 2006, 149, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Adam, A.A.; Hegab, M.S.; Refat, M.S.; Eldaroti, H.H. Proton-transfer and charge-transfer interactions between the antibiotic trimethoprim and several σ- and π-acceptors: A spectroscopic study. J. Mol. Struct. 2021, 1231, 129687. [Google Scholar] [CrossRef]
- Adam, A.A.; Saad, H.A.; Alsuhaibabi, A.M.; Refat, M.S.; Hegab, M.S. Charge-transfer chemistry of azithromycin, the antibiotic used worldwide to treat the coronavirus disease (COVID-19). Part I: Complexation with iodine in different solvents. J. Mol. Struct. 2021, 325, 115187. [Google Scholar] [CrossRef]
- Al-Humaidi, J.Y.; El-Sayed, M.Y.; Refat, M.S.; Altalhi, T.A.; Eldaroti, H.H. Spectrophotometric studies on the charge transfer interactions between thiazolidine as a donor and three π-acceptors: P-chloranil (CHL), DDQ and TCNQ. J. Mol. Struct. 2021, 333, 115928. [Google Scholar] [CrossRef]
- Khan, I.M.; Islam, M.; Shakya, S.; Alam, K.; Alam, N.; Shahid, M. Synthesis, characterization, antimicrobial and DNA binding properties of an organic charge transfer complex obtained from pyrazole and chloranilic acid. Bioorg. Chem. 2020, 99, 103779. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.M.; Shakya, S.; Akhtar, R.; Alam, K.; Islam, M.; Alam, N. Exploring interaction dynamics of designed organic cocrystal charge transfer complex of 2-hydroxypyridine and oxalic acid with human serum albumin: Single crystal, spectrophotometric, theoretical and antimicrobial studies. Bioorg. Chem. 2020, 100, 103872. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, A.; Bandyopadhyay, P.; Banerjee, S.; Mandal, N.C.; Singh, B. Synthesis, spectroscopic, theoretical and antimicrobial studies on molecular charge-transfer complex of 4-(2-thiazolylazo) resorcinol (TAR) with 3, 5-dinitrosalicylic acid, picric acid, and chloranilic acid. J. Mol. Liq. 2020, 299, 112217. [Google Scholar] [CrossRef]
- Kosower, E.M. The Solvent Sensitivity of the Charge-Transfer Band of Tropylium Iodide. J. Org. Chem. 1964, 29, 956. [Google Scholar] [CrossRef]
- Refat, M.S.; Ibrahim, O.B.; Saad, H.A.; Adam, A.M.A. Usefulness of charge–transfer complexation for the assessment of sympathomimetic drugs: Spectroscopic properties of drug ephedrine hydrochloride complexed with some π-acceptors. J. Mol. Struct. 2014, 1064, 58–69. [Google Scholar] [CrossRef]
- McCormick, D.B.; Li, H.-C.; Mackenzie, R.E. Spectral evidence for the interaction of riboflavin with aromatic hydrocarbons. Spectrochim. Acta 1967, 23, 2353–2358. [Google Scholar] [CrossRef]
- Czent-Gyorgyi, A.; Isenberg, I. On the electron-donating properties of indoles. Proc. Natl. Acad. Sci. USA 1960, 46, 1334. [Google Scholar] [CrossRef] [Green Version]
- Isenberg, I.; Czent-Gyorgyi, A. On charge transfer complexes between substances of biochemical interest. Proc. Natl. Acad. Sci. USA 1959, 45, 1229. [Google Scholar] [CrossRef] [Green Version]
- Fleischman, D.E.; Tollin, G. Molecular complexes of flavins and phenols I. Absorption spectra and properties in solution. Biochim. Biophys. Acta 1965, 94, 248–257. [Google Scholar] [CrossRef]
- Massy, V.; Palmer, G. Charge transfer complexes of lipoyl dehydrogenase and free flavins. J. Biol. Chem. 1962, 237, 2374. [Google Scholar] [CrossRef]
- Cilento, G.; Sanioto, D.L. Electron Transfer from Polycyclic Aromatic Hydrocarbons to Menadione. Ber. Bunesnges Physik. Chem. 1963, 67, 426. [Google Scholar] [CrossRef]
- Cilento, G.; Tedeschi, P.J. Pyridine Coenzymes: IV. Charge Transfer Interaction with the Indole Nucleus. Biol. Chem. 1961, 236, 907–910. [Google Scholar] [CrossRef]
- Wilcox, P.E.; Cohen, E.; Wen Tans, J. Amino acid composition of α-chymotrypsinogen, including estimation of asparagine and glutamine. Biol. Chem. 1957, 228, 999–1019. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Dallakyan, S. PyRx-Python Prescription v. 0.8; The Scripps Research Institute: La Jolla, CA, USA, 2008. [Google Scholar]
- Chu, C.-H.; Li, K.-M.; Lin, S.-W.; Chang, M.D.-T.; Jiang, T.-Y.; Sun, Y.-J. Crystal structures of starch binding domain from Rhizopus oryzae glucoamylase in complex with isomaltooligosaccharide: Insights into polysaccharide binding mechanism of CBM21 family. Proteins Struct. Funct. Bioinform. 2014, 82, 1079–1085. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; He, X.; Vanommeslaeghe, K.; MacKerell, A.D., Jr. Extension of the CHARMM General Force Field to sulfonyl containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulations of Liquids; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, P.J.; Brooks, B.R. New Spherical-Cutoff Methods for Long-Range Forces in Macromolecular Simulation. J. Comput. Chem. 1994, 15, 667–683. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 28–33. [Google Scholar] [CrossRef]
- DeLano, W.L. PyMOL; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]
- Zhurko, G.A.; Zhurko, D.A. Chemcraft—Graphical Program for Visualization of Quantum Chemistry Computations; Academic Version 1.5; Chemcraft: Ivanovo, Russia, 2004. [Google Scholar]
- Ilangovan, R.; Subha, V.; Ravindran, R.E.; Kirubanandan, S.; Renganathan, S. Chapter 2—Nanomaterials: Synthesis, physicochemical characterization, and biopharmaceutical applications. In Nanoscale Processing; Elsevier: Amsterdam, The Netherlands, 2021; pp. 33–70. [Google Scholar]
- El-Mossalamy, E.H.; Batouti, M.E.; Fetouh, H.A. The role of natural biological macromolecules: Deoxyribonucleic and ribonucleic acids in the formulation of new stable charge transfer complexes of thiophene Schiff bases for various life applications. Int. J. Biol. Macromol. 2021, 193, 1572–1586. [Google Scholar] [CrossRef]
- Abdallah, A.M.; Frag, E.Y.; Tamam, R.H.; Mohamed, G.G. Gliclazide charge transfer complexes with some benzoquinone acceptors: Synthesis, structural characterization, thermal analyses, DFT studies, evaluation of anticancer activity and utility for determination of gliclazide in pure and dosage forms. J. Mol. Struct. 2021, 1234, 130153. [Google Scholar] [CrossRef]
- Niranjani, S.; Nirmala, C.B.; Rajkumar, P.; Serdaroğlu, G.; Jayaprakash, N.; Venkatachalam, K. Synthesis, characterization, biological and DFT studies of charge-transfer complexes of antihyperlipidemic drug atorvastatin calcium with Iodine, Chloranil, and DDQ. J. Mol. Liq. 2022, 346, 117862. [Google Scholar] [CrossRef]
- Yu, Y.; Liang, G. Interaction mechanism of phenolic acids and zein: A spectrofluorometric and molecular dynamics investigation. J. Mol. Liq. 2022, 348, 118032. [Google Scholar] [CrossRef]
- Samir, A.; Salem, H.; Abdelkawy, M. Optimization of two charge transfer reactions for colorimetric determination of two beta 2 agonist drugs, salmeterol xinafoate and salbutamol, in pharmaceutical and biological samples. Spectrochim. Acta Mol. Biomol. Spectrosc. 2022, 269, 120747. [Google Scholar] [CrossRef]
- Al Rabiah, H.; Yousef, T.A.; Al-Gamal, A.; Homoda, A.M.; Mostafa, G.A. Tamoxifen charge transfer complexes with 2, 3-dichloro-5, 6-dicyano-1, 4-benzoquinone and 7, 7, 8, 8-tetracyanoquinodimethan: Synthesis, spectroscopic characterization and theoretical study. Bioorg. Chem. 2022, 120, 105603. [Google Scholar]
- Khalil, T.E.; Elbadawy, H.A.; Attia, A.A.; El-Sayed, D.S. Synthesis, spectroscopic, and computational studies on molecular charge-transfer complex of 2-((2-hydroxybenzylidene) amino)-2-(hydroxymethyl) propane-1, 3-diol with chloranilic acid: Potential antiviral activity simulation of CT-complex against SARS-CoV-2. J. Mol. Struct. 2022, 1251, 132010. [Google Scholar] [CrossRef] [PubMed]
- Niranjani, S.; Venkatachalam, K. Synthesis, spectroscopic, thermal, structural investigations and biological activity studies of charge-transfer complexes of atorvastatin calcium with dihydroxy-p-benzoquinone, quinalizarin and picric acid. J. Mol. Struct. 2020, 1219, 128564. [Google Scholar] [CrossRef]
- Adam, A.A.; Refat, M.S.; Altalhi, T.A.; Aldawsari, F.S.; Al-Hazmi, G.H. Liquid- and solid-state study of charge-transfer (CT) interaction between drug triamterene as a donor and tetracyanoethylene (TCNE) as an acceptor. J. Mol. Liq. 2021, 336, 116261. [Google Scholar] [CrossRef]
- Adam, A.A.; Refat, M.S. Analysis of charge-transfer complexes caused by the interaction of the antihypertensive drug valsartan with several acceptors in CH2Cl2 and CHCl3 solvents and correlations between their spectroscopic parameters. J. Mol. Liq. 2022, 348, 118466. [Google Scholar] [CrossRef]
- Usmana, R.; Khan, A.; Tang, H.; Ma, D.; Alsuhaibani, A.M.; Refat, M.S.; Adnan; Ara, N.; Fan, H.-J.S. Charge Transfer and Hydrogen Bonding Motifs in Organic Cocrystals Derived from Aromatic Diamines and TCNB. J. Mol. Struct. 2022, 1254, 132360. [Google Scholar] [CrossRef]
- Durgadevi, R.; Suvitha, A.; Arumanayagam, T. Growth, optical, electrical properties and DFT studies on piperidinium 4-nitrophenolate NLO single crystal in acetone. J. Cryst. Growth 2022, 582, 126512. [Google Scholar] [CrossRef]
- Coats, A.W.; Redfern, J.P. Kinetic Parameters from Thermogravimetric Data. Nat. Lett. 1964, 201, 68–69. [Google Scholar] [CrossRef]
- Horowitz, H.H.; Metzger, G.A. A New Analysis of Thermogravimetric Traces. Anal. Chem. 1963, 35, 1464–1468. [Google Scholar] [CrossRef]
- Akram, M.; Lal, H.; Shakya, S.; Kabir-ud-Din. Multispectroscopic and Computational Analysis Insight into the Interaction of Cationic Diester-Bonded Gemini Surfactants with Serine Protease α-Chymotrypsin. ACS Omega 2020, 5, 3624–3637. [Google Scholar] [CrossRef]
- Tseng, T.C.; Urban, C.; Wang, Y.; Otero, R.; Tait, S.L.; Alcamí, M.; Ecija, D.; Trelka, M.; Gallego, J.M.; Lin, N.; et al. Charge-transfer-induced structural rearrangements at both sides of organic/metal interfaces. Nat. Chem. 2010, 2, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Garrido, E.M.; Garrido, J.; Calheiros, R.; Marques, M.P.; Borges, F. Fluoxetine and norfluoxetine revisited: New insights into the electrochemical and spectroscopic properties. J. Phys. Chem. A 2009, 113, 9934–9944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.M.; Islam, M.; Shakya, S.; Alam, N.; Imtiaz, S.; Islam, M.R. Synthesis, spectroscopic characterization, antimicrobial activity, molecular docking and DFT studies of proton transfer (H-bonded) complex of 8-aminoquinoline (donor) with chloranilic acid (acceptor). J. Biomol. Struct. Dyn. 2021, 1–15. [Google Scholar] [CrossRef]
- Dunlop, B.W.; Nemeroff, C.B. The role of dopamine in the pathophysiology of depression. Arch. Gen. Psychiatry 2007, 64, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Peng, Z.; Anishchenko, I.; Cong, Q.; Baker, D.; Yang, J. Protein contact prediction using metagenome sequence data and residual neural networks. Bioinformatics 2020, 36, 41–48. [Google Scholar] [CrossRef]
- Chen, J.E.; Huang, C.C.; Ferrin, T.E. RRDistMaps: A UCSF Chimera tool for viewing and comparing protein distance maps. Bioinformatics 2015, 31, 1484–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, D.S.; Colwell, L.J.; Sheridan, R.; Hopf, T.A.; Pagnani, A.; Zecchina, R.; Sander, C. Protein 3D structure computed from evolutionary sequence variation. PLoS ONE 2011, 6, e28766. [Google Scholar] [CrossRef] [Green Version]
- Kavitha, R.; Nirmala, S.; Nithyabalaji, R.; Sribalan, R. Biological evaluation, molecular docking and DFT studies of charge transfer complexes of quinaldic acid with heterocyclic carboxylic acid. J. Mol. Struct. 2020, 1204, 127508. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Method | Parameter | r | ||||
|---|---|---|---|---|---|---|---|
| E (kJol−1) | A (s−1) | ΔS (J mol−1 K−1) | ΔH (kJ mol−1) | ΔG (kJ mol−1) | |||
| (SRX)(PA) | CR | 11.5 × 104 | 4.00 × 108 | −8.52 × 101 | 1.12 × 105 | 1.54 × 105 | 0.9990 |
| HM | 11.2 × 104 | 5.60 × 109 | −6.32 × 101 | 1.12 × 105 | 1.50 × 105 | 0.9989 | |
| (SRX)(DNB) | CR | 7.80 × 104 | 1.50 × 105 | −1.55 × 102 | 7.25 × 104 | 1.47 × 105 | 0.9980 |
| HM | 8.65 × 104 | 1.34 × 105 | −1.30 × 102 | 8.12 × 104 | 1.44 × 105 | 0.9989 | |
| (SRX)(pNBA) | CR | 6.38 × 104 | 1.32 × 104 | −1.72 × 102 | 5.90 × 104 | 1.51 × 105 | 0.9995 |
| HM | 7.23 × 104 | 1.22 × 104 | −1.56 × 102 | 6.71 × 104 | 1.54 × 105 | 0.9985 | |
| (SRX)(DCQ) | CR | 4.80 × 104 | 1.25 × 105 | −1.45 × 102 | 4.43 × 104 | 9.40 × 104 | 0.9943 |
| HM | 5.22 × 104 | 1.85 × 106 | −1.32 × 102 | 4.68 × 104 | 9.22 × 104 | 0.9987 | |
| (SRX)(DBQ) | CR | 5.77 × 104 | 5.12 × 103 | −1.85 × 102 | 5.22 × 104 | 1.45 × 105 | 0.9890 |
| HM | 6.35 × 104 | 2.75 × 104 | −1.72 × 102 | 5.90 × 104 | 1.40 × 105 | 0.9994 | |
| (SRX)(TCNQ) | CR | 11.1 × 104 | 6.22 × 108 | −8.14 × 101 | 9.72 × 104 | 1.33 × 105 | 0.9984 |
| HM | 11.8 × 104 | 5.50 × 109 | −6.35 × 101 | 1.12 × 105 | 1.42 × 105 | 0.9996 | |
| Receptor | Binding Free Energy (kcal/mol) | ||
|---|---|---|---|
| 6BQH | 6CM4 | 4ASZ | |
| SRX-PA | −7.8 | −9.2 | −8.4 |
| SRX-DNB | −6.8 | −8.3 | −6.5 |
| SRX-pNBA | −8.7 | −7.8 | −7.0 |
| SRX-DCQ | −7.5 | −9.5 | −7.4 |
| SRX-DBQ | −7.9 | −8.1 | −7.5 |
| SRX-TCNQ | −9.4 | −9.9 | −8.2 |
| SRX | −7.4 | −7.3 | −6.0 |
| Receptor | Binding Free Energy (kcal/mol) | Interactions | |
|---|---|---|---|
| H-Bond | Others | ||
| SRX-TCNQ | −9.9 | Tyr416 and Trp413 | Leu94, Trp100 (π-Alkyl); Phe189 (π-Sigma); Asp114 (π-Anion); Ile184 (Halogen-Fluorine) |
| SRX | −7.3 | Ser409 and Thr412 | Tpr100, Val91 (π-Alkyl); Tyr416 (π-Sigma) |
| CT Complex | Minimum SCF Energy (a.u.) | Dipole Moment (Debye) | Electronic Spatial Extent (a.u.) | ΔE (eV) |
|---|---|---|---|---|
| [(SRX)(PA}] | −1958.944644 | 10.500053 | 33,762.8991 | 2.7845 |
| [(SRX)(DNB)] | −1689.608194 | 9.644797 | 20,168.3034 | 3.4449 |
| [(SRX)(p-NBA)] | −1673.728419 | 11.524028 | 26,521.9908 | 3.3189 |
| [(SRX)(DCQ)] | −2788.736562 | 5.693616 | 19,344.1851 | 2.3924 |
| [(SRX)(DBQ)] | −7011.542112 | 5.965700 | 18,542.9710 | 2.4310 |
| [(SRX)(TCNQ)] | −1726.964350 | 5.607618 | 35,156.4199 | 1.8942 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsanie, W.F.; Alamri, A.S.; Alyami, H.; Alhomrani, M.; Shakya, S.; Habeeballah, H.; Alkhatabi, H.A.; Felimban, R.I.; Alzahrani, A.S.; Alhabeeb, A.A.; et al. Increasing the Efficacy of Seproxetine as an Antidepressant Using Charge–Transfer Complexes. Molecules 2022, 27, 3290. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103290
Alsanie WF, Alamri AS, Alyami H, Alhomrani M, Shakya S, Habeeballah H, Alkhatabi HA, Felimban RI, Alzahrani AS, Alhabeeb AA, et al. Increasing the Efficacy of Seproxetine as an Antidepressant Using Charge–Transfer Complexes. Molecules. 2022; 27(10):3290. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103290
Chicago/Turabian StyleAlsanie, Walaa F., Abdulhakeem S. Alamri, Hussain Alyami, Majid Alhomrani, Sonam Shakya, Hamza Habeeballah, Heba A. Alkhatabi, Raed I. Felimban, Ahmed S. Alzahrani, Abdulhameed Abdullah Alhabeeb, and et al. 2022. "Increasing the Efficacy of Seproxetine as an Antidepressant Using Charge–Transfer Complexes" Molecules 27, no. 10: 3290. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103290