
Histone Deacetylase Inhibitory Activity and Antiproliferative Potential of New [6]-Shogaol Derivatives

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
3. Conclusions
4. Experimental Section
4.1. General
4.2. Plant Material
4.3. Structural Modifications
4.3.1. Synthesis of the Immino Derivatives (4a–4e)
4.3.2. Synthesis of the Pyrazole Derivatives (5a–5k)
4.3.3. Synthesis of the Amino Derivatives (6a–6c, 7)
4.4. HDAC Activity Assay
4.5. MTT Assay
4.6. Molecular Docking Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Micelli:, C.; Rastelli, G. Histone deacetylases: Structural determinants of inhibitor selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, S.J.; Woster, P.M.; Shen, J.K.; Georg, G.; Wang, S. Epigenetics: Novel therapeutics targeting epigenetics. J. Med. Chem. 2015, 58, 523–524. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, S.; Yuan, H.; Hu, J.; Zhao, C.; Chai, R.; Cao, H. Design, synthesis and biological evaluation of nitro oxide donating N-hydroxycinnamamide derivatives as histone deacetylase inhibitors. Chem. Phar. Bull. 2014, 62, 1185–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, M.; Porcelloni, M.; Binaschi, M.; Fattori, D. Histone deacetylase inhibitors: From bench to clinic. J. Med. Chem. 2008, 51, 1505–1529. [Google Scholar] [CrossRef]
- Chen, W.; Dong, G.; Wu, Y.; Zhang, W.; Miao, C.; Sheng, C. Dual NAMPT/HDAC inhibitors as a new strategy for multitargeting antitumor drug discovery. ACS Med. Chem. Lett. 2018, 9, 34–38. [Google Scholar] [CrossRef]
- Manal, M.; Chandrasekar, M.J.N.; Priya, J.G.; Nanjan, M.J. Inhibitors of histone deacetylase as antitumor agents: A critical review. Bioorg. Chem. 2016, 67, 18–42. [Google Scholar] [CrossRef]
- Bertrand, P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef]
- Banerjee, S.; Adhikari, N.; Amin, S.A.; Jha, T. Histone deacetylase 8 (HDAC8) and its inhibitors with selectivity to other isoforms: An overview. Eur. J. Med. Chem. 2019, 164, 214–240. [Google Scholar] [CrossRef] [PubMed]
- Ververis, K.; Hiong, A.; Karagiannis, T.C.; Licciardi, P.V. Histone deacetylase inhibitors (HDACIs): Multitargeted anticancer agents. Biologics 2013, 7, 47–60. [Google Scholar] [PubMed] [Green Version]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Bailey, H.; Stenehjem, D.D.; Sharma, S. Panobinostat for the treatment of multiple myeloma: The evidence to date. J. Blood. Med. 2015, 6, 269–276. [Google Scholar] [PubMed] [Green Version]
- Zagni, C.; Floresta, G.; Monciino, G.; Rescifina, A. The search for potent, small-molecule HDACIs in cancer treatment: A decade after Vorinostat. Med. Res. Rev. 2017, 37, 1373–1428. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.M.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B.P. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem, J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Padige, G.; Negmeldin, A.T.; Pflum, M.K.H. Development of an ELISA-based HDAC activity assay for characterization of isoform-selective inhibitors. J. Biomol. Screen. 2015, 20, 1277–1285. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, O.A.; Heaney, M.L.; Schwartz, L.; Richardson, S.; Willim, R.; Cortelli, B.M.; Curly, T.; Moskowitz, C.; Portlock, C.; Horwitz, S.; et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J. Clin. Oncol. 2006, 24, 166–173. [Google Scholar] [CrossRef]
- Hanessian, S.; Vinci, V.; Auzzas, L.; Marzi, M.; Giannini, G. Exploring alternative Zn-binding groups in the design of HDAC inhibitors: Squaric acid, N-hydroxyurea, and oxazoline analogues of SAHA. Bioorg. Med. Chem. Lett. 2006, 16, 4784–4787. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T. Explorative study on isoform-selective histone deacetylase inhibitors. Chem. Phar. Bull. 2009, 57, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estiu, G.; Greenberg, E.; Harrison, C.B.; Kwiatkowski, N.P.; Mazitschek, R.; Bradner, J.E.; Wiest, O. Structural origin of selectivity in class II-selective histone deacetylase inhibitors. J. Med. Chem. 2008, 51, 2898–2906. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Anantharaju, P.G.; Reddy, B.D.; Padukudru, M.A.; Chitturi, C.M.K.; Vimalambike, M.G.; Madhunapantula, S.V. Naturally occurring benzoic acid derivatives retard cancer cell growth by inhibiting histone deacetylases (HDAC). Cancer Biol. Ther. 2017, 18, 492–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
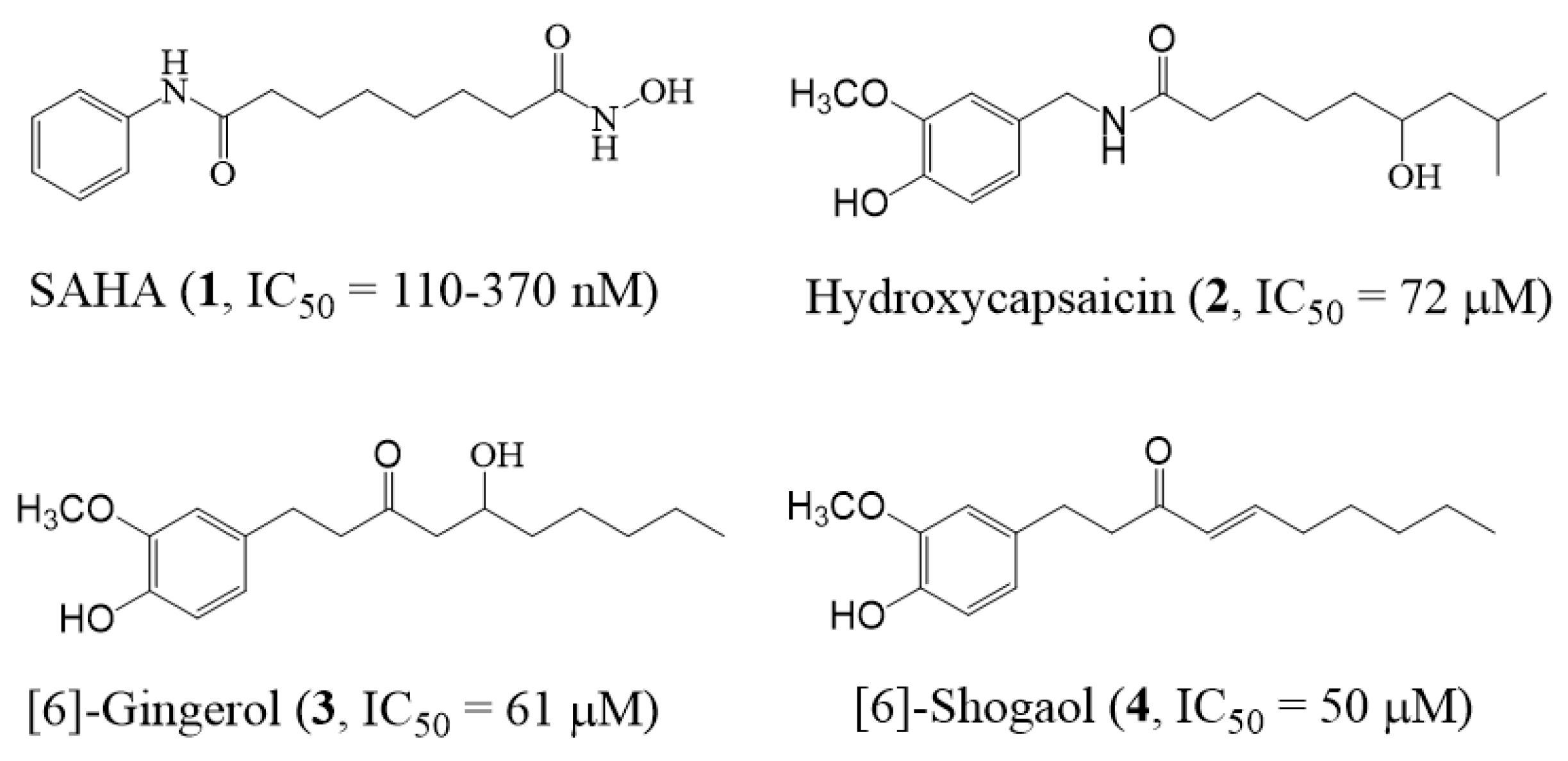
- Senawong, T.; Wongphakham, P.; Saiwichai, T.; Phaosir, C.; Kumboonma, P. Histone deacetylase (HDAC) inhibitory activity of hydroxycapsaicin, a synthetic derivative of capsaicin, and its cytotoxic effects against human colon cancer cell lines. Turk. J. Biol. 2015, 39, 1–10. [Google Scholar] [CrossRef]
- Kumboonma, P.; Senawong, T.; Saenglee, S.; Yenjai, C.; Phaosiri, C. Identification of phenolic compounds from Zingiber offinale and their derivatives as histone deacetylase inhibitors and antioxidant. Med. Chem. Res. 2017, 26, 650–661. [Google Scholar] [CrossRef]
- Suzuki, T.; Miyata, N. Non-hydroxamate histone deacetylase inhibitors. Curr. Med. Chem. 2005, 12, 2867–2880. [Google Scholar] [CrossRef]
- Tatar, G.B.; Erden, D.D.; Demir, A.S.; Dalkara, S.; Yelekci, K.; Yurter, H.E. Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: Activity and docking studies. Bioorg. Med. Chem. 2009, 17, 5219–5228. [Google Scholar] [CrossRef]
- Kumboonma, P.; Saenglee, S.; Senawong, T.; Phaosiri, C. Discovery of new capsaicin and dihydrocapsaicin derivatives as histone deacetylase inhibitors and molecular docking studies. Org. Commun. 2021, 14, 133–143. [Google Scholar] [CrossRef]
- Shukla, Y.; Singh, M. Cancer preventive properties of ginger: A brief review. Food Chem. Toxic. 2007, 45, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.H.H.; Gonda, T.; Hunyadi, A. Medicinal chemistry inspired by ginger: Exploring the chemical space around 6-gingerol. RSC Adv. 2021, 11, 26687–26699. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Doharey, P.K.; Kumar, V.; Saxena, J.K.; Siddiqi, M.I.; Rathaur, S.; Narender, T. Synthesis, molecular docking and brugia malayi thymidylate kinase (bmtmk) enzyme inhibition study of novel derivatives of [6]-shogaol. Eur. J. Med. Chem. 2015, 93, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Kumnerdkhonkaen, P.; Saenglee, S.; Asgar, M.A.; Senawong, G.; Khongsukwiwat, K.; Senawong, T. Antiproliferative activities and phenolic acid content of water and ethanolic extracts of the powdered formula of Houttuynia cordata Thunb. fermented broth and Phyllanthus emblica Linn. fruit. BMC Complement Altern. Med. 2018, 18, 130–132. [Google Scholar] [CrossRef] [Green Version]
- Asgar, M.A.; Senawong, G.; Sripa, B.; Senawong, T. Synergistic anticancer effects of cisplatin and histone deacetylase inhibitors (SAHA and TSA) on cholangiocarcinoma cell lines. Int. J. Oncol. 2016, 48, 409–420. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | %HDAC Inhibition | Compounds | %HDAC Inhibition |
|---|---|---|---|
| TSA | 92 | 5a | 74 |
| 4 | 86 | 5b | 56 |
| 4a | 73 | 5c | 52 |
| 4b | 73 | 5d | 67 |
| 4c * | 80 | 5e | 54 |
| 4d * | 80 | 5f | 77 |
| 4e | 73 | 5g | 60 |
| 6a | 54 | 5h | 68 |
| 6b | 64 | 5i | 77 |
| 6c | 53 | 5j * | 85 |
| 7 | 63 | 5k * | 83 |
| Cp | HDAC1 | HDAC2 | ||||
| ∆G * | Ki ** | Binding Residues | ∆G * | Ki ** | Binding Residues | |
| TSA | −8.12 | 1.12 | Zn, His28, Asp99, Gly149, Phe205 | −8.75 | 0.39 | Zn, Gly32, His33, Asp104, Gly154 |
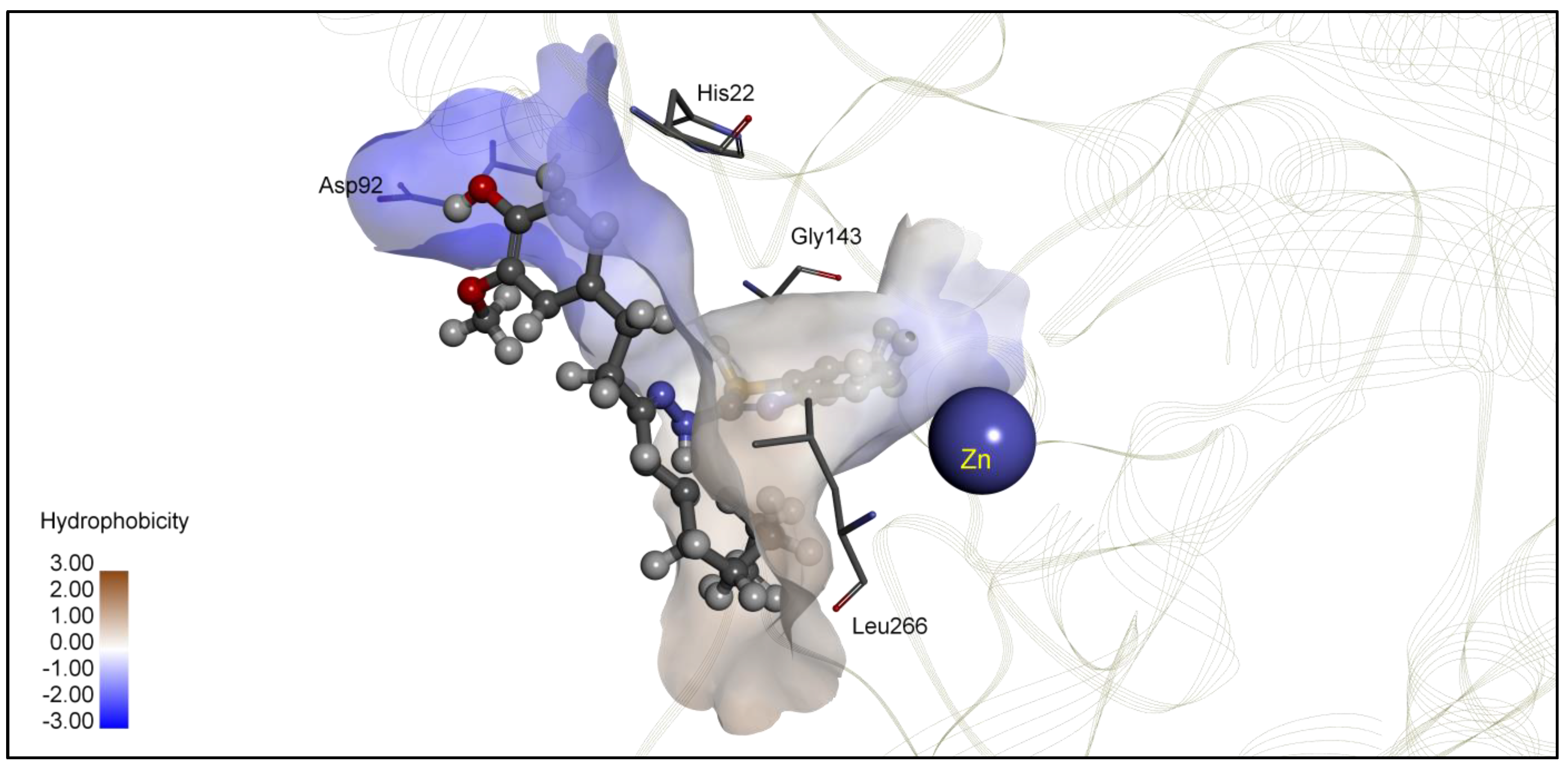
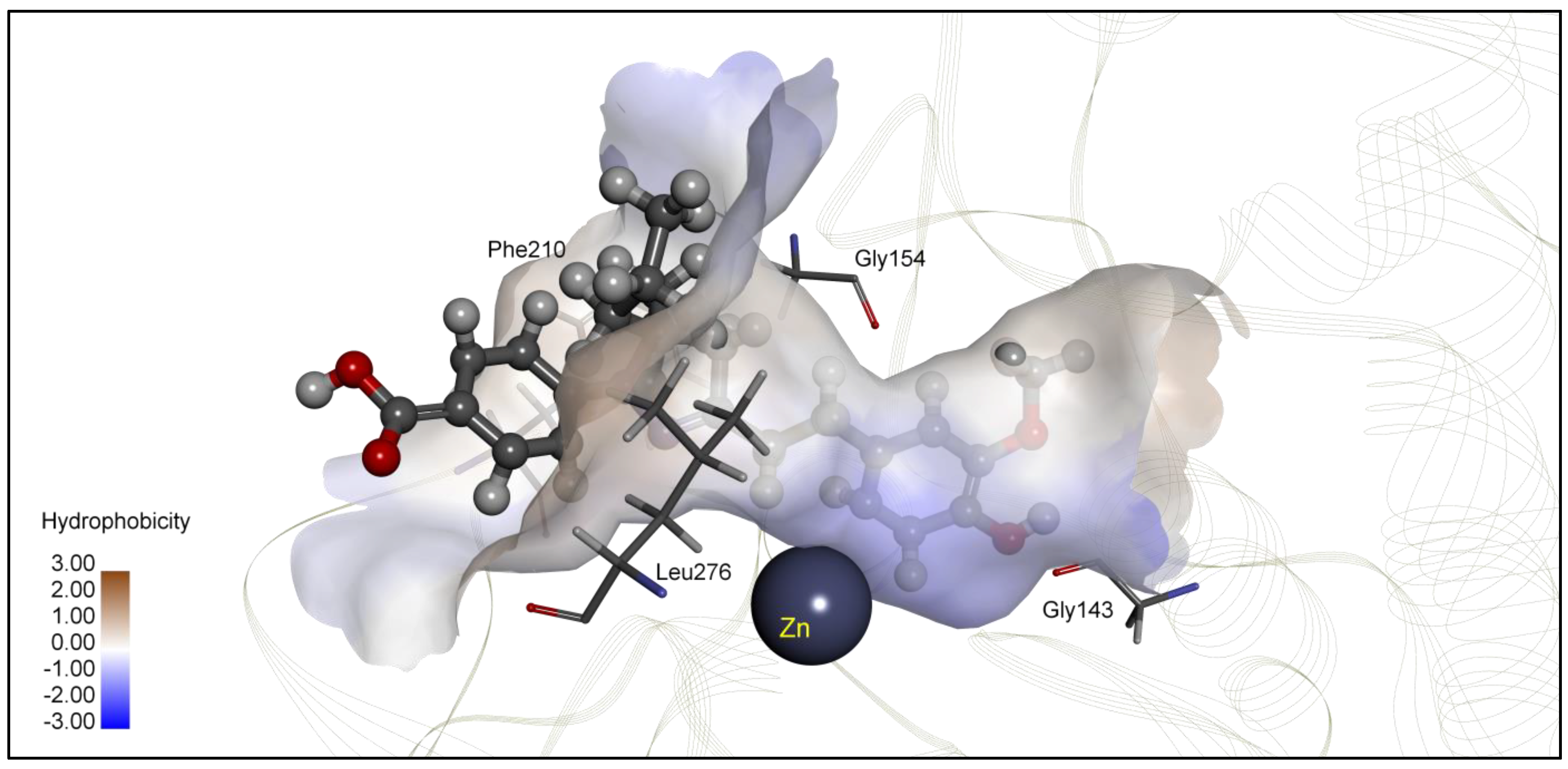
| 4c | −6.80 | 10.36 | Glu98, Asp99, Glu271 | −6.60 | 14.45 | Gly154, Tyr209, Phe210, Leu276 |
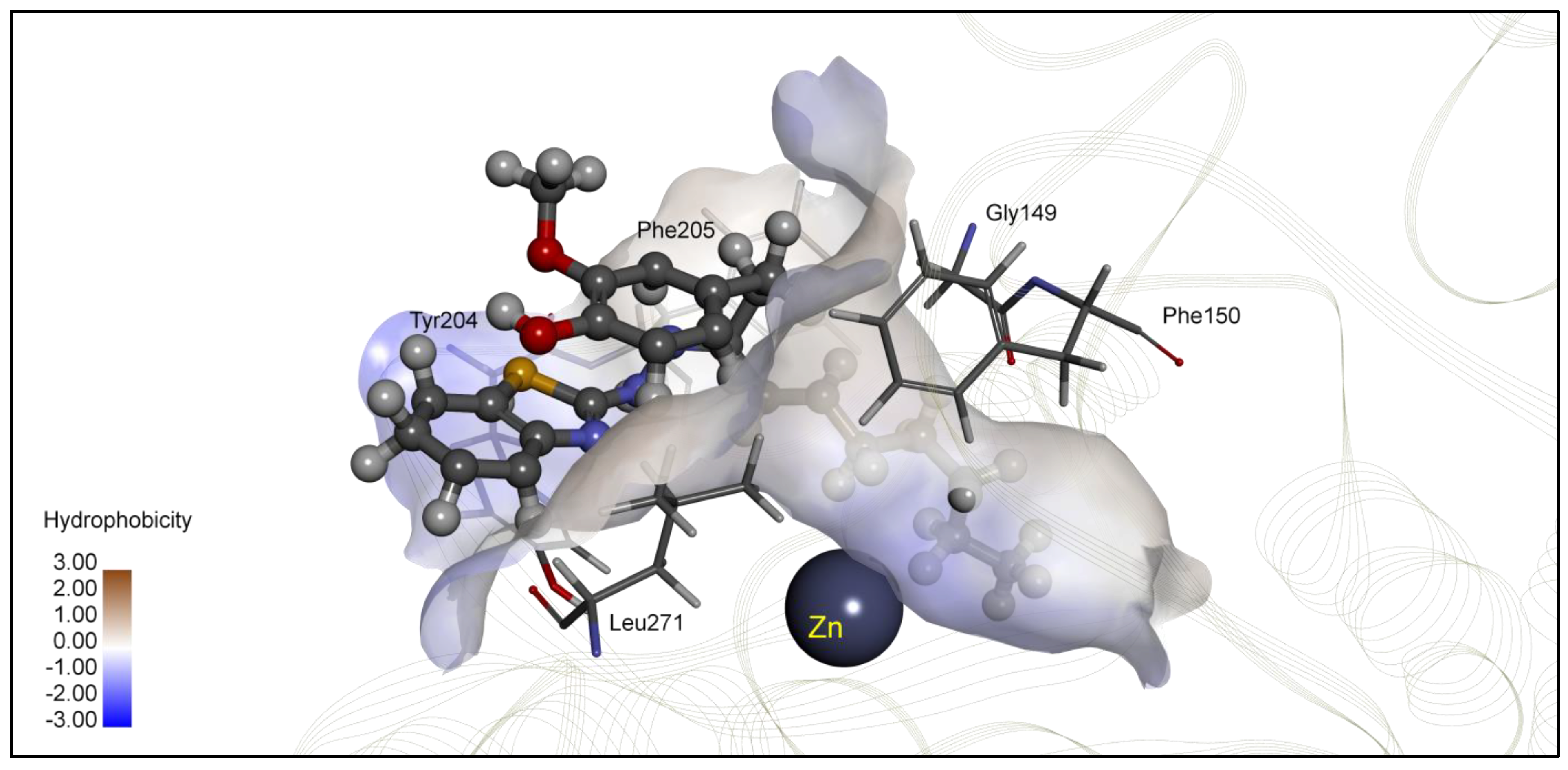
| 4d | −8.31 | 0.81 | Zn, Gly149, Tyr204, Phe205, Leu271 | −7.65 | 2.48 | Gly32, Thr309, Ile310 |
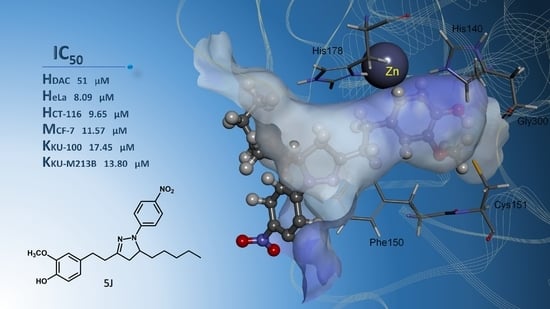
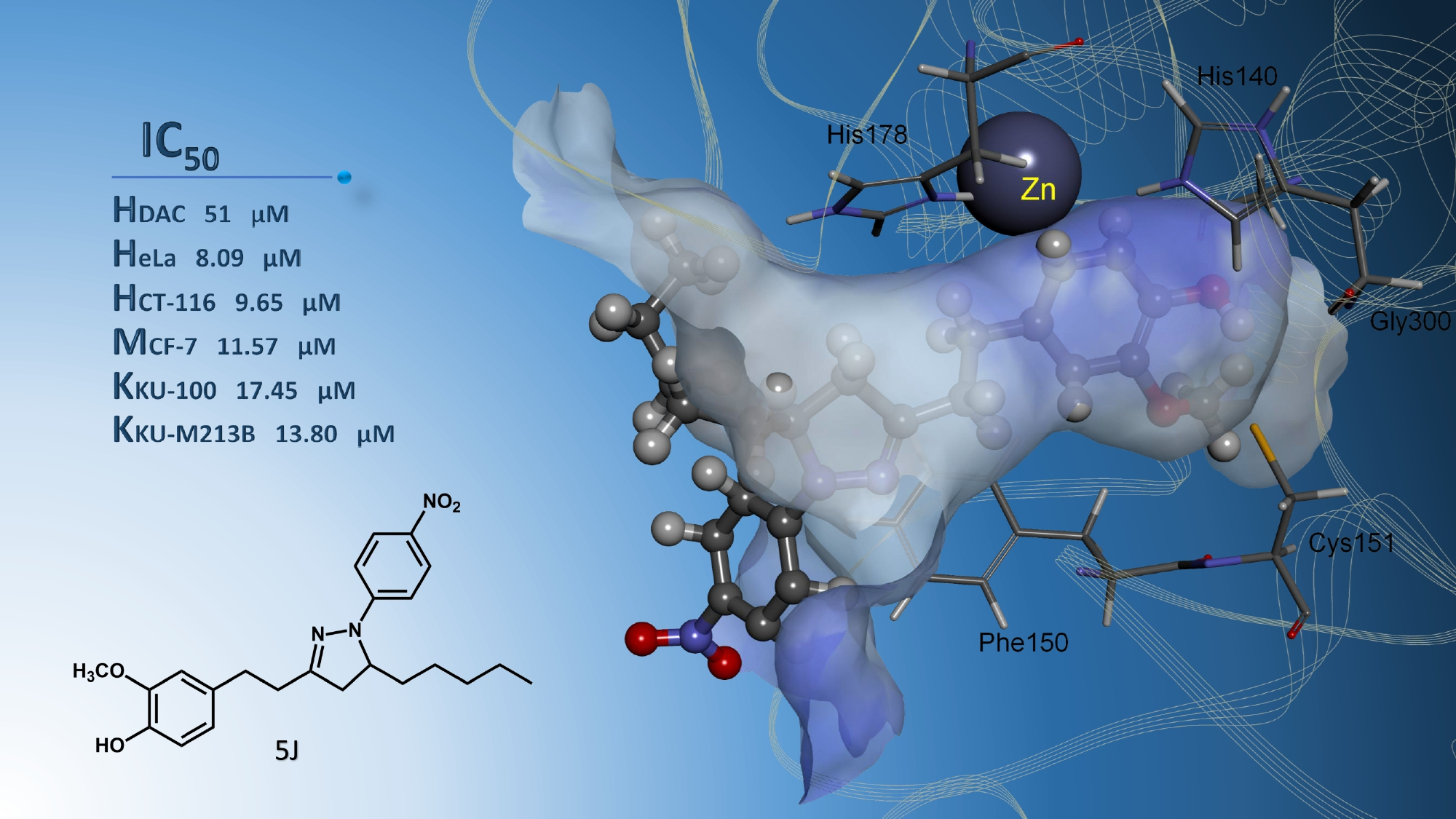
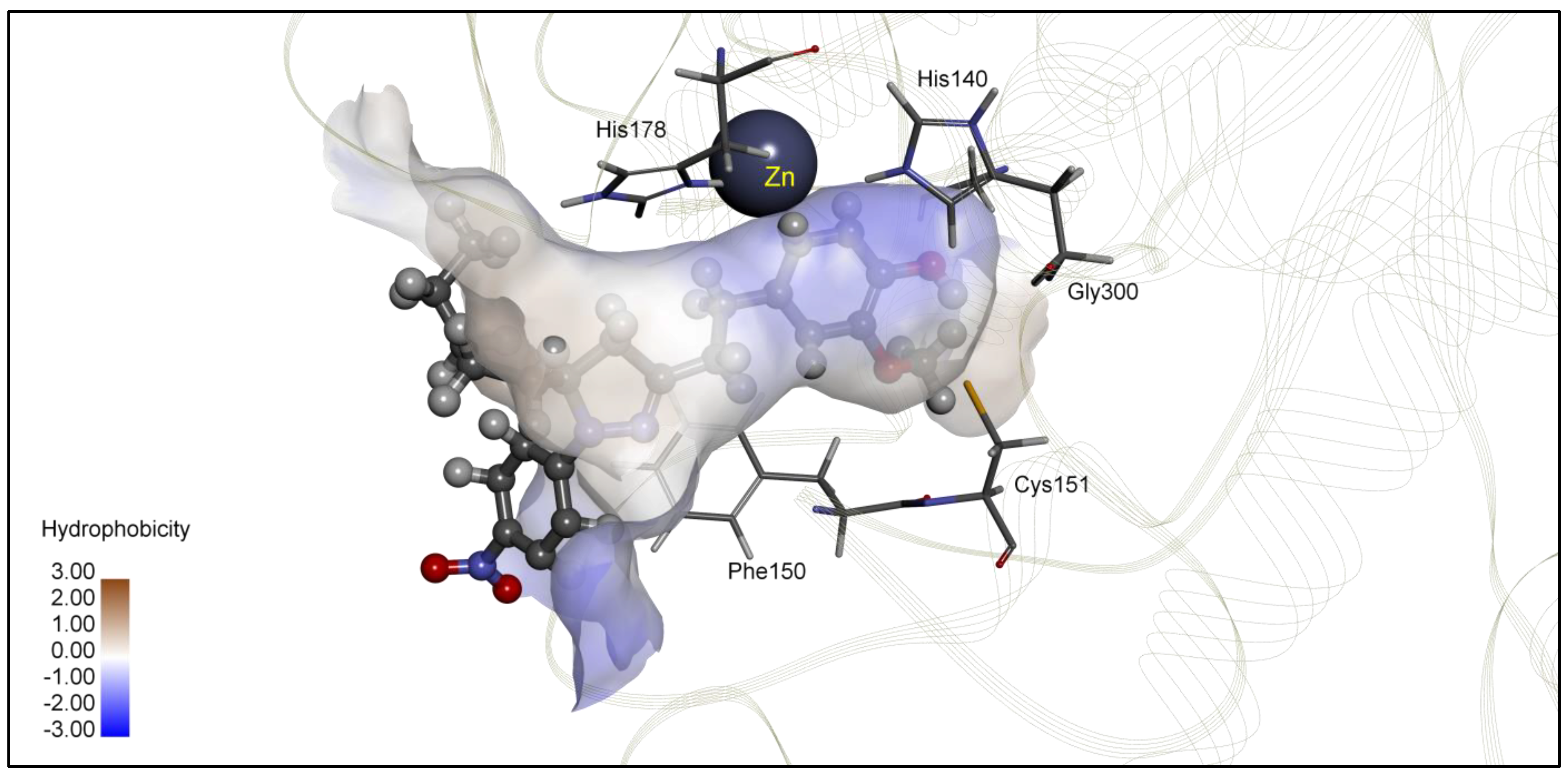
| 5j | −8.24 | 0.92 | Zn, Phe150, Cys151, Gly300, | −7.05 | 6.79 | Asn26, Tyr27, His38, Arg311 |
| 5k | −6.02 | 38.39 | Tyr201, Leu211, Pro227, Lys361 | −7.72 | 2.28 | Zn, Gly143, Gly154, Leu276 |
| Cp | HDAC3 | HDAC8 | ||||
| ∆G * | Ki ** | Binding Residues | ∆G * | Ki ** | Binding Residues | |
| TSA | −8.23 | 0.93 | Zn, His22, Gly143, Phe144, His172, Phe200 | −8.85 | 0.33 | Zn, His143, Phe152, Phe207, Phe208 |
| 4c | −7.60 | 2.67 | Pro23, Asp93, Arg265, Leu266 | −7.12 | 5.36 | Phe152, Gly206, Phe207, Met274 |
| 4d | −9.18 | 0.19 | Zn, His22, Asp92, Gly143, Phe200 | −7.99 | 1.40 | Phe152, Gly206, Phe207, Met274 |
| 5j | −7.42 | 3.64 | Pro23, Phe199, Leu266 | −7.45 | 3.47 | Lys33, Pro273, Asn307, Leu308 |
| 5k | −6.69 | 12.42 | Thr298, Glu300, Tyr331 | −6.95 | 8.11 | Lys33, Asn307, Leu308 |
| Cpd | IC50 Values (Mean ± SD; n = 3 (μM)) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vero Cells | HeLa Cells | HCT116 Cells | MCF-7 Cells | |||||||||
| 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | |
| Cisplatin | 42.86 ± 2.39 | 12.73 ± 0.63 | 6.55 ± 0.81 | 17.07 ± 1.00 | 9.97 ± 0.34 | 6.45 ± 0.12 | 20.98 ± 2.56 | 7.61 ± 0.61 | 4.98 ± 0.77 | 29.17 ± 1.35 | 13.75 ± 0.54 | 10.42 ± 0.25 |
| 4 | 37.63 ± 0.94 | 35.96 ± 0.95 | 23.34 ± 0.24 | 16.86 ± 0.80 | 9.26 ± 0.46 | 8.10 ± 0.29 | 17.30 ± 0.57 | 12.37 ± 0.15 | 12.30 ± 0.13 | 24.78 ± 0.66 | 16.54 ± 0.15 | 13.39 ± 0.07 |
| 4c | 69.47± 3.94 | 49.31 ± 0.18 | 45.17 ± 0.49 | 49.31 ±1.27 | 33.35 ±1.09 | 29.31 ±0.88 | 20.19 ±0.46 | 18.42 ±0.32 | 17.25 ±0.23 | 46.42 ±1.30 | 39.40 ±0.68 | 27.07 ±0.47 |
| 4d | 169.6 ± 6.98 | 69.97 ± 1.27 | 60.15 ± 1.69 | 51.56 ± 1.92 | 37.23 ± 1.33 | 33.66 ± 0.76 | 40.08 ± 1.47 | 28.28 ± 0.79 | 26.63 ± 0.15 | 103.5 ± 3.17 | 57.81 ± 2.37 | 25.52 ± 1.15 |
| 5j | 41.63 ± 2.99 | 32.20 ± 1.31 | 26.08 ± 0.47 | 26.83 ± 0.62 | 11.59 ± 0.60 | 8.09 ± 0.03 | 42.72 ± 2.23 | 10.18 ± 0.43 | 9.65 ± 0.17 | 27.85 ± 0.86 | 13.75 ± 0.56 | 11.57 ± 0.44 |
| 5k | 143.0 ± 2.15 | 127.6 ± 1.73 | 116.7 ± 1.13 | 83.82 ± 1.83 | 56.78 ± 1.24 | 23.14 ± 0.76 | 94.44 ± 2.70 | 39.22 ± 1.06 | 34.47 ± 0.66 | 56.61 ± 3.21 | 50.54 ± 1.64 | 31.76 ± 0.88 |
| Cpd | IC50 Values (Mean ± SD; n = 3 (µM)) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| H-69 Cells | KKU-100 Cells | KKU-M213B Cells | |||||||
| 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | |
| Cisplatin | >20 | >20 | 15.80 ± 0.35 | 17.32 ± 0.12 | 11.18 ± 0.14 | 7.57 ± 0.07 | >20 | 18.56 ± 0.57 | 12.98 ± 0.61 |
| 4 | 38.35 ± 0.84 | 31.98 ± 0.55 | 24.78 ± 0.24 | 27.64 ± 0.26 | 18.92 ± 0.48 | 17.04 ± 0.04 | 17.95 ± 0.05 | 12.92 ± 0.47 | 9.91 ± 0.17 |
| 4c | 40.03 ± 1.57 | 30.63 ± 0.82 | 28.72 ± 0.36 | 36.82 ± 0.89 | 29.70 ± 0.96 | 19.20 ± 0.41 | 39.00 ± 0.27 | 28.98 ± 0.26 | 25.94 ± 0.71 |
| 4d | 51.42 ± 1.51 | 41.44 ± 0.61 | 29.40 ± 0.89 | 41.63 ± 1.24 | 31.50 ± 0.80 | 25.75 ± 0.20 | 39.84 ± 0.29 | 20.60 ± 0.18 | 18.18 ± 0.75 |
| 5j | 44.06 ± 0.89 | 34.63 ± 0.41 | 26.37 ± 0.43 | 27.22 ± 0.59 | 22.43 ± 0.65 | 17.45 ± 0.14 | 23.86 ± 0.85 | 19.30 ± 0.90 | 13.80 ± 0.16 |
| 5k | 68.71 ± 1.15 | 52.67 ± 1.73 | 47.56 ± 0.73 | 68.14 ± 1.98 | 38.90 ± 0.65 | 26.99 ± 0.08 | 63.16 ± 2.52 | 35.68 ± 1.40 | 29.13 ± 1.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phaosiri, C.; Yenjai, C.; Senawong, T.; Senawong, G.; Saenglee, S.; Somsakeesit, L.-o.; Kumboonma, P. Histone Deacetylase Inhibitory Activity and Antiproliferative Potential of New [6]-Shogaol Derivatives. Molecules 2022, 27, 3332. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103332
Phaosiri C, Yenjai C, Senawong T, Senawong G, Saenglee S, Somsakeesit L-o, Kumboonma P. Histone Deacetylase Inhibitory Activity and Antiproliferative Potential of New [6]-Shogaol Derivatives. Molecules. 2022; 27(10):3332. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103332
Chicago/Turabian StylePhaosiri, Chanokbhorn, Chavi Yenjai, Thanaset Senawong, Gulsiri Senawong, Somprasong Saenglee, La-or Somsakeesit, and Pakit Kumboonma. 2022. "Histone Deacetylase Inhibitory Activity and Antiproliferative Potential of New [6]-Shogaol Derivatives" Molecules 27, no. 10: 3332. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103332