
1,2σ3λ3-Oxaphosphetanes and Their P-Chalcogenides—A Combined Experimental and Theoretical Study

 , and
, and
Abstract
:1. Introduction
2. Results
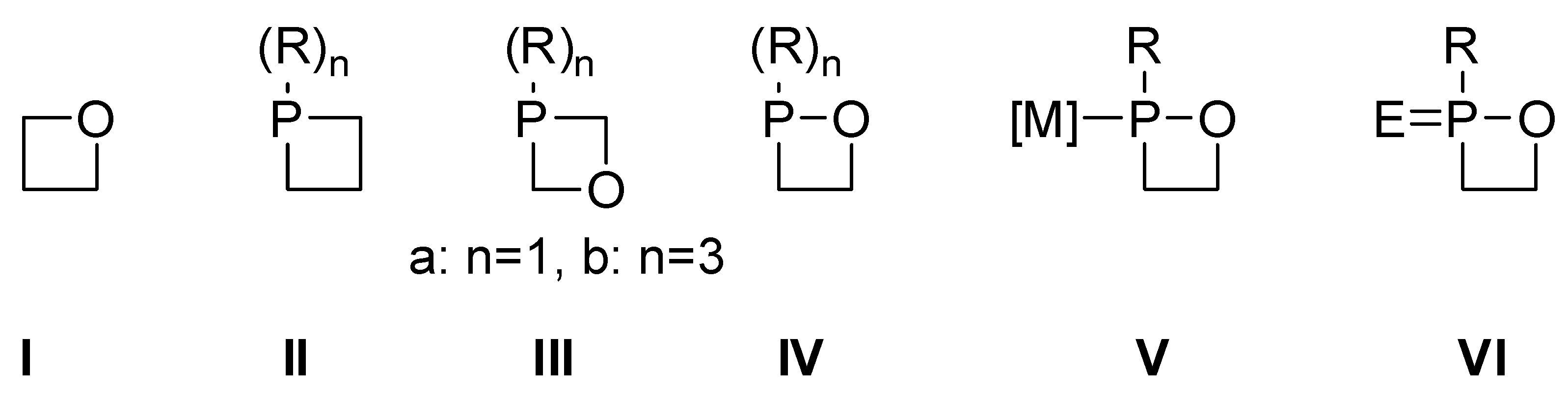
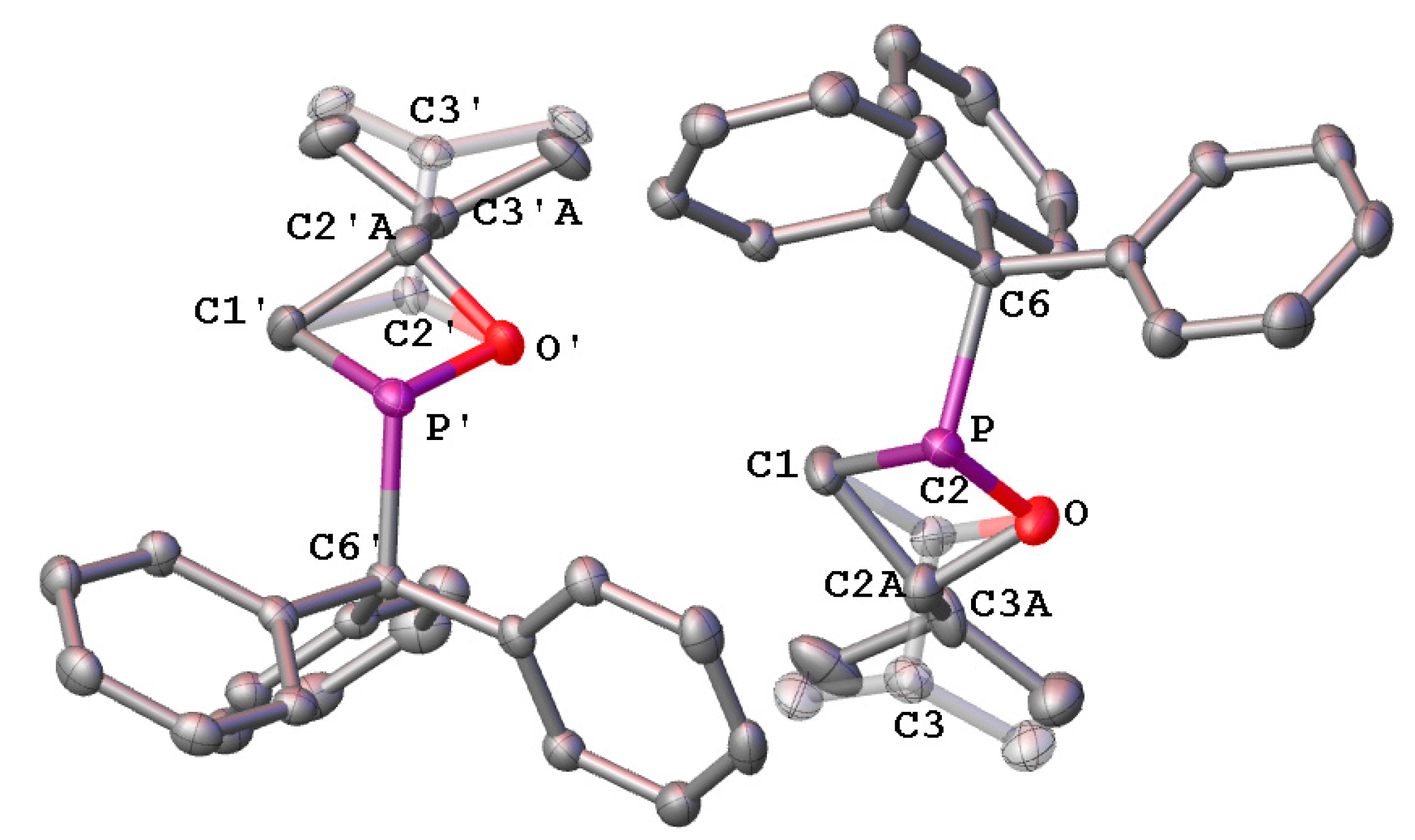
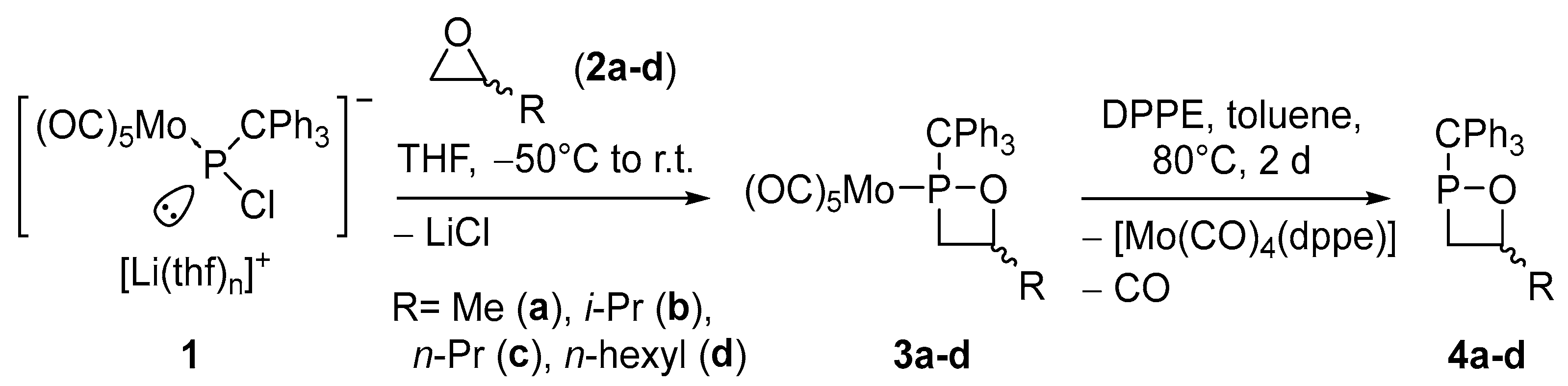
2.1. Synthesis and Spectroscopic Characterization of 1,2σ3λ3-Oxaphosphetanes
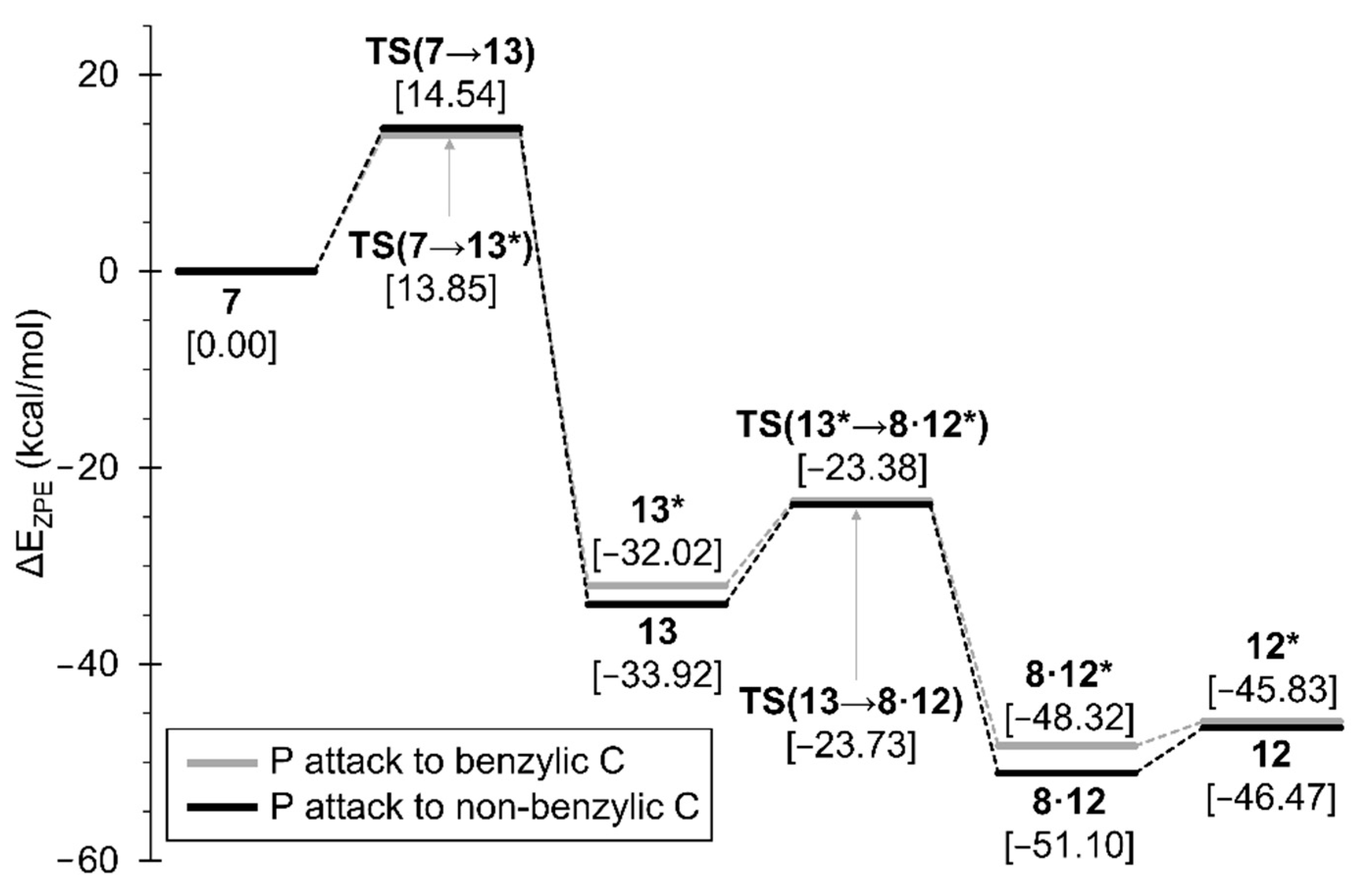
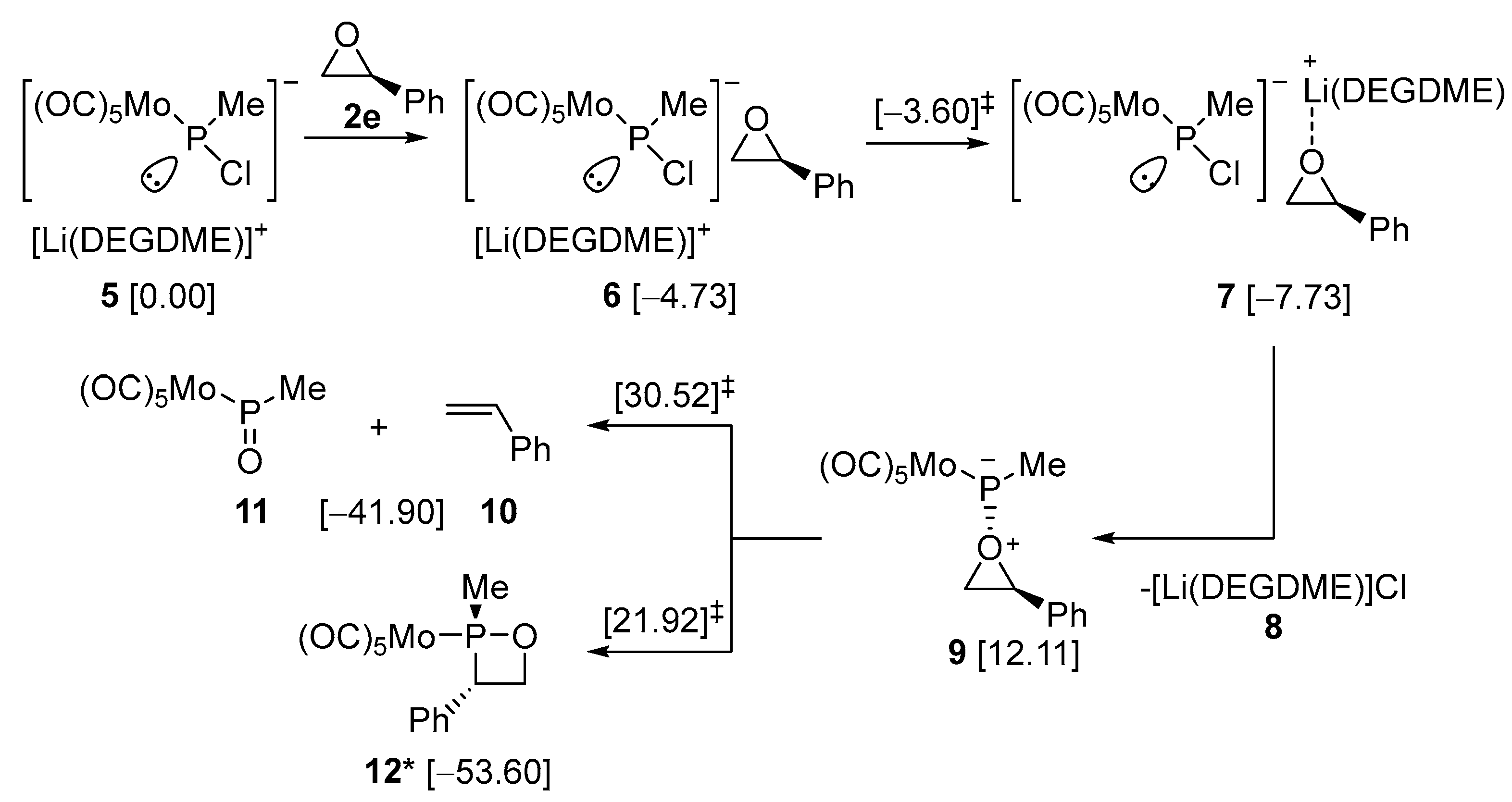
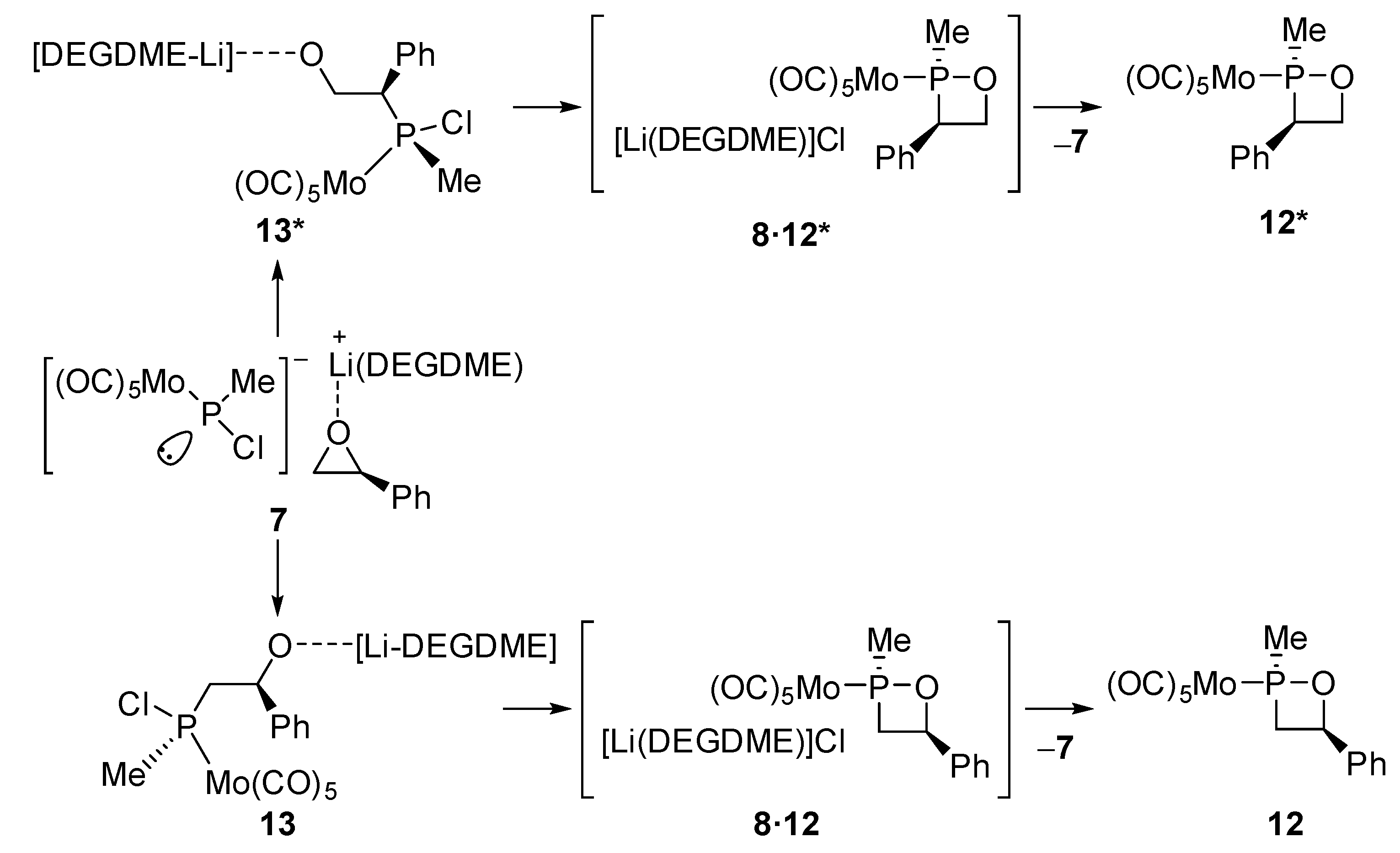
2.2. DFT-Based Mechanistic Proposal
2.3. Synthesis of 1,2-Oxaphosphetane P-Chalcogenides
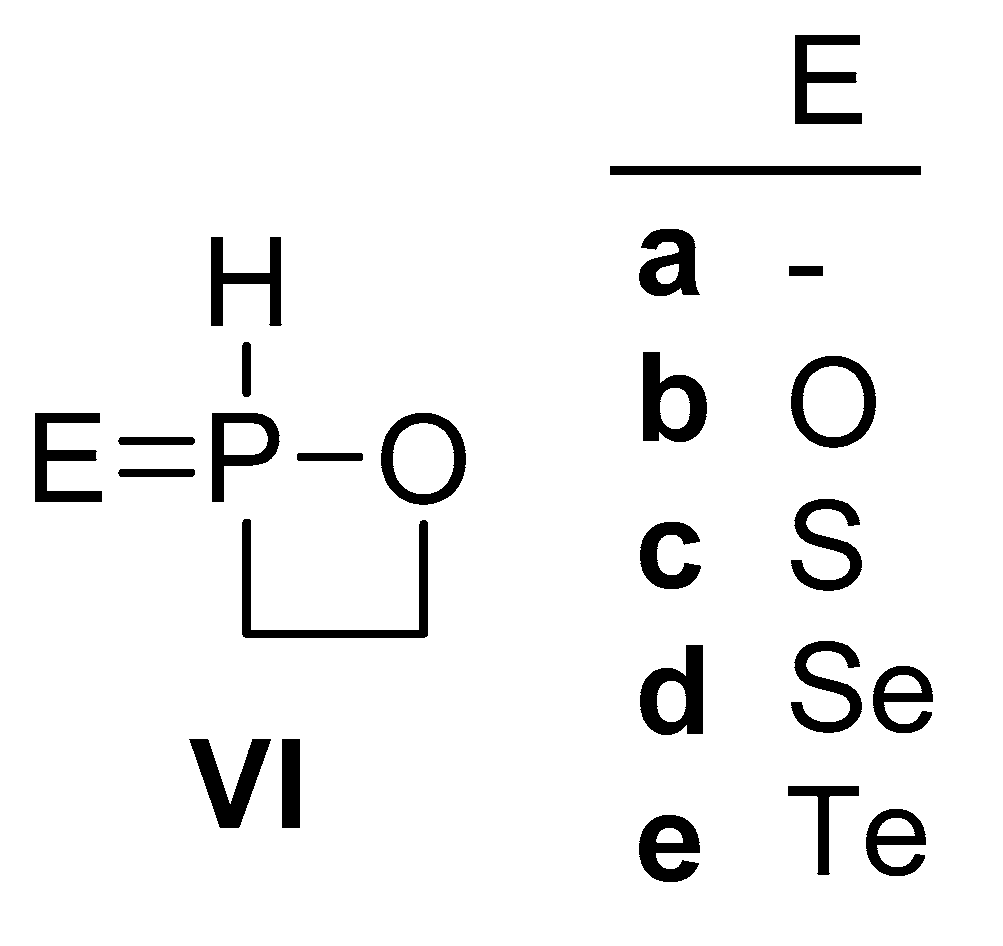
2.4. Ring Strain Energy of Model 1,2-Oxaphosphetane Derivatives
3. Materials and Methods
3.1. Synthetic Details
3.2. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, G.; Shynkaruk, O.; Lui, M.W.; Rivard, E. Small Inorganic Rings in the 21st Century: From Fleeting Intermediates to Novel Isolable Entities. Chem. Rev. 2014, 114, 7815–7880. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.A.; Croft, R.A.; Davis, O.A.; Doran, R.; Morgan, K.F. Oxetanes: Recent Advances in Synthesis, Reactivity, and Medicinal Chemistry. Chem. Rev. 2016, 116, 12150–12233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.; Wurm, F.R. Aliphatic Polyethers: Classical Polymers for the 21st Century. Macromol. Rapid Commun. 2015, 36, 1147–1165. [Google Scholar] [CrossRef] [PubMed]
- Marinetti, A.; Carmichael, D. Synthesis and Properties of Phosphetanes. Chem. Rev. 2002, 102, 201–230. [Google Scholar] [CrossRef]
- Zhao, W.; Yan, P.K.; Radosevich, A.T. A Phosphetane Catalyzes Deoxygenative Condensation of α-Keto Esters and Carboxylic Acids via PIII/PV=O Redox Cycling. J. Am. Chem. Soc. 2015, 137, 616–619. [Google Scholar] [CrossRef]
- Reichl, K.D.; Dunn, N.L.; Fastuca, N.J.; Radosevich, A.T. Biphilic Organophosphorus Catalysis: Regioselective Reductive Transposition of Allylic Bromides via PIII/PV Redox Cycling. J. Am. Chem. Soc. 2015, 137, 5292–5295. [Google Scholar] [CrossRef] [Green Version]
- Nykaza, T.V.; Harrison, T.S.; Ghosh, A.; Putnik, R.A.; Radosevich, A.T. A Biphilic Phosphetane Catalyzes N–N Bond-Forming Cadogan Heterocyclization via PIII/PV=O Redox Cycling. J. Am. Chem. Soc. 2017, 139, 6839–6842. [Google Scholar] [CrossRef] [Green Version]
- Kaboudin, B.; Haghighat, H.; Yokomatsu, T. Synthesis of a New Class of Phosphinic Acids: Synthesis of Novel Four-Membered Cyclic Oxaphosphetanes by Intramolecular Mitsunobu Reaction of Bis(α-hydroxyalkyl)phosphinic Acids. Synthesis 2011, 2011, 3185–3189. [Google Scholar] [CrossRef]
- Wittig, G.; Geissler, G. Zur Reaktionsweise des Pentaphenyl-phosphors und einiger Derivate. Justus Liebigs Ann. Chem. 1953, 580, 44–57. [Google Scholar] [CrossRef]
- Hinz, A.; Labbow, R.; Rennick, C.; Schulz, A.; Goicoechea, J.M. HPCO—A Phosphorus-Containing Analogue of Isocyanic Acid. Angew. Chem. Int. Ed. 2017, 56, 3911–3915. [Google Scholar] [CrossRef] [Green Version]
- Wittig, G.; Schöllkopf, U. Über Triphenyl-phosphin-methylene als olefinbildende Reagenzien (I. Mitteil). Chem. Ber. 1954, 87, 1318–1330. [Google Scholar] [CrossRef]
- Vedejs, E.; Meier, G.P.; Snoble, K.A.J. Low-temperature characterization of the intermediates in the Wittig reaction. J. Am. Chem. Soc. 1981, 103, 2823–2831. [Google Scholar] [CrossRef]
- Wittig, G.; Haag, W. Über Triphenyl-phosphinmethylene als olefinbildende Reagenzien (II. Mitteil.1). Chem. Ber. 1955, 88, 1654–1666. [Google Scholar] [CrossRef]
- Espinosa Ferao, A. On the Mechanism of Trimethylphosphine-Mediated Reductive Dimerization of Ketones. Inorg. Chem. 2018, 57, 8058–8064. [Google Scholar] [CrossRef]
- Dieckbreder, U.; Lork, E.; Röschenthaler, G.-V.; Kolomeitsev, A.A. First P-trifluoromethylated ylides. Heteroat. Chem. 1996, 7, 281–284. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Iyama, Y.; Mochizuki, E.; Oshima, T. First isolation and characterization of 1,2-oxaphosphetanes with three phenyl groups at the phosphorus atom in typical Wittig reaction using cyclopropylidenetriphenylphosphorane. Tetrahedron Lett. 2005, 46, 8949–8952. [Google Scholar] [CrossRef]
- Kawashima, T.; Kato, K.; Okazaki, R. Synthesis, Structure, and Thermolysis of a 3-Methoxycarbonyl-1, 2 λ5-oxaphosphetane. Angew. Chem. Int. Ed. Engl. 1993, 32, 869–870. [Google Scholar] [CrossRef]
- Caughlan, C.N.; Ramirez, F.; Pilot, J.F.; Smith, C.P. Crystal and molecular structure of a four-membered cyclic oxyphosphorane with pentavalent phosphorus, PO2(C6H5)2(CF3)4C3H2. J. Am. Chem. Soc. 1971, 93, 5229–5235. [Google Scholar] [CrossRef]
- Dianova, E.N.; Zabotina, E.Y.; Akhmethkhaova, I.Z.; Samuilov, Y.D. 5-Methyl-2-phenyl-1,2,3-diazaphosphole in reaction with 2,5-bis(methoxycarbonyl)-3,4-diphenylcyclopentadiene. Zh. Obs. Khim. 1991, 64, 1063–1066. [Google Scholar]
- Kyri, A.W.; Schnakenburg, G.; Streubel, R. A novel route to C-unsubstituted 1,2-oxaphosphetane and 1,2-oxaphospholane complexes. Chem. Commun. 2016, 52, 8593–8595. [Google Scholar] [CrossRef]
- Kyri, A.W. Investigations on 1,2-Oxaphosphetane Complexes. Ph.D. Thesis, University of Bonn, Bonn, Germany, 2017. [Google Scholar]
- Kyri, A.W.; Gleim, F.; García Alcaraz, A.; Schnakenburg, G.; Espinosa Ferao, A.; Streubel, R. “Low-coordinate” 1,2-oxaphosphetanes—A new opportunity in coordination and main group chemistry. Chem. Commun. 2018, 54, 7123–7126. [Google Scholar] [CrossRef] [PubMed]
- Espinosa Ferao, A.; Deschamps, B.; Mathey, F. Towards functional phospholide ions: Synthesis of a 3-ethoxycarbonyl derivative. Bull. Soc. Chim. Fr. 1993, 130, 695–699. [Google Scholar]
- Regitz, M.; Scherer, H.; Illger, W.; Eckes, H. Detection of Alkylideneoxophosphoranes by Cycloaddition of Carbonyl Compounds. Angew. Chem. Int. Ed. Engl. 1973, 12, 1010–1011. [Google Scholar] [CrossRef]
- Kawashima, T.; Inamoto, N. The Correct Structure of Cyclic Adducts of (Diphenylmethylene)oxophenylphosphorane with Aromatic Aldehydes. Bull. Chem. Soc. Jpn. 1991, 64, 713–715. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, S.; Yoshifuji, M.; Okazaki, R.; Inamoto, N. Phosphinidenes and Related Intermediates. V. Reactions of Phosphinylidenes with cis- and trans-Stilbene Oxides. Bull. Chem. Soc. Jpn. 1976, 49, 1173–1174. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, T.; Nakayama, S.; Yoshifuji, M.; Okazaki, R.; Inamoto, N. Revised Structure of 2-Cyclohexyl-3,4-diphenyl-1,2-oxaphosphetane 2-Oxide. Bull. Chem. Soc. Jpn. 1991, 64, 711–712. [Google Scholar] [CrossRef]
- Hafez, T.S.; El-Khoshnieh, Y.O.; Mahran, M.R.; Atta, S.M.S. Organophosphorus Chemistry, 20.1 the Behaviour of Certain γ-Pyrone Derivatives toward 2,4-BIS-(4-Methoxyphenyl)-1,3,2,4-Dithiaphosphetan-2,4-Disulphide (Lawesson Reagent). Phosphorus. Sulfur. Silicon Relat. Elem. 1991, 56, 165–171. [Google Scholar] [CrossRef]
- Kawashima, T.; Takami, H.; Okazaki, R. Synthesis and Thermolysis of Tetracoordinate 1,2-Oxaphosphetanes Stabilized by Steric Protection. Chem. Lett. 1994, 23, 1487–1490. [Google Scholar] [CrossRef]
- Dabrowska, U.; Dabrowski, J. IR-Spektren und Struktur von α-β-ungesättigten Carbonylverbindungen, XXI. Thione aus der Reaktion von Alkylaminocrotonestern mit Phosphorpentasulfid. Chem. Ber. 1976, 109, 1779–1786. [Google Scholar] [CrossRef]
- Kyri, A.W.; Nesterov, V.; Schnakenburg, G.; Streubel, R. Synthesis and Reaction of the First 1,2-Oxaphosphetane Complexes. Angew. Chem. Int. Ed. 2014, 53, 10809–10812. [Google Scholar] [CrossRef]
- Espinosa Ferao, A.; García Alcaraz, A.; Zaragoza Noguera, S.; Streubel, R. Terminal Phosphinidene Complex Adducts with Neutral and Anionic O-Donors and Halides and the Search for a Differentiating Bonding Descriptor. Inorg. Chem. 2020, 59, 12829–12841. [Google Scholar] [CrossRef] [PubMed]
- Espinosa Ferao, A.; Streubel, R. 1,2-Thiaphosphetanes: The Quest for Wittig-Type Ring Cleavage, Rearrangement, and Sulfur Atom Transfer. Inorg. Chem. 2020, 59, 3110–3117. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Sugino, M.; Matsukawa, S.; Nakamoto, M.; Akiba, K. First Isolation and Characterization of an Anti-Apicophilic Spirophosphorane Bearing an Oxaphosphetane Ring: A Model for the Possible Reactive Intermediate in the Wittig Reaction. J. Am. Chem. Soc. 2002, 124, 7674–7675. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Murai, T. P-Chiral Phosphinoselenoic Chlorides and Phosphinochalcogenoselenoic Acid Esters: Synthesis, Characterization, and Conformational Studies. J. Org. Chem. 2005, 70, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Rey, A.; Espinosa Ferao, A.; Streubel, R. Quantum Chemical Calculations on CHOP Derivatives—Spanning the Chemical Space of Phosphinidenes, Phosphaketenes, Oxaphosphirenes, and COP–Isomers. Molecules 2018, 23, 3341. [Google Scholar] [CrossRef] [Green Version]
- Espinosa Ferao, A. Kinetic energy density per electron as quick insight into ring strain energies. Tetrahedron Lett. 2016, 57, 5616–5619. [Google Scholar] [CrossRef]
- Streubel, R.; Faßbender, J.; Schnakenburg, G.; Espinosa Ferao, A. Formation of Transient and Stable 1,3-Dipole Complexes with P,S,C and S,P,C Ligand Skeletons. Organometallics 2015, 34, 3103–3106. [Google Scholar] [CrossRef]
- Villalba Franco, J.M.; Schnakenburg, G.; Sasamori, T.; Espinosa Ferao, A.; Streubel, R. Stimuli-Responsive Frustrated Lewis-Pair-Type Reactivity of a Tungsten Iminoazaphosphiridine Complex. Chem.—A Eur. J. 2015, 21, 9650–9655. [Google Scholar] [CrossRef]
- Albrecht, C.; Schneider, E.; Engeser, M.; Schnakenburg, G.; Espinosa, A.; Streubel, R. Synthesis and DFT calculations of spirooxaphosphirane complexes. Dalt. Trans. 2013, 42, 8897–8906. [Google Scholar] [CrossRef]
- Espinosa, A.; Gómez, C.; Streubel, R. Single Electron Transfer-Mediated Selective endo- and exocyclic Bond Cleavage Processes in Azaphosphiridine Chromium(0) Complexes: A Computational Study. Inorg. Chem. 2012, 51, 7250–7256. [Google Scholar] [CrossRef]
- Schulten, C.; von Frantzius, G.; Schnakenburg, G.; Streubel, R. Deoxygenation of isocyanates via transient electrophilic terminal phosphinidene complexes: Are strained P-heterocycles involved? Heteroat. Chem. 2011, 22, 275–286. [Google Scholar] [CrossRef]
- Espinosa, A.; Streubel, R. Computational Studies on Azaphosphiridines, or How to Effect Ring-Opening Processes through Selective Bond Activation. Chem.—A Eur. J. 2011, 17, 3166–3178. [Google Scholar] [CrossRef] [PubMed]
- Krahe, O.; Neese, F.; Streubel, R. The Quest for Ring Opening of Oxaphosphirane Complexes: A Coupled-Cluster and Density Functional Study of CH3PO Isomers and Their Cr(CO)5 Complexes. Chem. Eur. J. 2009, 15, 2594–2601. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J. Chem. Phys. 2015, 143, 54107. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys. Chem. Chem. Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals—Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2011, 7, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M. RI-MP2: First derivatives and global consistency. Theor. Chem. Acc. 1997, 97, 331–340. [Google Scholar] [CrossRef]
- Bernholdt, D.E.; Harrison, R.J. Large-scale correlated electronic structure calculations: The RI-MP2 method on parallel computers. Chem. Phys. Lett. 1996, 250, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Caldeweyher, E.; Bannwarth, C.; Grimme, S. Extension of the D3 dispersion coefficient model. J. Chem. Phys. 2017, 147, 34112. [Google Scholar] [CrossRef] [PubMed]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef]
- Pople, J.A.; Head-Gordon, M.; Raghavachari, K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
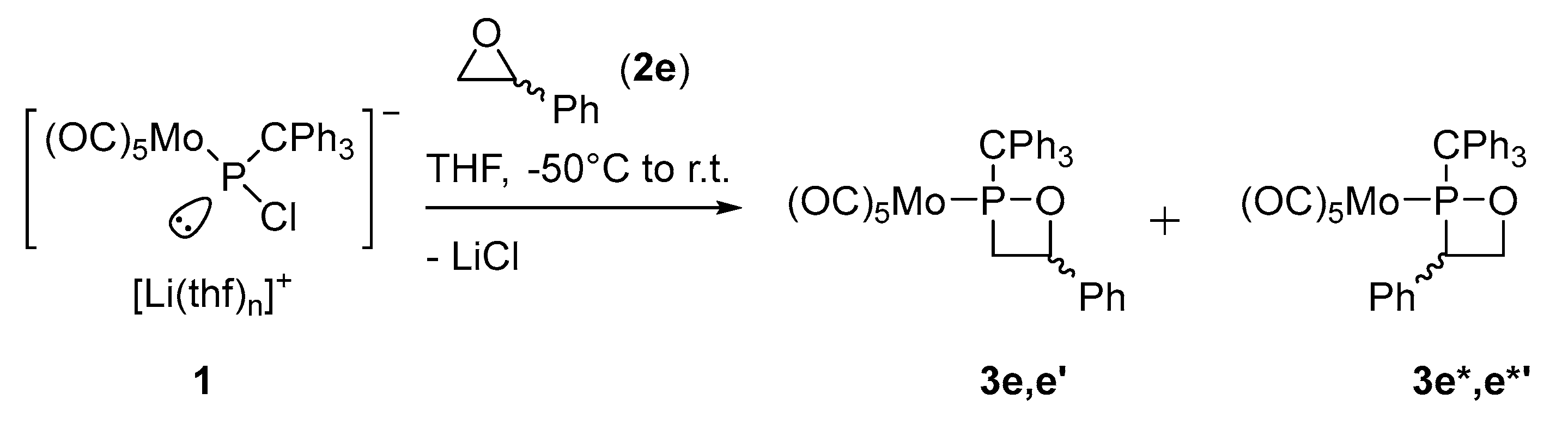{kind=link}
{kind=link}
{kind=link}
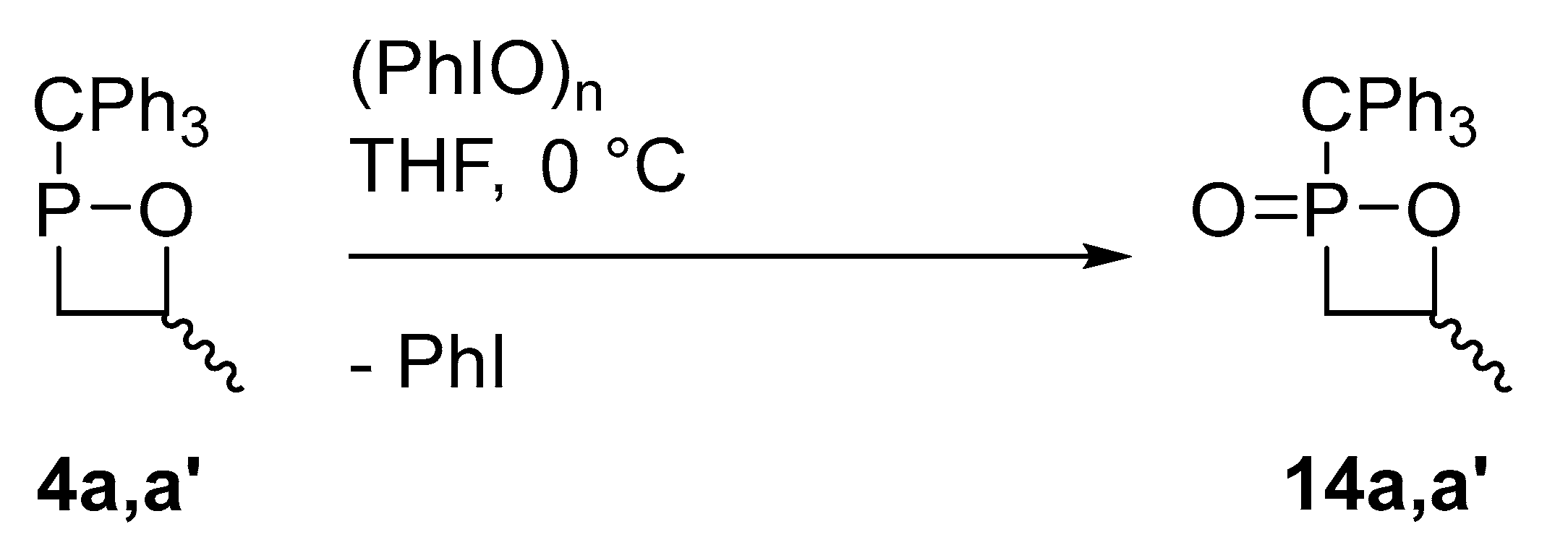{kind=link}
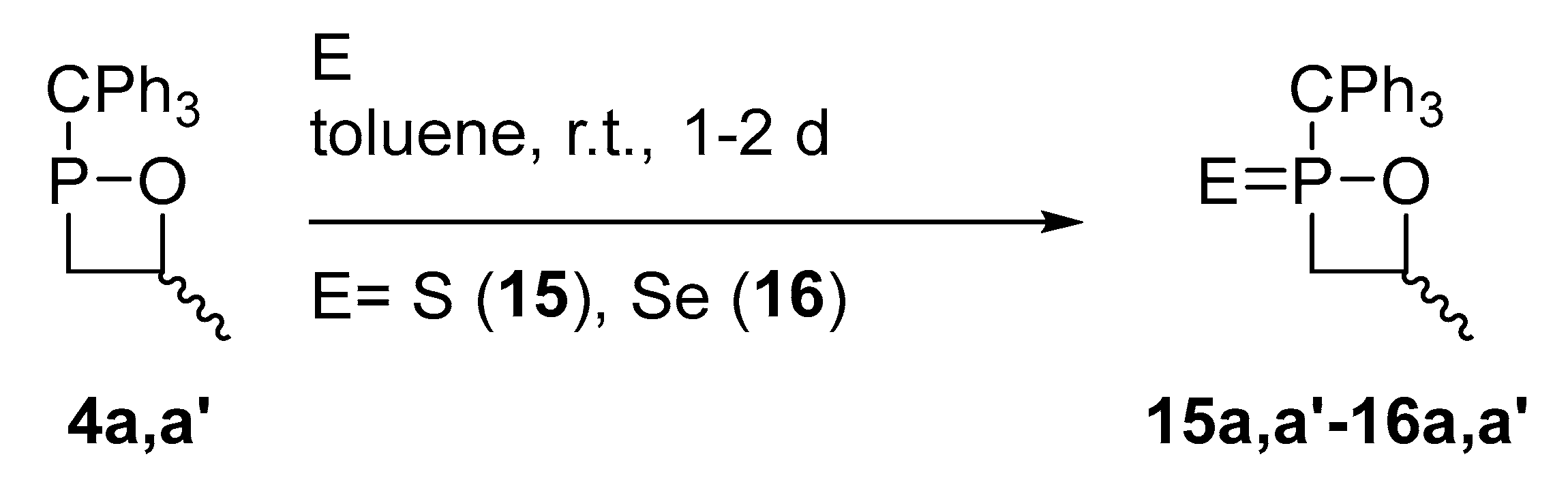{kind=link}
{kind=link}
| 3a,a’ | 3b,b’ | 3c,c’ | 3d,d’ | |
|---|---|---|---|---|
| Ratio | 51:49 | 50:50 | 50:50 | 48:52 |
| δ(P) | 185.6/207.2 | 183.5/206.1 | 187.6/208.8 | 187.3/208.7 |
| δ(CH2) | 2.92/2.96 | 2.90/2.35 | 2.91/2.95 | 2.90/2.98 |
| δ(CH2*) | 3.01/3.18 | 2.90/2.99 | 3.00/3.12 | 2.98/3.13 |
| δ(CH) | 5.35/4.69 | 4.54/4.97 | 4.48/5.13 | 5.12/4.48 |
| δ(CH2) | 41.7/39.9 | 37.5/39.9 | 40.6/38.5 | 38.5/40.5 |
| 1J(P-C) | 18.3/21.7 | 21.5/18.5 | 18.3/21.4 | 21.5/8.3 |
| δ(CPh3) | 68.0/67.2 | 67.1/67.8 | 67.2/67.9 | 67.1/67.9 |
| 1J(P-C) | 10.6/9.0 | 9.3/10.7 | 9.3/10.7 | 9.4/10.7 |
| δ(CH) | 77.7/82.0 | 85.9/90.1 | 80.9/85.0 | 81.1/85.2 |
| 2J(P-C) | 11.7/11.7 | 11.3/11.2 | 11.6/11.4 | 11.6/11.5 |
| 4a,a’ | 4b,b’ | 4c,c’ | 4d,d’ | |
|---|---|---|---|---|
| Ratio | 42:58 | 34:66 | 39:61 | 18:82 |
| δ(P) | 163.7/199.0 1 | 161.2/196.7 1 | 166.2/199.0 2 | 166.3/199.4 3 |
| 3e | 3e’ | 3e* | 3e*’ | |
|---|---|---|---|---|
| Ratio | 40 | 45 | 10 | 5 |
| δ(P) | 191.5 | 210.9 | 237.3 | 244.9 |
| δ(CH2) | 39.7 | 40.4 | 75.9 | 76.2 |
| nJ(P-C) | 18.8 | 21.7 | 13.1 | 13.6 |
| 4a,a’ 1 | 14a,a’ 1 | 15a,a’ 2 | 16a,a’ 2 | |
|---|---|---|---|---|
| δ(P) | 163.7/199.0 | 62.1/63.5 | 115.8/120.0 | 116.1/121.5 |
| δ(CH2) | 2.22/2.45 | 2.53/2.56 | 2.49/2.22 | 2.62/2.43 |
| δ(CH2*) | 2.69/2.48 | 2.75/2.96 | 2.49/2.72 | 2.75/3.00 |
| δ(CH) | 5.12/4.62 | 4.25/5.04 | 4.78/4.05 | 4.88/4.26 |
| δ(CH2) | 33.7/31.2 | 39.5/38.9 | 44.8/44.4 | 45.6/44.6 |
| 1J(P-C) | 13.6/7.9 | 60.8/64.0 | 51.5/49.8 | 44.6/43.5 |
| δ(CPh3) | 63.4/62.9 | 65,6/66.2 | 70.4/69.8 | 70.4/69.8 |
| 1J(P-C) | 52.3/50.7 | 73.3/71.9 | 46.1/47.8 | 33.4/35.4 |
| δ(CH) | 77.6/83.0 | 72.1/74.3 | 75.3/74.5 | 76.1/76.1 |
| 2J(P-C) | 4.6/2.2 | 20.2/20.2 | 19.4/19.9 | 19.4/19.4 |
| VIa | VIb | Vic | Vid | Vie |
|---|---|---|---|---|
| 18.95 | 19.57 | 19.93 | 20.35 | 20.59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gleim, F.; Alcaraz, A.G.; Schnakenburg, G.; Ferao, A.E.; Streubel, R. 1,2σ3λ3-Oxaphosphetanes and Their P-Chalcogenides—A Combined Experimental and Theoretical Study. Molecules 2022, 27, 3345. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103345
Gleim F, Alcaraz AG, Schnakenburg G, Ferao AE, Streubel R. 1,2σ3λ3-Oxaphosphetanes and Their P-Chalcogenides—A Combined Experimental and Theoretical Study. Molecules. 2022; 27(10):3345. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103345
Chicago/Turabian StyleGleim, Florian, Antonio García Alcaraz, Gregor Schnakenburg, Arturo Espinosa Ferao, and Rainer Streubel. 2022. "1,2σ3λ3-Oxaphosphetanes and Their P-Chalcogenides—A Combined Experimental and Theoretical Study" Molecules 27, no. 10: 3345. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103345