
Discovery of Dipyridamole Analogues with Enhanced Metabolic Stability for the Treatment of Idiopathic Pulmonary Fibrosis


Abstract
:1. Introduction
2. Results
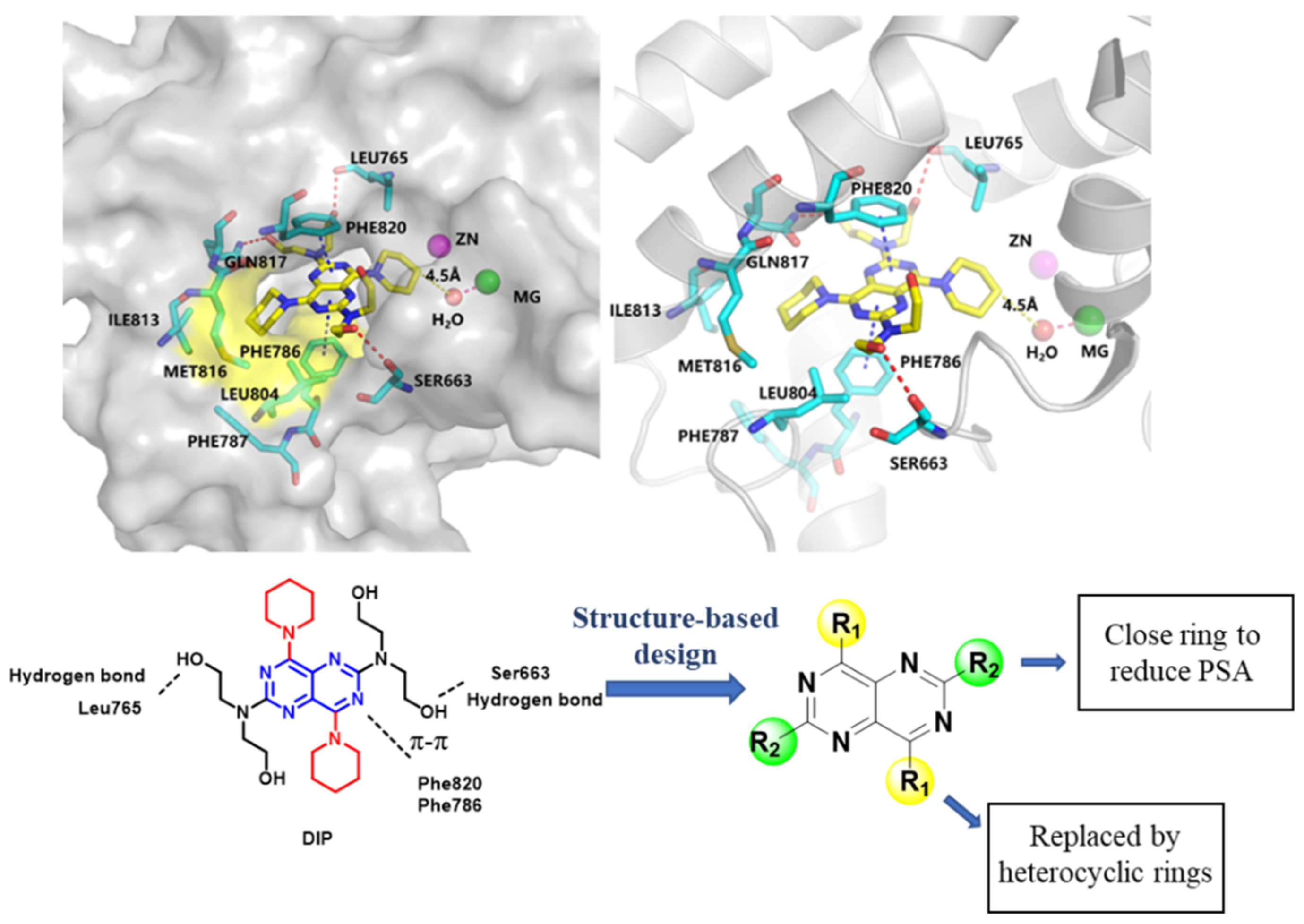
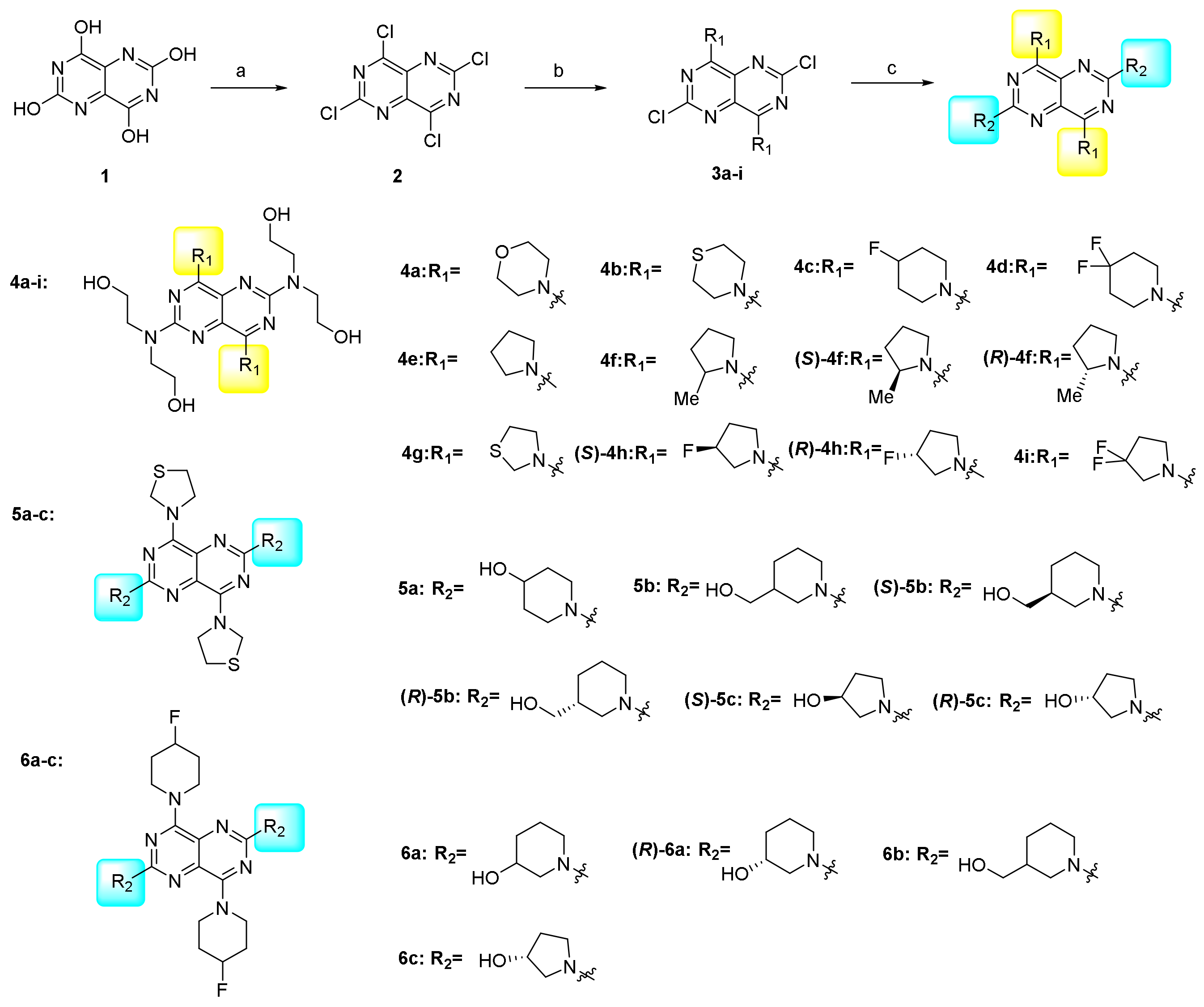
2.1. Chemistry
2.2. Inhibitory Activity of PDE5 Inhibitors
2.3. Metabolic Stability of Typical Compounds in Rat Liver Microsomes
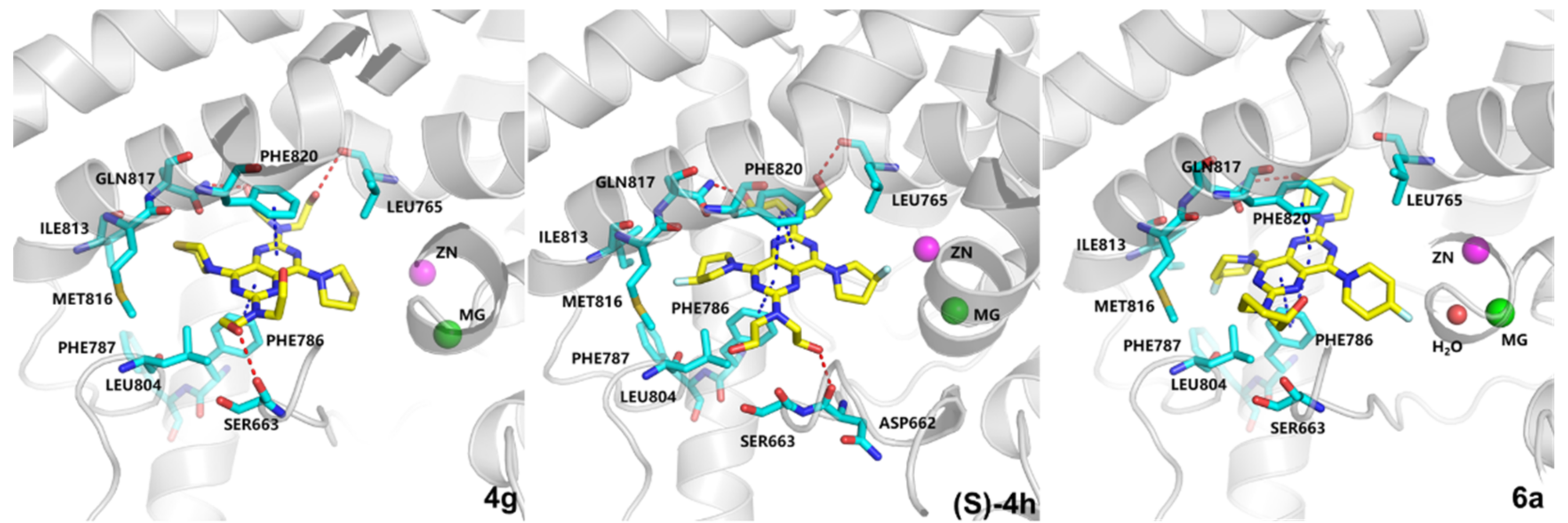
2.4. Molecular Docking
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for Synthesis of Intermediate Compounds 2
3.1.2. General Procedure for Synthesis of Intermediate Compounds 3a–i
3.1.3. General Procedure for Synthesis of Target Compounds 4a–i
3.1.4. General Procedure for Synthesis of Target Compounds 5a–c, 6a–c
3.1.5. General Procedure for Synthesis of Intermediate Compounds 7, 8a and 8b
3.1.6. General Procedure for Synthesis of Target Compounds 9a and 9b
3.2. In Vitro Assay for PDE5 Inhibitors
3.2.1. Protein Expression and Purification
3.2.2. Enzymatic Assay of PDE5A
3.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Raghu, G.; Chen, S.-Y.; Hou, Q.; Yeh, W.-S.; Collard, H.R. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18-64 years old. Eur. Respir. J. 2016, 48, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERSARS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [Green Version]
- Sergew, A.; Brown, K.K. Advances in the treatment of idiopathic pulmonary fibrosis. Expert Opin. Emerg. Drugs 2015, 20, 537–552. [Google Scholar] [CrossRef]
- Thomson, C.C.; Duggal, A.; Bice, T.; Lederer, D.J.; Wilson, K.C.; Raghu, G. 2018 Clinical Practice Guideline Summary for Clinicians: Diagnosis of Idiopathic Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2019, 16, 285–290. [Google Scholar] [CrossRef]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Yip, S.; Benavente, O. Antiplatelet Agents for Stroke Prevention. Neurotherapeutics 2011, 8, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresele, P.; Momi, S.; Falcinelli, E. Anti-platelet therapy: Phosphodiesterase inhibitors. Br. J. Clin. Pharmacol. 2011, 72, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Saigal, A.; Zhao, W.; Amini, P.; Tamburino, A.M.; Raghavan, S.; Hoek, M. Identification of the Molecular Basis of Anti-fibrotic Effects of Soluble Guanylate Cyclase Activator Using the Human Lung Fibroblast Phosphoproteome. J. bioRxiv 2020, 1, 1–26. [Google Scholar] [CrossRef]
- Zisman, D.A.; Schwarz, M.; Anstrom, K.J.; Collard, H.R.; Flaherty, K.R.; Hunninghake, G.W.; Idiopathic Pulm Fibrosis, C. A Controlled Trial of Sildenafil in Advanced Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2010, 363, 620–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, Z.; Liu, S.; Sun, J.; Chen, Z.; Jiang, M.; Zhang, Q.; Wei, Y.; Wang, X.; Huang, Y.Y.; et al. Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm. Sin. B 2020, 10, 1205–1215. [Google Scholar] [CrossRef]
- Luo, H.B.; Wu, Y.N.; Li, Z.; Tian, Y.J.; Zhou, Q. Application of dipyridamoletable or pharmaceutically acceptable salt thereof in preparation of medicine for preventing and/ or treating lung inflammation. Faming Zhuanli Shenqing Gongkai Shuomingshu CN 111388476A, 10 July 2020. (In Chinese). [Google Scholar]
- Wu, Y.; Zhou, Q.; Zhang, T.; Li, Z.; Chen, Y.-P.; Zhang, P.; Yu, Y.-F.; Geng, H.; Tian, Y.-J.; Zhang, C.; et al. Discovery of Potent, Selective, and Orally Bioavailable Inhibitors against Phosphodiesterase-9, a Novel Target for the Treatment of Vascular Dementia. J. Med. Chem. 2019, 62, 4218–4224. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Q.; Jiang, M.-Y.; Huang, Y.-Y.; Zhu, Z.; Han, C.; Tian, Y.-J.; Zhang, B.; Luo, H.-B. Discovery of Potent Phosphodiesterase-9 Inhibitors for the Treatment of Hepatic Fibrosis. J. Med. Chem. 2021, 64, 9537–9549. [Google Scholar] [CrossRef]
- Northen, J.S.; Boyle, F.T.; Clegg, W.; Curtin, N.J.; Edwards, A.J.; Griffin, R.J.; Golding, B.T. Controlled stepwise conversion of 2,4,6,8-tetrachloropyrimido [5,4-d]pyrimidine into 2,4,6,8-tetrasubstituted pyrimido [5,4-d]pyrimidines. J. Chem. Soc. Perkin Trans. 2002, 1, 108–115. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Curtin, N.J.; Barlow, H.C.; Bowman, K.J.; Calvert, A.H.; Davison, R.; Golding, B.T.; Huang, B.; Loughlin, P.J.; Newell, D.R.; Smith, P.G.; et al. Resistance-Modifying Agents. 11. Pyrimido [5,4-d]pyrimidine Modulators of Antitumor Drug Activity. Synthesis and Structure−Activity Relationships for Nucleoside Transport Inhibition and Binding to α1-Acid Glycoprotein. J. Med. Chem. 2004, 47, 4905–4922. [Google Scholar] [CrossRef] [PubMed]
- Shang, N.N.; Shao, Y.X.; Cai, Y.H.; Guan, M.; Huang, M.; Cui, W.; He, L.; Yu, Y.J.; Huang, L.; Li, Z.; et al. Discovery of 3-(4-hydroxybenzyl)-1-(thiophen-2-yl)chromeno [2,3-c]pyrrol-9(2H)-one as a phosphodiesterase-5 inhibitor and its complex crystal structure. Biochem. Pharmacol. 2014, 89, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.-X.; Huang, M.; Cui, W.; Feng, L.-J.; Wu, Y.; Cai, Y.; Li, Z.; Zhu, X.; Liu, P.; Wan, Y.; et al. Discovery of a Phosphodiesterase 9A Inhibitor as a Potential Hypoglycemic Agent. J. Med. Chem. 2014, 57, 10304–10313. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Comp. | R1 | R2 | Inhibition Ratio (%) | IC50 (nM) a | |
| 0.5 μM | 0.1 μM | ||||
| DIP |  |  | 84 | 17 | 235 ± 35 |
| 4a |  | | 40 | 0 | - |
| 4b |  | | 75 | 53 | 113 ± 11 |
| 4c |  | | 62 | 31 | 343 ± 18 |
| 4d |  | | 72 | 41 | 362 ± 81 |
| 4e |  | | 47 | 7 | - |
| 4f |  | | 60 | 14 | 478 ± 3 |
| (S)-4f |  | | 42 | 16 | - |
| (R)-4f |  | | 44 | 27 | - |
| 4g |  | | 93 | 60 | 64 ± 16 |
| (S)-4h |  | | 55 | 34 | 332 ± 56 |
| (R)-4h |  | | 44 | 27 | - |
| 4i |  | | 61 | 24 | 353 ± 57 |
 | |||||
|---|---|---|---|---|---|
| Comp. | R1 | R2 | Inhibition Ratio (%) | IC50 (nM) a | |
| 0.5 μM | 0.1 μM | ||||
| 5a |  |  | 32 | 29 | - |
| 5b | |  | 79 | 52 | 167 ± 41 |
| (S)-5b | |  | 34 | 13 | - |
| (R)-5b | |  | - | - | 109 ± 20 |
| (S)-5c | |  | 49 | 36 | - |
| (R)-5c | |  | 74 | 45 | 198 ± 45 |
| 6a |  |  | 48 | 7 | - |
| (R)-6a | |  | 36 | 8 | - |
| 6b | |  | 7 | 0 | - |
| 6c | |  | 20 | 5 | - |
 | ||||
|---|---|---|---|---|
| Comp. | R1 | Inhibition Ratio (%) | IC50 (nM) a | |
| 0.5 μM | 0.1 μM | |||
| 9a |  | 91 | 45 | 100 ± 1 |
| 9b |  | 81 | 33 | 94 ± 9 |
| Comp. | PDE5 IC50 (nM) | T1/2 in RLM (min) |
|---|---|---|
| DIP | 235 | 7 |
| 4b | 113 | 7 |
| 4c | 343 | 44 |
| 4d | 362 | 19 |
| 4g | 64 | 5 |
| (S)-4h | 332 | 67 |
| 9a | 100 | 4 |
| 9b | 94 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, M.-X.; Chen, Y.-Q.; Liu, R.-D.; Huang, Y.; Zhang, C. Discovery of Dipyridamole Analogues with Enhanced Metabolic Stability for the Treatment of Idiopathic Pulmonary Fibrosis. Molecules 2022, 27, 3452. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113452
Huang M-X, Chen Y-Q, Liu R-D, Huang Y, Zhang C. Discovery of Dipyridamole Analogues with Enhanced Metabolic Stability for the Treatment of Idiopathic Pulmonary Fibrosis. Molecules. 2022; 27(11):3452. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113452
Chicago/Turabian StyleHuang, Meng-Xing, Yan-Quan Chen, Run-Duo Liu, Yue Huang, and Chen Zhang. 2022. "Discovery of Dipyridamole Analogues with Enhanced Metabolic Stability for the Treatment of Idiopathic Pulmonary Fibrosis" Molecules 27, no. 11: 3452. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113452