
One-Step, Low-Cost, Operator-Friendly, and Scalable Procedure to Synthetize Highly Pure N-(4-ethoxyphenyl)-retinamide in Quantitative Yield without Purification Work-Up



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
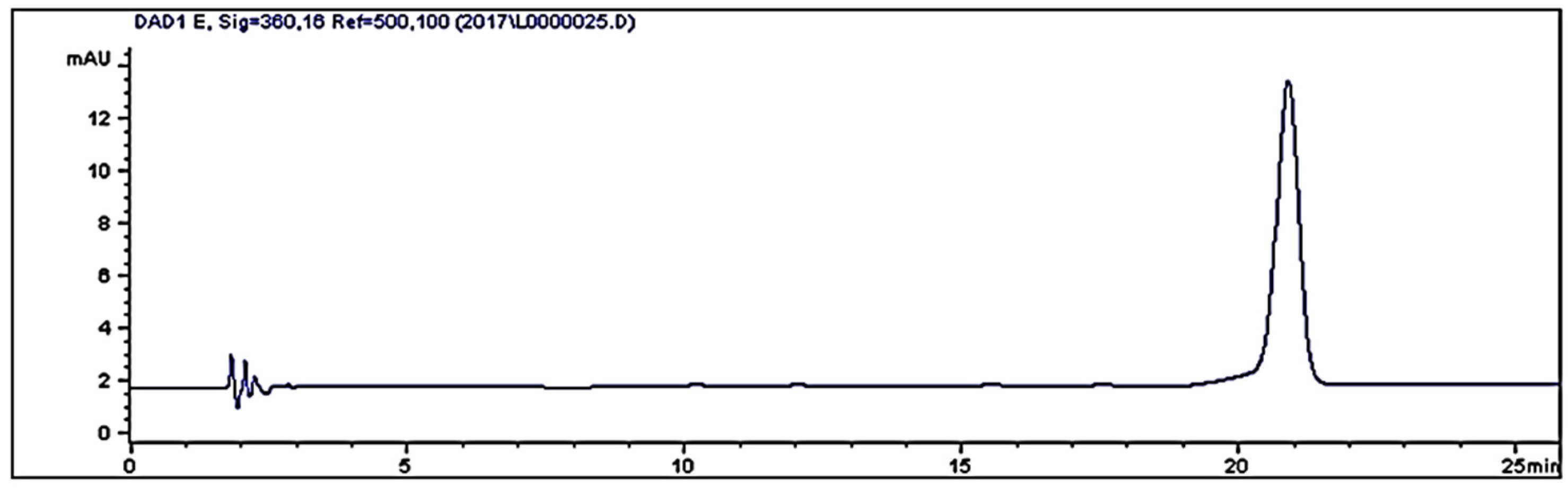
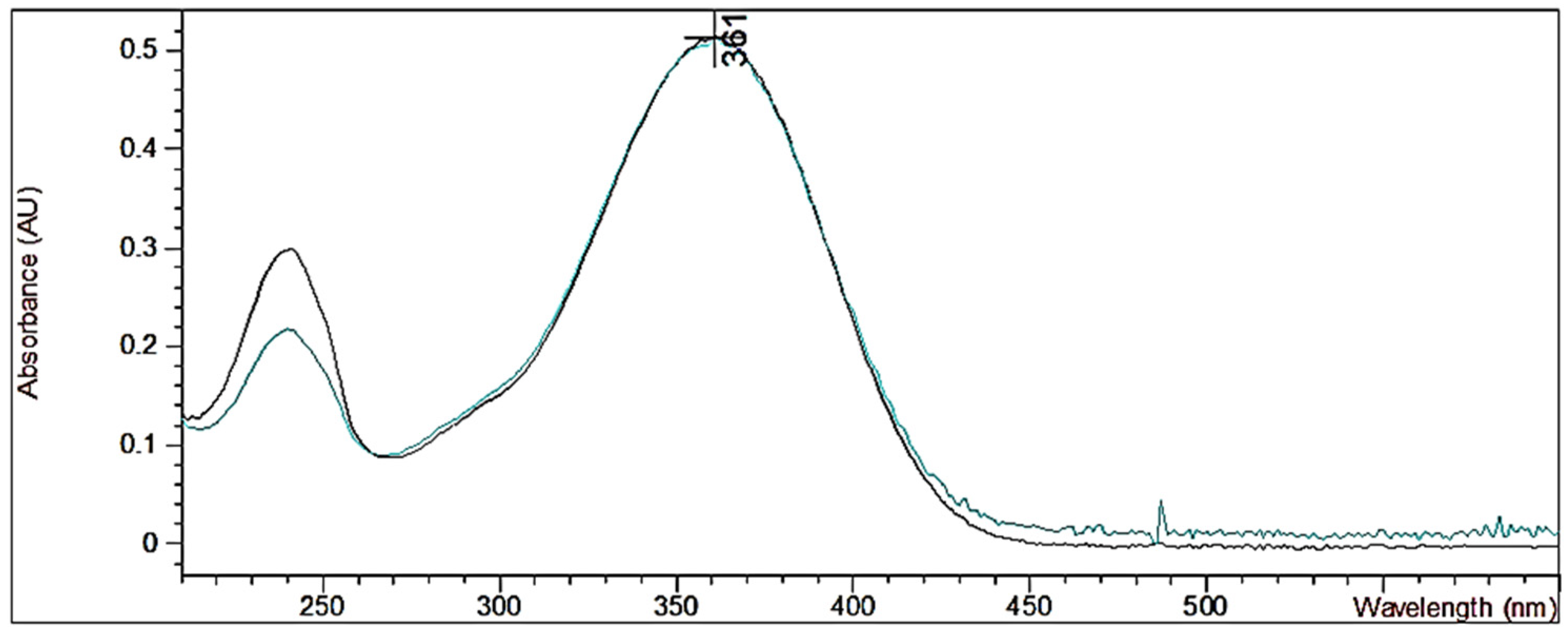
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals and Instruments
3.2. Procedure for the Preparation and Isolation of N-(4-ethoxyphenyl)-retinamide (4-EPR)
3.3. TLC
3.4. HPLC Analyses
3.5. UV–Vis Analyses
3.6. FTIR Spectra
3.7. 1H and 13C NMR Analyses
3.8. GC-MS Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kelloff, G.J.; Crowell, J.A.; Boone, C.W.; Steele, V.E.; Lubet, R.A.; Greenwald, P.; Alberts, D.S.; Covey, J.M.; Doody, L.A.; Knapp, G.G.; et al. Clinical development plan: N-(4-hydroxyphenyl)retinamide. J. Cell. Biochem. Suppl. 1994, 20, 176–196. [Google Scholar] [PubMed]
- Reynolds, C.P.; Lemons, R.S. Retinoid Therapy of Childhood Cancer. Hematol. Oncol. Clin. N. Am. 2001, 15, 867–910. [Google Scholar] [CrossRef]
- Delia, D.; Aiello, A.; Lombardi, L.; Pelicci, P.G.; Grignani, F.; Grignani, F.; Formelli, F.; Menard, S.; Costa, A.; Veronesi, U.; et al. N-(4-Hydroxyphenyl)Retinamide Induces Apoptosis of Malignant Hemopoietic Cell Lines Including Those Unresponsive to Retinoic Acid1. Cancer Res. 1993, 53, 6036–6041. [Google Scholar]
- Kazmi, S.M.; Plante, R.K.; Visconti, V.; Lau, C.Y. Comparison of N-(4-hydroxyphenyl)retinamide and all-trans-retinoic acid in the regulation of retinoid receptor-mediated gene expression in human breast cancer cell lines. Cancer Res. 1996, 56, 1056–1062. [Google Scholar]
- Reynolds, C.P.; Wang, Y.; Melton, L.J.; Einhorn, P.A.; Slamon, D.J.; Maurer, B.J. Retinoic-Acid-Resistant Neuroblastoma Cell Lines Show Altered MYC Regulation and High Sensitivity to Fenretinide. Med. Pediatric Oncol. 2000, 35, 597–602. [Google Scholar] [CrossRef]
- Sheikh, M.S.; Shao, Z.-M.; Li, X.-S.; Ordonez, J.V.; Conley, B.A.; Wu, S.; Dawson, M.I.; Han, Q.-X.; Chao, W.; Quick, T.; et al. N-(4-Hydroxyphenyl)Retinamide (4-HPR)-Mediated Biological Actions Involve Retinoid Receptor-Independent Pathways in Human Breast Carcinoma. Carcinogenesis 1995, 16, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Supino, R.; Crosti, M.; Clerici, M.; Warlters, A.; Cleris, L.; Zunino, F.; Formelli, F. Induction of Apoptosis by Fenretinide (4HPR) in Human Ovarian Carcinoma Cells and Its Association with Retinoic Acid Receptor Expression. Int. J. Cancer 1996, 65, 491–497. [Google Scholar] [CrossRef]
- Formelli, F.; Clerici, M.; Campa, T.; Di Mauro, M.G.; Magni, A.; Mascotti, G.; Moglia, D.; De Palo, G.; Costa, A.; Veronesi, U. Five-Year Administration of Fenretinide: Pharmacokinetics and Effects on Plasma Retinol Concentrations. J. Clin. Oncol. 1993, 11, 2036–2042. [Google Scholar] [CrossRef] [PubMed]
- Delia, D.; Aiello, A.; Meroni, L.; Nicolini, M.; Reed, J.C.; Pierotti, M.A. Role of Antioxidants and Intracellular Free Radicals in Retinamide-Induced Cell Death. Carcinogenesis 1997, 18, 943–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oridate, N.; Suzuki, S.; Higuchi, M.; Mitchell, M.F.; Hong, W.K.; Lotan, R. Involvement of Reactive Oxygen Species in N-(4-Hydroxyphenyl)Retinamide-Induced Apoptosis in Cervical Carcinoma Cells. J. Natl. Cancer Inst. 1997, 89, 1191–1198. [Google Scholar] [CrossRef]
- Maurer, B.J.; Melton, L.; Billups, C.; Cabot, M.C.; Reynolds, C.P. Synergistic Cytotoxicity in Solid Tumor Cell Lines Between N-(4-Hydroxyphenyl)Retinamide and Modulators of Ceramide Metabolism. J. Natl. Cancer Inst. 2000, 92, 1897–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, B.; Metelitsa, L.; Seeger, R.; Cabot, M.; Reynolds, C. Increase of Ceramide and Induction of Mixed Apoptosis/Necrosis by N-(4-Hydroxyphenyl)-Retinamide in Neuroblastoma Cell Lines. J. Natl. Cancer Inst. 1999, 91, 1138–1146. [Google Scholar] [CrossRef]
- O’Donnell, P.; Guo, W.-X.; Reynolds, C.; Maurer, B. N-(4-Hydroxyphenyl)Retinamide Increases Ceramide and Is Cytotoxic to Acute Lymphoblastic Leukemia Cell Lines, but Not to NoN-Malignant Lymphocytes. Leukemia 2002, 16, 902–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, L.R.; Nelson, E.C. Separation of Vitamin A and Retinyl Esters by Reversed-Phase High-Performance Liquid Chromatography. J. Chromatogr. A 1984, 294, 466–470. [Google Scholar] [CrossRef]
- Connor, M.J.; Lindae, M.L.; Lowe, N.J. Pharmacokinetics of Topically Applied Radiolabeled Retinoids in Hairless Mouse Epidermis and Dermis After Single Applications. J. Investig. Dermatol. 1985, 84, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Cullum, M.E.; Zile, M.H. Metabolism of all-trans-retinoic acid and all-trans-retinyl acetate. Demonstration of common physiological metabolites in rat small intestinal mucosa and circulation. J. Biol. Chem. 1985, 260, 10590–10596. [Google Scholar] [CrossRef]
- Cullum, M.E.; Zile, M.H. Quantitation of Biological Retinoids by High-Pressure Liquid Chromatography: Primary Internal Standardization Using Tritiated Retinoids. Anal. Biochem. 1986, 153, 23–32. [Google Scholar] [CrossRef]
- Frolik, C.A.; Roller, P.P.; Roberts, A.B.; Sporn, M.B. In Vitro and in Vivo Metabolism of All-Trans- and 13-Cis-Retinoic Acid in Hamsters. Identification of 13-Cis-4-Oxoretinoic Acid. J. Biol. Chem. 1980, 255, 8057–8062. [Google Scholar] [CrossRef]
- Frolik, C.A.; Tavela, T.E.; Peck, G.L.; Sporn, M.B. High-Pressure Liquid Chromatographic Determination of 13-Cis-Retinoic Acid and All-Trans-Retinoic Acid in Human Plasma. Anal. Biochem. 1978, 86, 743–750. [Google Scholar] [CrossRef]
- Furr, H.C.; Amédée-Manesme, O.; Olson, J.A. Gradient reversed-phase high-performance liquid chromatographic separation of naturally occurring retinoids. J. Chromatogr. B Biomed. Sci. Appl. 1984, 309, 299–307. [Google Scholar] [CrossRef]
- Hultin, T.A.; Mehta, R.G.; Moon, R.C. Simple High-Performance Liquid Chromatographic Method for the Separation of Retinoids Including N-(4-Hydroxyphenyl)-All-Trans-Retinamide. J. Chromatogr. B Biomed. Sci. Appl. 1985, 341, 187–192. [Google Scholar] [CrossRef]
- Ito, Y.L.; Zile, M.; Ahrens, H.; DeLuca, H.F. Liquid-gel partition chromatography of vitamin A compounds; formation of retinoic acid from retinyl acetate in vivo. J. Lipid Res. 1974, 15, 517–524. [Google Scholar] [CrossRef]
- McCormick, A.M.; Napoli, J.L. Identification of 5,6-epoxyretinoic acid as an endogenous retinol metabolite. J. Biologic. Chem. 1982, 257, 1730–1735. [Google Scholar] [CrossRef]
- McCormick, A.M.; Napoli, J.L.; DeLuca, H.F. High-Pressure Liquid Chromatography of Vitamin A Metabolites and Analogs. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1980; Volume 67, pp. 220–233. [Google Scholar] [CrossRef]
- McCormick, A.M.; Napoli, J.L.; Yoshizawa, S.; DeLuca, H.F. 5,6-Epoxyretinoic Acid Is a Physiological Metabolite of Retinoic Acid in the Rat. Biochem. J. 1980, 186, 475–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, J.L.; Khalil, H.; McCormick, A.M. Metabolism of 5,6-Epoxyretinoic Acid in Vivo: Isolation of a Major Intestinal Metabolite. Biochemistry 1982, 21, 1942–1949. [Google Scholar] [CrossRef]
- Roberts, A.; Nichols, M.; Frolik, C.; Newton, D.; Sporn, M. Assay of Retinoids in Biological Samples by Reverse-Phase High-Pressure Liquid Chromatography. Cancer Res. 1978, 38, 3327–3332. [Google Scholar]
- Silva, D.P., Jr.; DeLuca, H.F. Metabolism of Retinoic Acid in Vivo in the Vitamin A-Deficient Rat. Biochem. J. 1982, 206, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaresan, P.R.; Bhat, P.V. IoN-pair high-pressure liquid chromatography of cis-trans isomers of retinoic acid in tissues of vitamin A-sufficient rats. J. Lipid Res. 1982, 23, 448–455. [Google Scholar] [CrossRef]
- Vahlquist, A. Vitamin A in Human Skin: I. Detection and Identification of Retinoids in Normal Epidermis. J. Investig. Dermatol. 1982, 79, 89–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss, R. Chromatography of Retinoids. J. Chromatogr. B Biomed. Sci. Appl. 1990, 531, 481–508. [Google Scholar] [CrossRef]
- Evans, J.E.; McCaffery, P. HPLC/MS(N) analysis of retinoids. Methods Mol. Biol. 2010, 652, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Wyss, R.; Bucheli, F. Quantitative Analysis of Retinoids in Biological Fluids by High-Performance Liquid Chromatography Using Column Switching. II. Simultaneous Determination of Etretinate, Acitretin and 13-Cis-Acitretin in Plasma. J. Chromatogr. 1988, 431, 297–307. [Google Scholar] [CrossRef]
- Vratilova, J.; Frgala, T.; Maurer, B.J.; Patrick Reynolds, C. Liquid Chromatography Method for Quantifying N-(4-Hydroxyphenyl)Retinamide and N-(4-Methoxyphenyl)Retinamide in Tissues. J. Chromatogr. B 2004, 808, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.E.; Min, H.K. Analysis of fenretinide and its metabolites in human plasma by liquid chromatography–tandem mass spectrometry and its application to clinical pharmacokinetics. J. Pharm. Biomed. Anal. 2017, 132, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Curley, R.W., Jr. Solid Phase Synthesis of Arylretinamides. WO 2003003987 A2, 16 January 2003. [Google Scholar]
- Maryanoff, C.A. Process for Preparing Retinoyl Chlorides. EP 261911 A2, 30 March 1988. [Google Scholar]
- Koenig, H.; Peh, J.; Scholz, H.; Paust, J. Amides of Vitamin A Acid. DE 2300107 A1, 11 July 1974. [Google Scholar]
- Mershon, S.M.; Anding, A.L.; Chapman, J.S.; Clagett-Dame, M.; Stonerock, L.A.; Curley, R.W. Solid Phase-Assisted Synthesis and Screening of a Small Library of N-(4-Hydroxyphenyl)Retinamide (4-HPR) Analogs. Bioorg. Med. Chem. Lett. 2007, 17, 836–840. [Google Scholar] [CrossRef] [Green Version]
- Campos-Sandoval, J.A.; Redondo, C.; Kinsella, G.K.; Pal, A.; Jones, G.; Eyre, G.S.; Hirst, S.C.; Findlay, J.B.C. Fenretinide Derivatives Act as Disrupters of Interactions of Serum Retinol Binding Protein (SRBP) with Transthyretin and the SRBP Receptor. J. Med. Chem. 2011, 54, 4378–4387. [Google Scholar] [CrossRef] [Green Version]
- ABClabtory. Available online: http://www.abclabtory.com (accessed on 1 June 2022).
- Aurora Building Blocks 6. Available online: https://www.aurorafinechemicals.com (accessed on 1 June 2022).
- Alfei, S.; Castellaro, S. Synthesis and Characterization of Polyester-Based Dendrimers Containing Peripheral Arginine or Mixed Amino Acids as Potential Vectors for Gene and Drug Delivery. Macromol. Res. 2017, 25, 1172–1186. [Google Scholar] [CrossRef]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/hexachloroacetone#section=Safety-and-Hazards (accessed on 1 June 2022).
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Tetrafluorophthalic-anhydride#section=Safety-and-Hazards (accessed on 1 June 2022).
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/65578 (accessed on 1 June 2022).
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/24386#section=Safety-and-Hazards (accessed on 1 June 2022).






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfei, S.; Zuccari, G. One-Step, Low-Cost, Operator-Friendly, and Scalable Procedure to Synthetize Highly Pure N-(4-ethoxyphenyl)-retinamide in Quantitative Yield without Purification Work-Up. Molecules 2022, 27, 3632. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113632
Alfei S, Zuccari G. One-Step, Low-Cost, Operator-Friendly, and Scalable Procedure to Synthetize Highly Pure N-(4-ethoxyphenyl)-retinamide in Quantitative Yield without Purification Work-Up. Molecules. 2022; 27(11):3632. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113632
Chicago/Turabian StyleAlfei, Silvana, and Guendalina Zuccari. 2022. "One-Step, Low-Cost, Operator-Friendly, and Scalable Procedure to Synthetize Highly Pure N-(4-ethoxyphenyl)-retinamide in Quantitative Yield without Purification Work-Up" Molecules 27, no. 11: 3632. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113632