
Structures of Dimer-of-Dimers Type Defect Cubane Tetranuclear Copper(II) Complexes with Novel Dinucleating Ligands



Abstract
:1. Introduction
2. Results and Discussion
2.1. Preparation
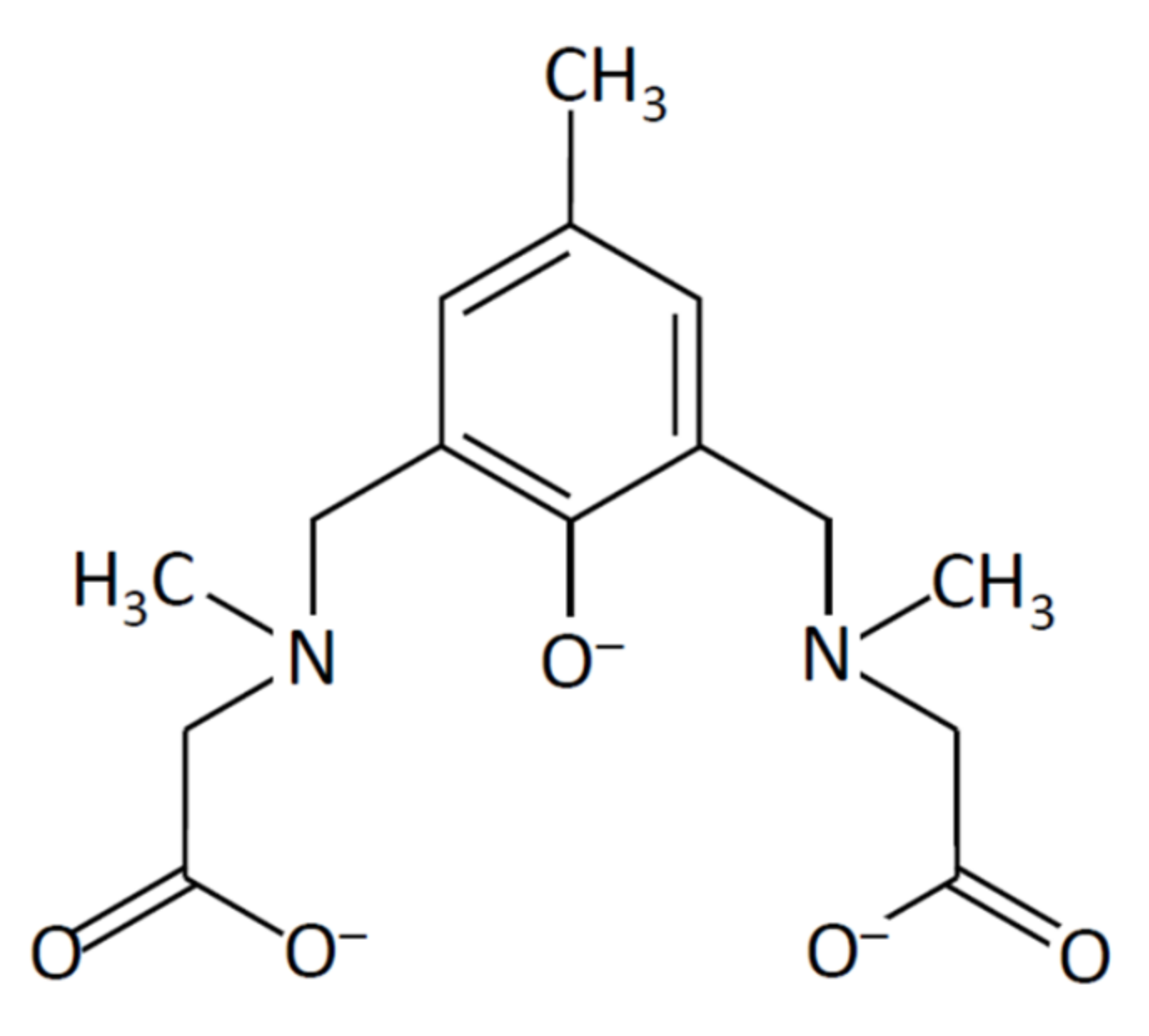
2.1.1. Preparation of a Dinucleating Ligand
2.1.2. Preparation of Copper(II) Complexes
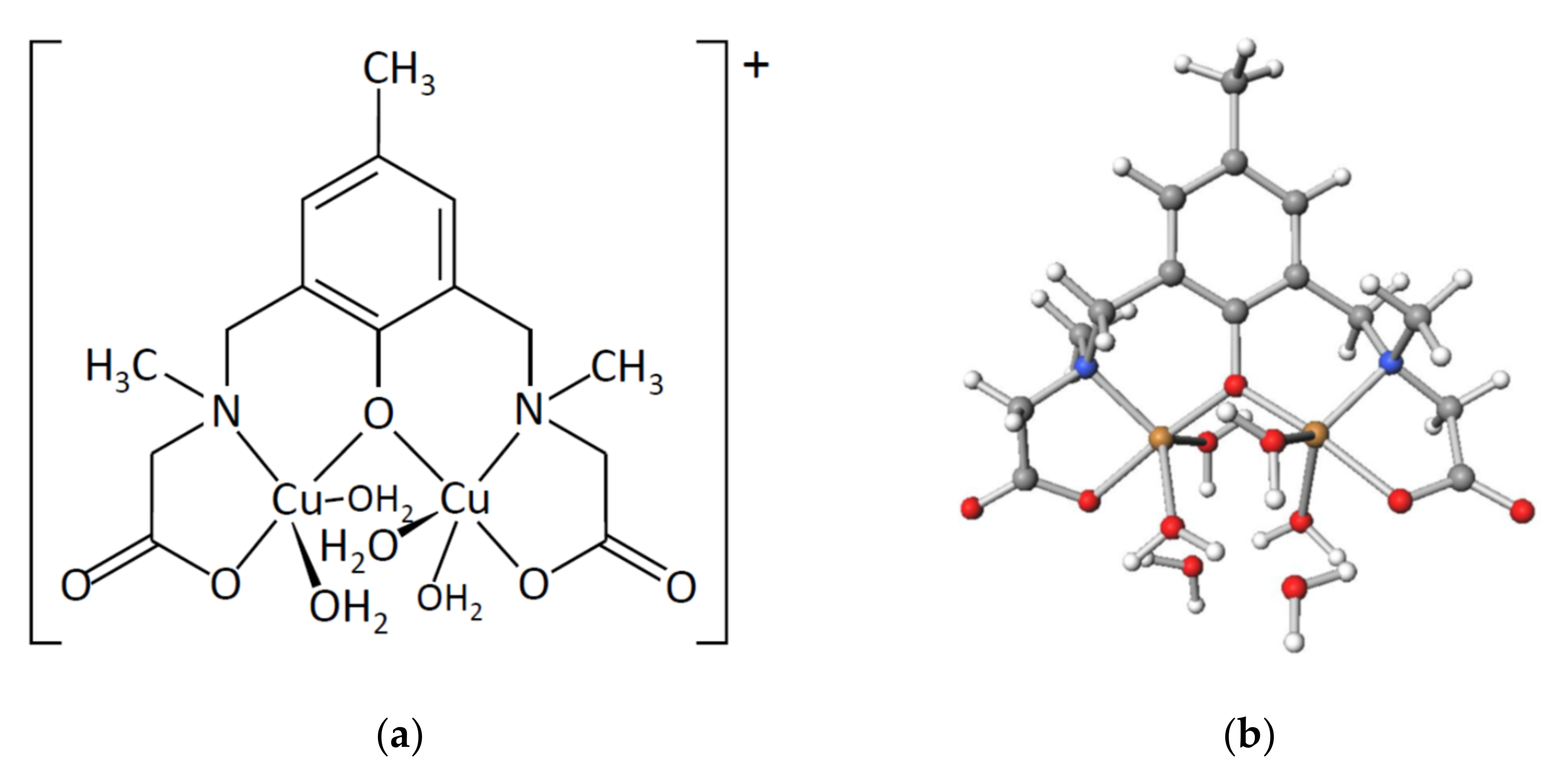
2.2. Crystal Structures of Copper(II) Complexes
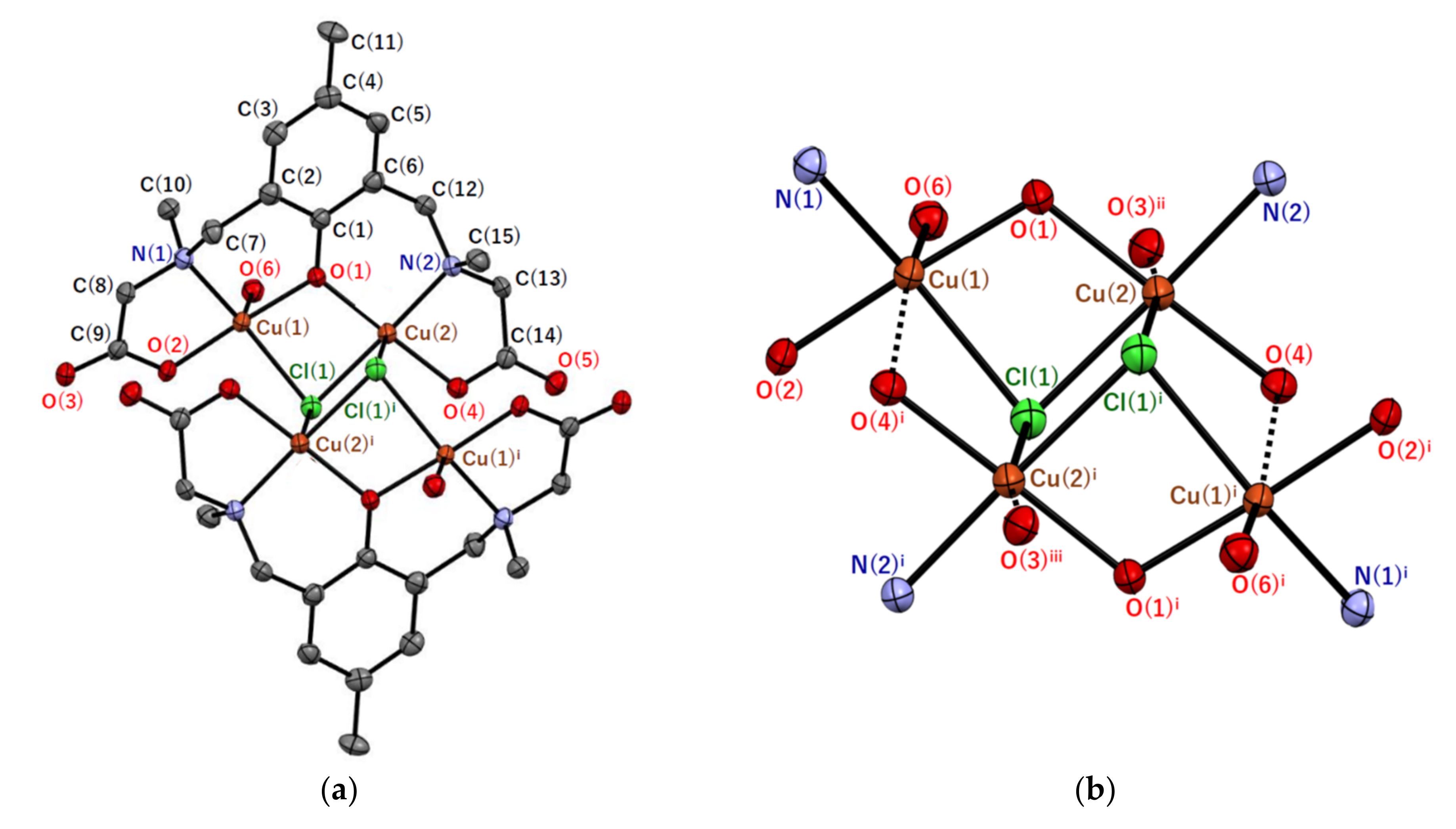
2.2.1. Crystal Structures of [Cu4(sym-cmp)2Cl2(H2O)2]·2.4CH3OH·1.8H2O (1′)
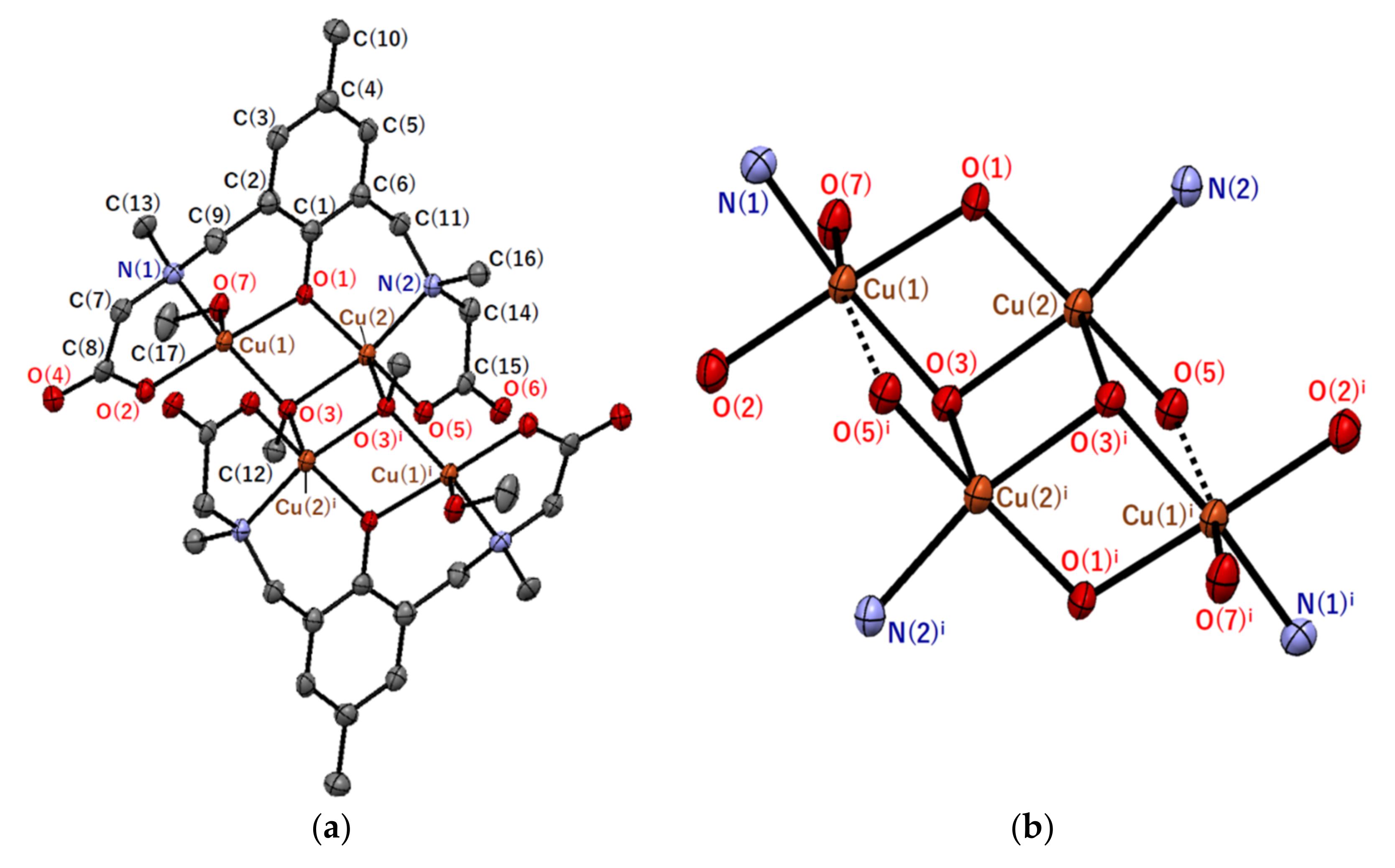
2.2.2. Crystal Structures of [Cu4(sym-cmp)2(CH3O)2(CH3OH)2]·2C3H7OH·2CH3OH (2)
2.3. Magnetic Properties
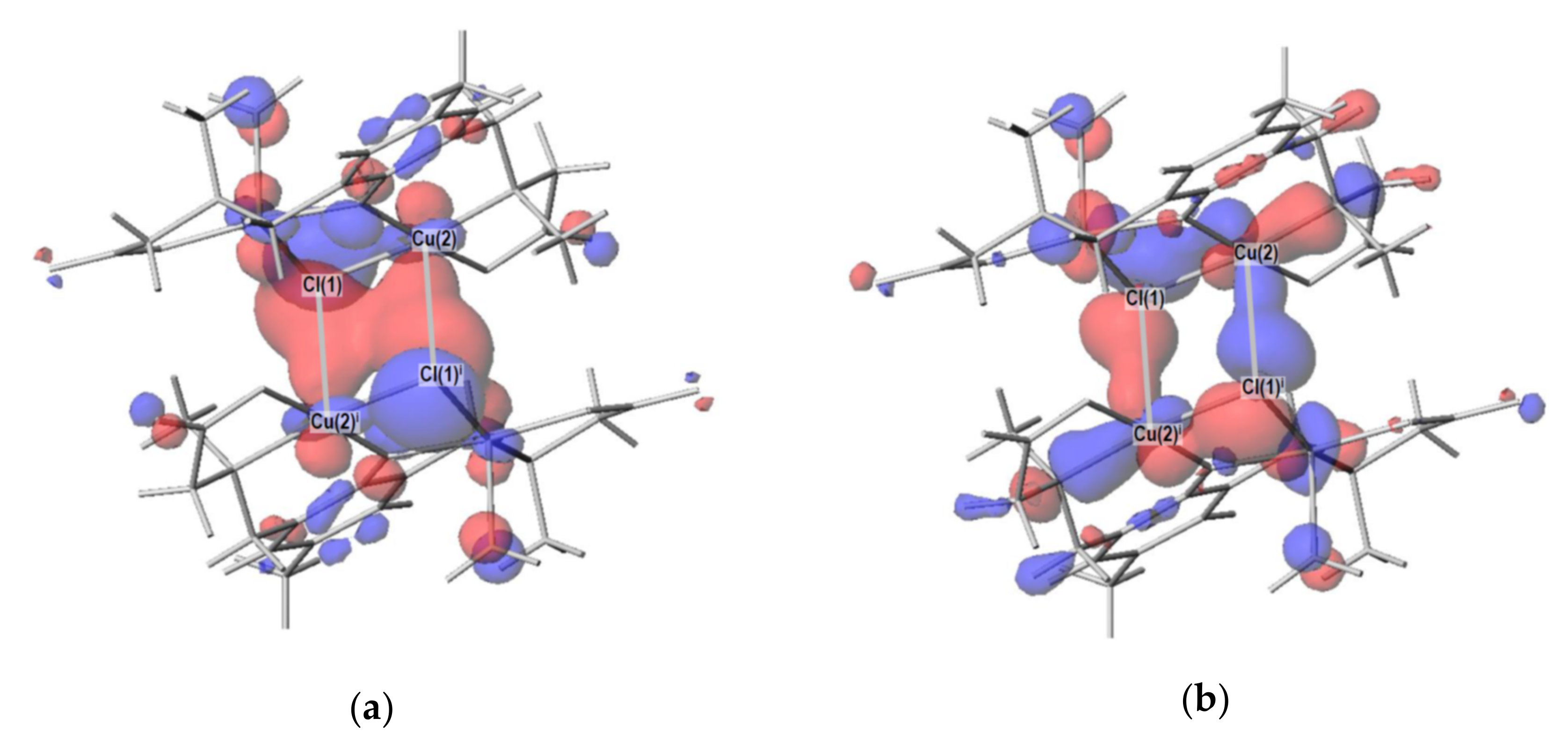
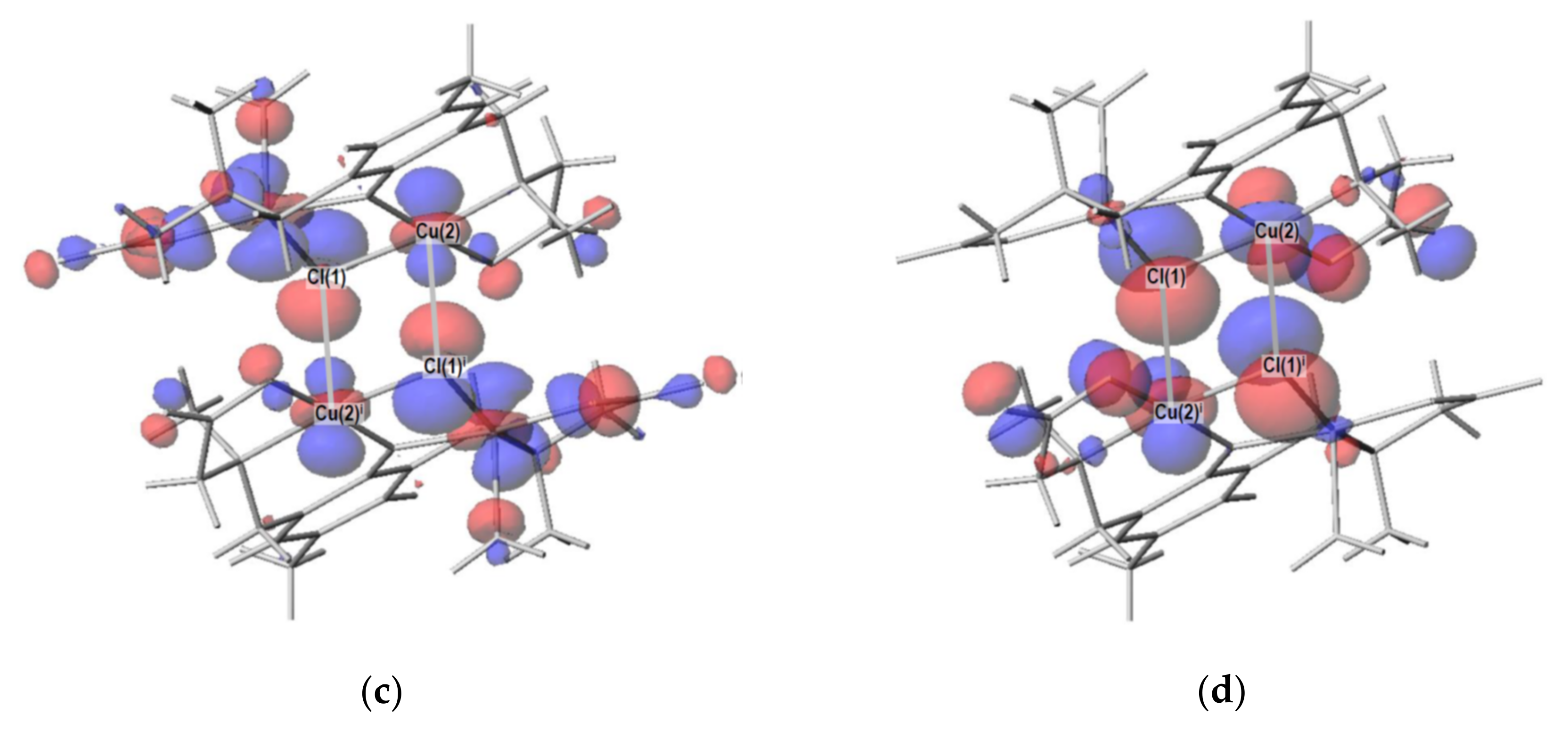
2.4. Density Functional Theory (DFT) Calculation
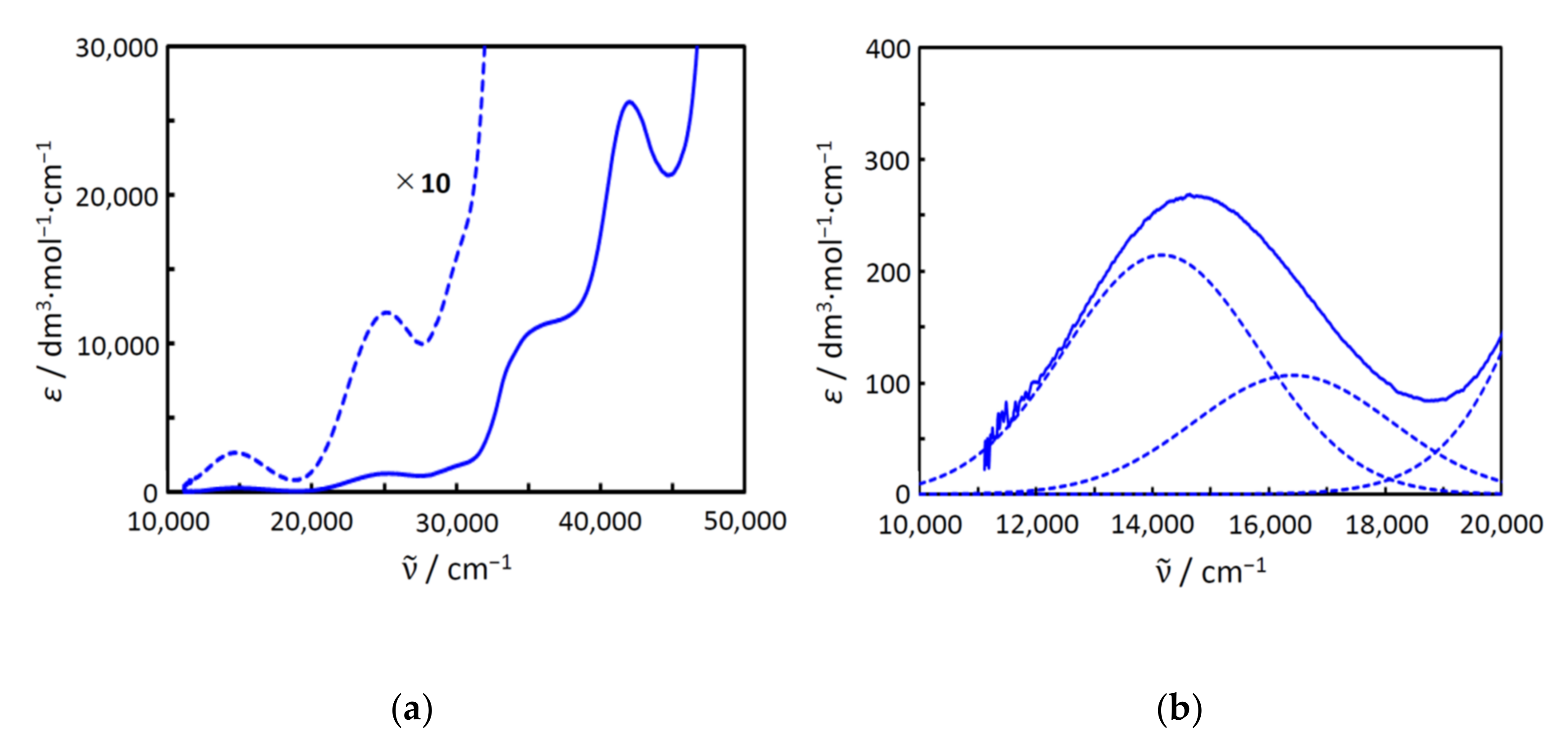
2.5. Electronic Spectra and Structure in Aqueous Solution
2.6. Structures of Metal Complexes and Ligand Design
3. Materials and Methods
3.1. Measurements
3.2. Materials
3.3. Preparations
3.4. Crystallography
3.5. Computation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kaim, W.; Schwederski, B. Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life; Wiley: Chichester, UK, 1991. [Google Scholar]
- Cowan, J.A. Inorganic Biochemistry; VCH: New York, NY, USA, 1993. [Google Scholar]
- Lippard, S.J.; Berg, J.M. Principles of Bioinorganic Chemistry; University Science Books: Mill Valley, CA, USA, 1994. [Google Scholar]
- Suzuki, M.; Kanatomi, H.; Murase, I. Synthesis and properties of binuclear cobalt(II) oxygen aduct with 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-methylphenol. Chem. Lett. 1981, 10, 1745–1748. [Google Scholar] [CrossRef]
- Murch, B.P.; Boyle, P.D.; Que, L., Jr. Structures of binuclear and tetranuclear iron(III) complexes as models for ferritin core formation. J. Am. Chem. Soc. 1985, 107, 6728–6729. [Google Scholar] [CrossRef]
- Murch, B.P.; Bradley, F.C.; Boyle, P.D.; Papaefthymiou, V.; Que, L., Jr. Iron-oxo aggregates. Crystal structures and solution characterization of 2-hydroxy-1,3-xylylenediaminetetraacetic acid complexes. J. Am. Chem. Soc. 1987, 109, 7993–8003. [Google Scholar] [CrossRef]
- Kazama, A.; Wada, A.; Sakiyama, H.; Hossain, M.J.; Nishida, Y. Synthesis of water-soluble dinuclear metal complexes [metal = cobalt(II) and nickel(II)] and their behavior in solution. Inorg. Chim. Acta 2008, 361, 2918–2922. [Google Scholar] [CrossRef]
- Jahn, H.A.; Teller, E. Stability of polyatomic molecules in degenerate electronic states I—Orbital degeneracy. Proc. R. Soc. Lond. Ser. A-Math. Phys. Sci. 1937, 161, 220–235. [Google Scholar]
- Tandon, S.S.; Thompson, L.K.; Bridson, J.N.; Bubenik, M. Synthesis and structural and magnetic properties of mononuclear, dinuclear, and tetranuclear copper(II) complexes of a 17-membered macrocyclic ligand (HM3), capable of forming endogenous phenoxide and pyridazino bridges. X-ray crystal structures of [Cu2(M3)(μ2-OMe)(NO3)2], [Cu4(M3)2(μ3-OMe)2(μ2-Cl)2Cl2], [Cu4(M3)2(μ3-OEt)2(μ2-N3)2(N3)2](MeOH), [Cu4(M3)2(μ3-OMe)2(NCS)4](DMF), and [Cu(M3)(NCS)2]. Inorg. Chem. 1993, 32, 4621–4631. [Google Scholar]
- Koikawa, M.; Yamashita, H.; Tokii, T. Crystal structures and magnetic properties of tetranuclear copper(II) complexes of N-(2-hydroxymethylphenyl)salicylideneimine with a defective double-cubane core. Inorg. Chim. Acta 2004, 357, 2635–2642. [Google Scholar] [CrossRef]
- Li, X.; Cheng, D.; Lin, J.; Li, Z.; Zheng, Y. Di-, tetra-, and hexanuclear hydroxy-bridged copper(II) cluster compounds: Syntheses, structures, and properties. Cryst. Growth Des. 2008, 8, 2853–2861. [Google Scholar] [CrossRef]
- Hatfield, W.E.; Inman, G.W. Spin-spin coupling in magnetically condensed complexes. IX. Exchange coupling constants for tetranuclear Schiff’s base complexes of copper(II). Inorg. Chem. 1969, 8, 1376–1378. [Google Scholar] [CrossRef]
- The IUPAC Compendium of Chemical Terminology (Gold Book Version 2.3.3). Available online: https://goldbook.iupac.org/ (accessed on 24 December 2021).
- Allen, G.C.; Hush, N.S. Reflectance spectrum and electronic states of the CuCl53– ion in a number of crystal lattices. Inorg. Chem. 1967, 6, 4–8. [Google Scholar] [CrossRef]
- Tone, K.; Sakiyama, H.; Mikuriya, M.; Yamasaki, M.; Nishida, Y. Magnetic behavior of dinuclear cobalt(II) complexes assumed to be caused by a paramagnetic impurity can be explained by tilts of local distortion axes. Inorg. Chem. Commun. 2007, 10, 944–947. [Google Scholar] [CrossRef]
- Sakiyama, H.; Kato, M.; Sasaki, S.; Tasaki, M.; Asato, E.; Koikawa, M. Synthesis and magnetic properties of a dinuclear manganese(II) complex with two manganese(II) ions of C2-twisted octahedral geometry. Polyhedron 2016, 111, 32–37. [Google Scholar] [CrossRef]
- Sakiyama, H.; Takahata, S.; Kashimoto, N.; Mitsuhashi, R.; Mikuriya, M. Crystal structure of a dinuclear magnesium(II) complex with 4-chloro-2,6-bis[(2-hydroxyethyl)methylaminomethyl]phenolate. X-ray. Struct. Anal. Online 2017, 33, 75–76. [Google Scholar] [CrossRef]
- Deutsch, M.; Claiser, N.; Gillet, J.-M.; Lecomte, C.; Sakiyama, H.; Tone, K.; Souhassou, M. d-Orbital orientation in a dimer cobalt complex: Link to magnetic properties? Acta Cryst. 2011, B67, 324–332. [Google Scholar] [CrossRef]
- Ridier, K.; Gillon, B.; Gukasov, A.; Chaboussant, G.; Cousson, A.; Luneau, D.; Borta, A.; Jacquot, J.-F.; Checa, R.; Chiba, Y.; et al. Polarized neutron diffraction as a tool for mapping molecular magnetic anisotropy: Local susceptibility tensors in CoII complexes. Chem. Eur. J. 2016, 22, 724–735. [Google Scholar] [CrossRef]
- Sakiyama, H.; Tone, K.; Yamasaki, M.; Mikuriya, M. Electronic spectrum and magnetic properties of a dinuclear nickel(II) complex with two nickel(II) ions of C2-twisted octahedral geometry. Inorg. Chim. Acta 2011, 365, 183–189. [Google Scholar] [CrossRef]
- Sakiyama, H.; Chiba, Y.; Tone, K.; Yamasaki, M.; Mikuriya, M.; Krzystek, J.; Ozarowski, A. Magnetic properties of a dinuclear nickel(II) complex with 2,6-bis[(2-hydroxyethyl)methylaminomethyl]-4-methylphenolate. Inorg. Chem. 2017, 56, 138–146. [Google Scholar] [CrossRef]
- Pan, Y.; Rao, C.Y.; Tan, X.L.; Ling, Y.; Singh, A.; Kumar, A.; Li, B.H.; Liu, J.Q. Cobalt-seamed C-methylpyrogallol[4]arene nanocapsules-derived magnetic carbon cubes as advanced adsorbent toward drug contaminant removal. Chem. Eng. J. 2021, 133857. [Google Scholar] [CrossRef]
- Zhong, Y.Y.; Chen, C.; Liu, S.; Lu, C.Y.; Liu, D.; Pan, Y.; Sakiyama, H.; Muddassir, M.; Liu, J.Q. A new magnetic adsorbent of eggshell-zeolitic imidazolate framework for highly efficient removal of norfloxacin. Dalton Trans. 2021, 50, 18016–18026. [Google Scholar] [CrossRef]
- Sun, Y.M.; Jiang, X.D.; Liu, Y.W.; Liu, D.; Chen, C.; Lu, C.Y.; Zhuang, S.Z.; Kumar, A.; Liu, J.Q. Recent advances in Cu(II)/Cu(I)-MOFs based nano-platforms for developing new nano-medicines. J. Inorg. Biochem. 2021, 225, 111599. [Google Scholar] [CrossRef]
- Liu., Y.W.; Zhou, L.Y.; Dong, Y.; Wang, R.; Pan, Y.; Zhuang, S.Z.; Liu, D.; Liu, J.Q. Recent developments on MOF-based platforms for antibacterial therapy. RSC Med. Chem. 2021, 12, 915–928. [Google Scholar] [CrossRef]
- Ding, Q.J.; Liu, Y.W.; Shi, C.C.; Xiao, J.F.; Dai, W.; Liu, D.; Chen, H.Y.; Li, B.H.; Liu, J.Q. Applications of ROS-InducedZr-MOFs platform in multimodal synergistic therapy. Mini-Rev. Med. Chem. 2021, 21, 1718–1733. [Google Scholar] [CrossRef]
- Qiu, Y.Z.; Tan, G.J.; Fang, Y.Q.; Liu, S.; Zhou, Y.B.; Kumar, A.; Trivedi, M.; Liu, D.; Liu, J.Q. Biomedical applications of metal–organic framework (MOF)-based nano-enzymes. New J. Chem. 2021, 45, 20987–21000. [Google Scholar] [CrossRef]
- Wang, J.; Rao, C.Y.; Lu, L.; Zhang, S.L.; Muddassir, M.; Liu, J.Q. Efficient photocatalytic degradation of methyl violet using two new 3D MOFs directed by different carboxylate spacers. Cryst. Eng. Comm. 2021, 23, 741–747. [Google Scholar] [CrossRef]
- Liu, J.Q.; Luo, Z.D.; Pan, Y.; Singh, A.K.; Trivedi, M.; Kumar, A. Recent developments in luminescent coordination polymers: Designing strategies, sensing application and theoretical evidences. Coord. Chem. Rev. 2020, 406, 213145. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Advances in Electronic Structure Theory; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom–Atom 1 | Distance/Å | Atom–Atom 1 | Distance/Å |
|---|---|---|---|
| Cu(1)–Cl(1) | 2.3638(14) | Cu(1)–O(1) | 1.937(3) |
| Cu(1)–O(2) | 1.926(3) | Cu(1)–O(6) | 2.214(4) |
| Cu(1)–N(1) | 1.998(4) | Cu(1)–O(4)i | 3.208(4) |
| Cu(2)–Cl(1) | 2.3113(14) | Cu(2)–O(1) | 1.942(3) |
| Cu(2)–O(4) | 1.936(3) | Cu(2)–N(2) | 2.003(4) |
| Cu(2)–Cl(1)i | 2.8012(14) | Cu(2)–O(3)ii | 2.804(4) |
| Cu(1)···Cu(2) | 3.1274(8) | Cu(1)···Cu(1)i | 5.8824(12) |
| Cu(1)···Cu(2)i | 3.8024(9) | Cu(2)···Cu(2)i | 3.7248(13) |
| Atom–Atom–Atom 1 | Angle/° | Atom–Atom–Atom 1 | Angle/° |
|---|---|---|---|
| Cl(1)–Cu(1)–O(1) | 83.38(11) | Cl(1)–Cu(1)–O(2) | 94.87(10) |
| Cl(1)–Cu(1)–O(6) | 94.72(10) | Cl(1)–Cu(1)–N(1) | 161.10(12) |
| Cl(1)–Cu(1)–O(4)i | 77.29(7) | O(1)–Cu(1)–O(2) | 173.59(15) |
| O(1)–Cu(1)–O(6) | 90.83(14) | O(1)–Cu(1)–N(1) | 94.06(15) |
| O(1)–Cu(1)–O(4)i | 92.89(12) | O(2)–Cu(1)–O(6) | 95.46(14) |
| O(2)–Cu(1)–N(1) | 85.61(15) | O(2)–Cu(1)–O(4)i | 80.71(12) |
| O(6)–Cu(1)–N(1) | 104.06(15) | O(6)–Cu(1)–O(4)i | 170.73(12) |
| N(1)–Cu(1)–O(4)i | 84.16(13) | Cl(1)–Cu(2)–O(1) | 84.68(11) |
| Cl(1)–Cu(2)–O(4) | 95.38(11) | Cl(1)–Cu(2)–N(2) | 172.22(13) |
| Cl(1)–Cu(2)–Cl(1)i | 86.96(5) | Cl(1)–Cu(2)–O(3)ii | 84.35(8) |
| O(1)–Cu(2)–O(4) | 175.93(15) | O(1)–Cu(2)–N(2) | 94.72(16) |
| O(1)–Cu(2)–Cl(1)i | 89.05(11) | O(1)i–Cu(2)–O(3)ii | 130.04(9) |
| O(4)–Cu(2)–N(2) | 84.66(15) | O(4)–Cu(2)–Cl(1)i | 95.02(11) |
| O(4)–Cu(2)–O(3)ii | 88.47(12) | N(2)–Cu(2)–Cl(1)i | 100.79(12) |
| N(2)–Cu(2)–O(3)ii | 87.87(14) | Cl(1)i–Cu(2)–O(3)ii | 170.91(8) |
| Cu(1)–Cl(1)–Cu(2) | 83.96(5) | Cu(1)–O(1)–Cu(2) | 107.46(17) |
| Cu(1)–Cl(1)–Cu(2)i | 94.44(4) | Cu(2)–Cl(1)–Cu(2)i | 93.04(5) |
| Atom–Atom 1 | Distance/Å | Atom–Atom 1 | Distance/Å |
|---|---|---|---|
| Cu(1)–O(1) | 1.930(3) | Cu(1)–O(2) | 1.933(3) |
| Cu(1)–O(3) | 1.981(3) | Cu(1)–O(7) | 2.331(3) |
| Cu(1)–N(1) | 2.008(4) | Cu(1)–O(5)i | 2.856(3) |
| Cu(2)–O(1) | 1.930(3) | Cu(2)–O(3) | 1.999(3) |
| Cu(2)–O(5) | 1.936(3) | Cu(2)–N(2) | 2.019(4) |
| Cu(2)–O(3)i | 2.305(3) | Cu(2)···O(6)ii | 3.3478(3) |
| Cu(1)···Cu(2) | 3.0138(8) | Cu(1)···Cu(1)i | 5.5123(12) |
| Cu(1)···Cu(2)i | 3.3743(9) | Cu(2)···Cu(2)i | 3.2483(11) |
| Atom–Atom–Atom 1 | Angle/° | Atom–Atom–Atom 1 | Angle/° |
|---|---|---|---|
| O(1)–Cu(1)–O(2) | 174.14(13) | O(1)–Cu(1)–O(3) | 79.22(13) |
| O(1)–Cu(1)–O(7) | 88.52(13) | O(1)–Cu(1)–N(1) | 93.40(14) |
| O(1)–Cu(1)–O(5)i | 89.28(12) | O(2)–Cu(1)–O(3) | 100.38(14) |
| O(2)–Cu(1)–O(7) | 97.33(13) | O(2)–Cu(1)–N(1) | 85.53(14) |
| O(2)–Cu(1)–O(5)i | 84.96(11) | O(3)–Cu(1)–O(7) | 90.78(13) |
| O(3)–Cu(1)–N(1) | 164.26(14) | O(3)–Cu(1)–O(5)i | 76.54(11) |
| O(7)–Cu(1)–N(1) | 103.00(14) | O(7)–Cu(1)–O(5)i | 167.32(11) |
| N(1)–Cu(1)–O(5)i | 89.59(13) | O(1)–Cu(2)–O(3) | 78.76(13) |
| O(1)–Cu(2)–O(5) | 170.63(13) | O(1)–Cu(2)–N(2) | 93.49(14) |
| O(1)–Cu(2)–O(3)i | 96.27(13) | O(3)–Cu(2)–O(5) | 99.19(13) |
| O(3)–Cu(2)–N(2) | 164.05(15) | O(3)–Cu(2)–O(3)i | 82.25(13) |
| O(5)–Cu(2)–N(2) | 86.18(14) | O(5)–Cu(2)–O(3)i | 92.48(12) |
| N(2)–Cu(2)–O(3)i | 112.69(13) | Cu(1)–O(1)–Cu(2) | 102.66(15) |
| Cu(1)–O(3)–Cu(2) | 98.45(14) | Cu(1)–O(3)–Cu(2)i | 103.63(13) |
| Cu(2)–O(3)–Cu(2)i | 97.75(12) |
| Complex | 1′ | 2 |
|---|---|---|
| Empirical formula 1 | C16.2H27.6ClCu2N2O8.1 | C21H38Cu2N2O9 |
| Formula weight 1 | 542.53 | 589.61 |
| Crystal system | Monoclinic | triclinic |
| Space group | C2/c | |
| a/Å | 27.0693(14) | 8.5827(6) |
| b/Å | 13.2690(5) | 13.0679(8) |
| c/Å | 13.1356(7) | 13.2862(7) |
| α/° | 90 | 115.021(6) |
| β/° | 100.422(5) | 102.208(5) |
| γ/° | 90 | 115.021(6) |
| V/Å 3 | 4640.2(4) | 1263.54(15) |
| Z 1 | 8 | 2 |
| Crystal dimensions/mm | 0.070 × 0.050 × 0.030 | 0.130 × 0.057 × 0.038 |
| T/K | 100 | 100 |
| λ/Å | 0.71073 | 0.71073 |
| ρcalcd/g·cm−3 | 1.553 | 1.645 |
| µ/mm−1 | 1.990 | 1.734 |
| F(000) | 2229 | 616 |
| 2θmax/◦ | 55 | 55 |
| No. of reflections measured | 9520 | 16014 |
| No. of independent reflections | 9520 (Rint = 0.0623) | 5771 (Rint = 0.0770) |
| Data/restraints/parameters | 9520/3/296 | 5771/45/341 |
| R1 2 [I > 2.00 σ(I)] | 0.0575 | 0.0674 |
| wR2 3 (all reflections) | 0.1664 | 0.1635 |
| Goodness of fit indicator | 1.020 | 0.992 |
| Highest peak, deepest hole/e Å−3 | 1.787, −0.647 | 1.669, −1.068 |
| CCDC deposition number | 2130618 | 2130619 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoshikawa, R.; Mitsuhashi, R.; Asato, E.; Liu, J.; Sakiyama, H. Structures of Dimer-of-Dimers Type Defect Cubane Tetranuclear Copper(II) Complexes with Novel Dinucleating Ligands. Molecules 2022, 27, 576. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27020576
Hoshikawa R, Mitsuhashi R, Asato E, Liu J, Sakiyama H. Structures of Dimer-of-Dimers Type Defect Cubane Tetranuclear Copper(II) Complexes with Novel Dinucleating Ligands. Molecules. 2022; 27(2):576. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27020576
Chicago/Turabian StyleHoshikawa, Ryusei, Ryoji Mitsuhashi, Eiji Asato, Jianqiang Liu, and Hiroshi Sakiyama. 2022. "Structures of Dimer-of-Dimers Type Defect Cubane Tetranuclear Copper(II) Complexes with Novel Dinucleating Ligands" Molecules 27, no. 2: 576. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27020576