
Functionalized Crystalline N-Trimethyltriindoles: Counterintuitive Influence of Peripheral Substituents on Their Semiconducting Properties

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
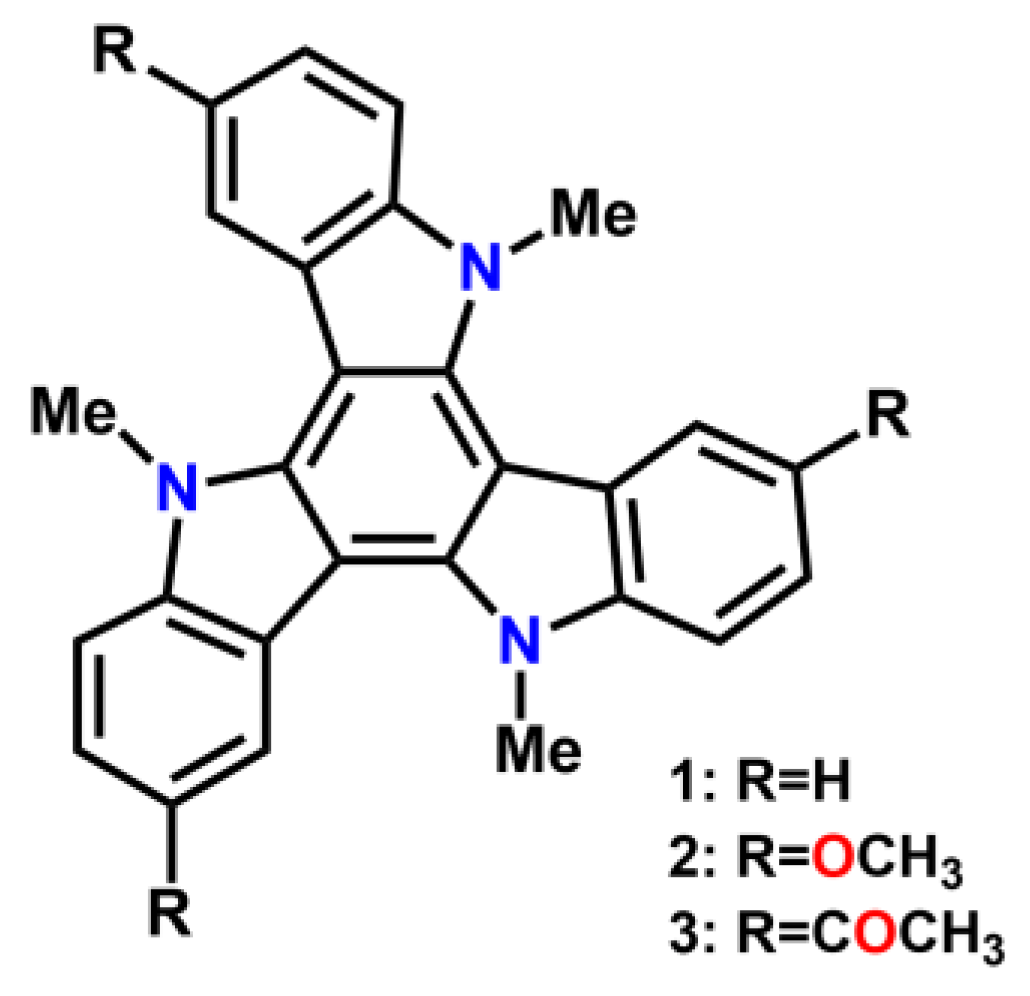
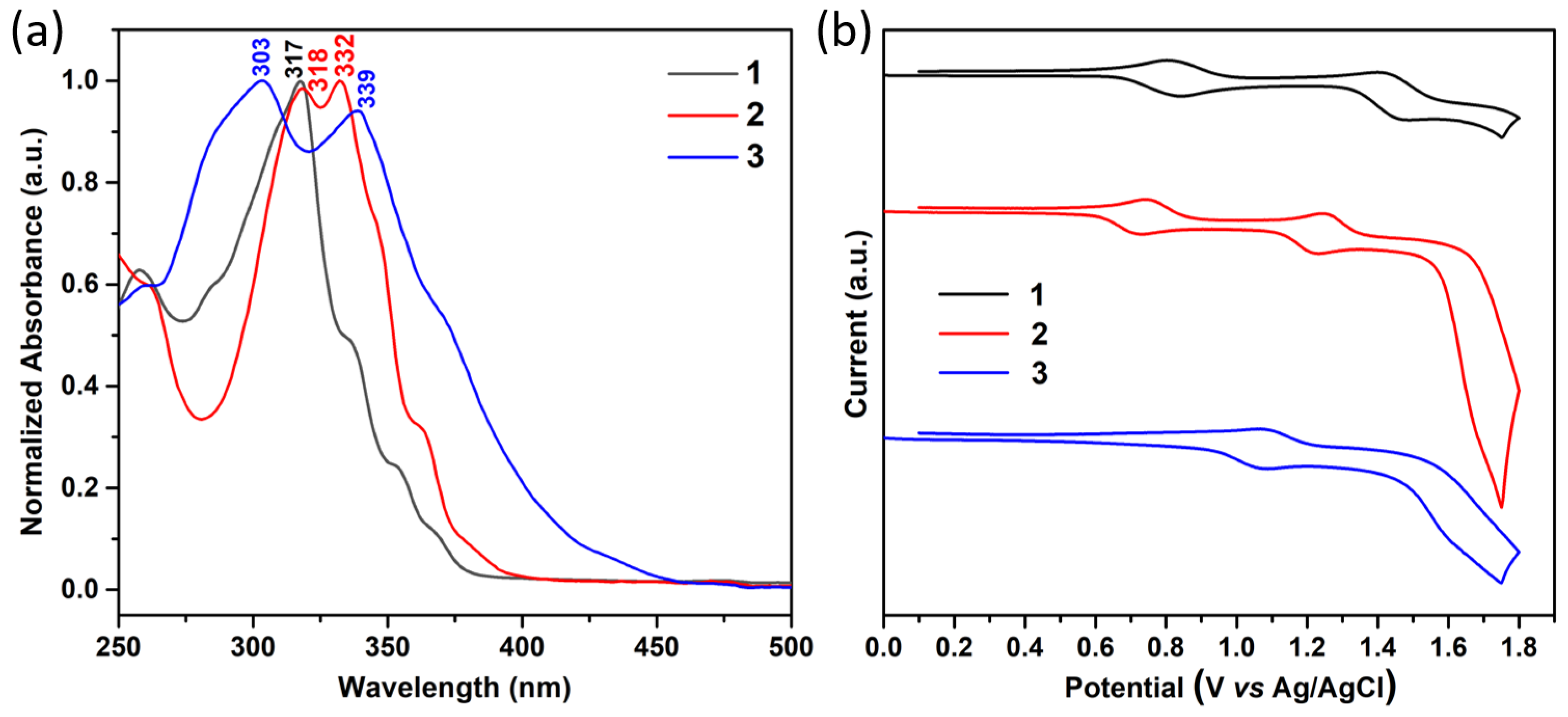
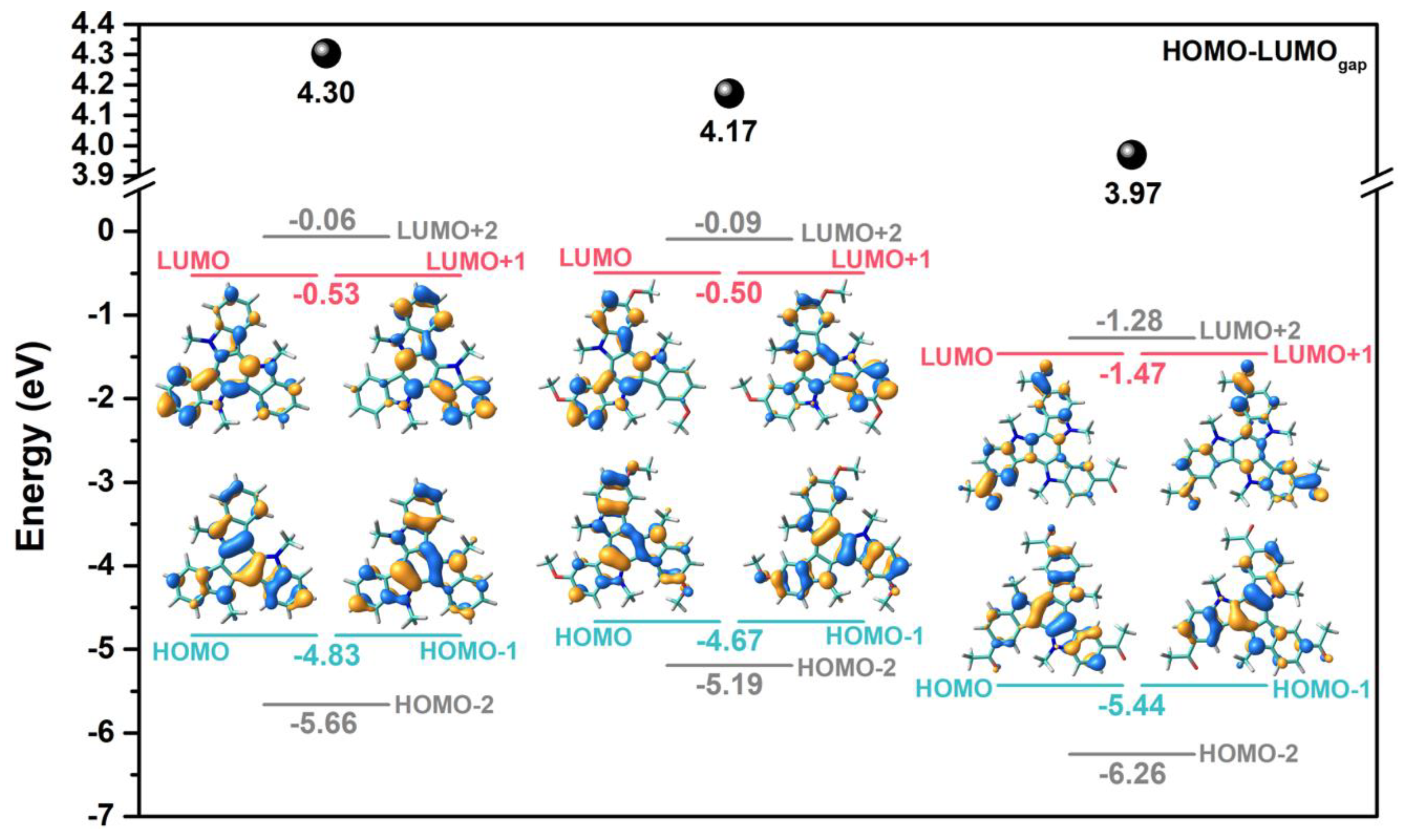
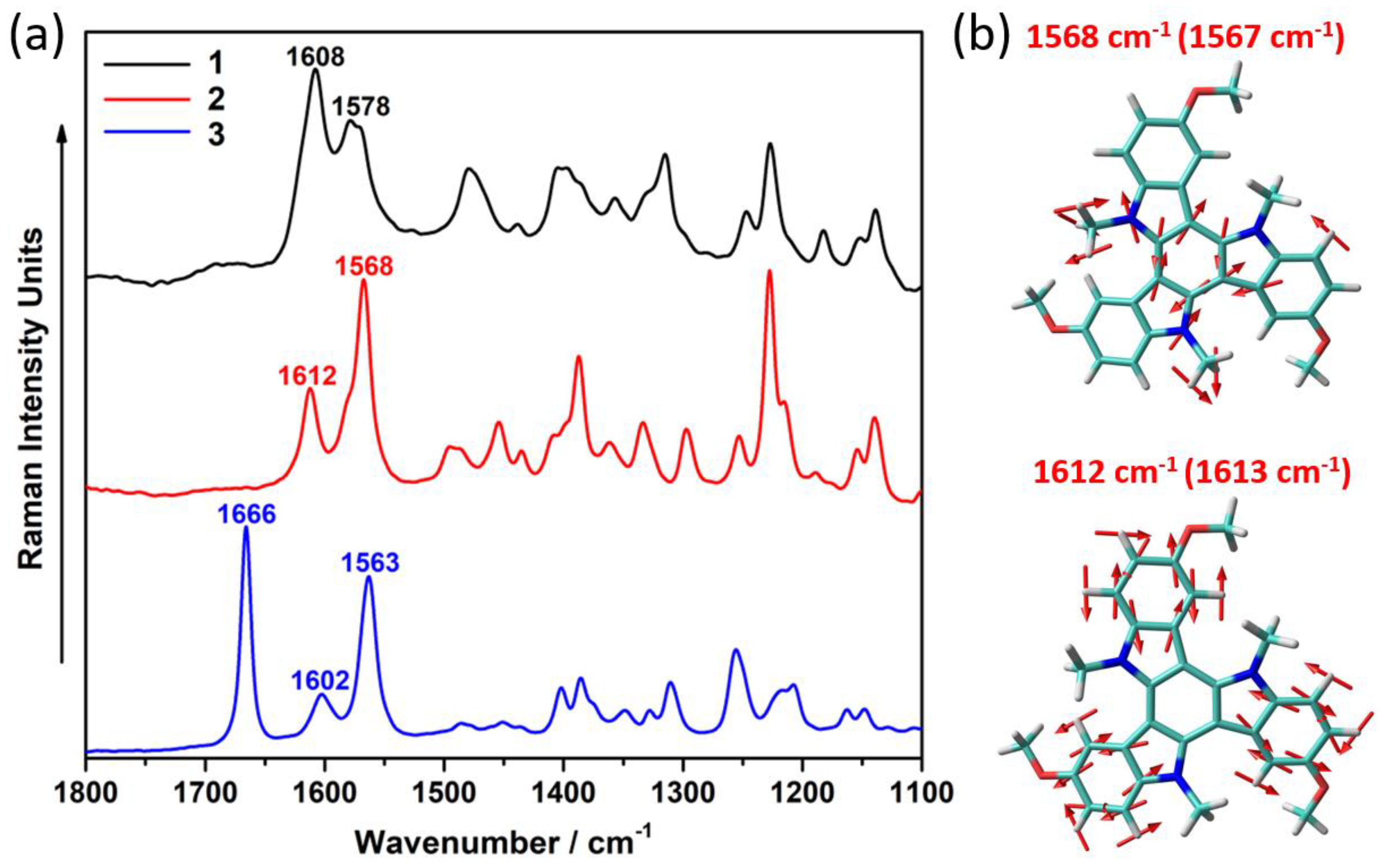
2.1. Synthesis and Electronic Properties
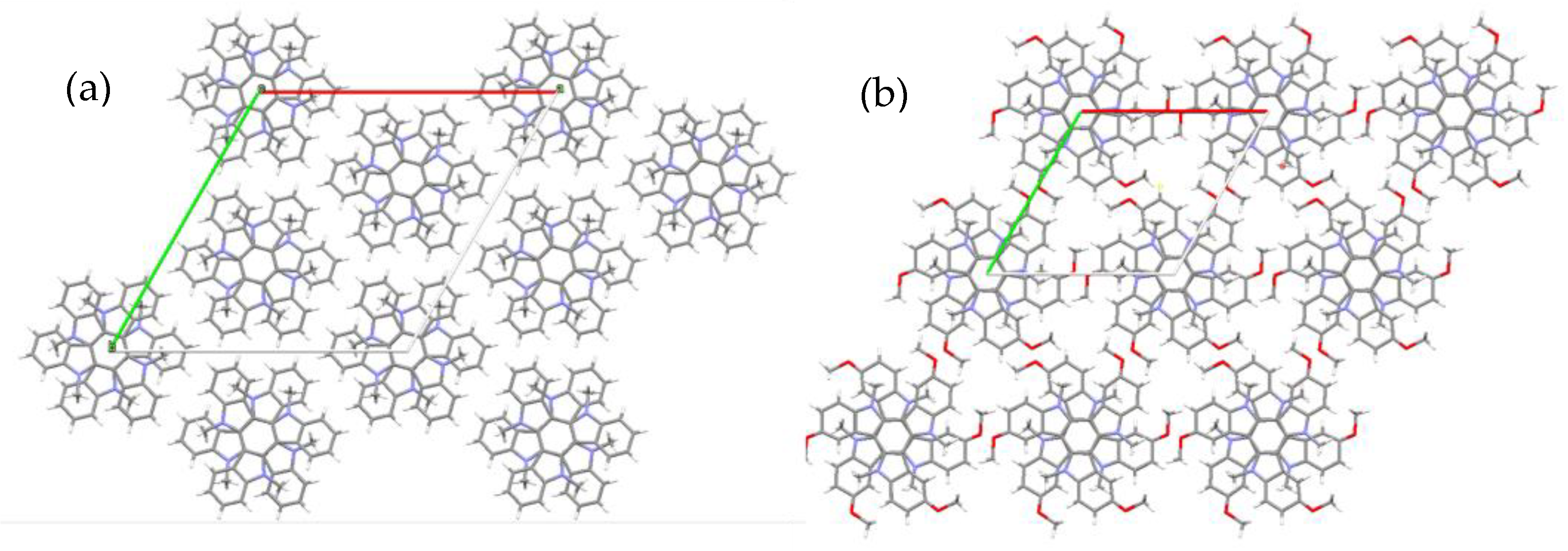
2.2. Crystal Structure
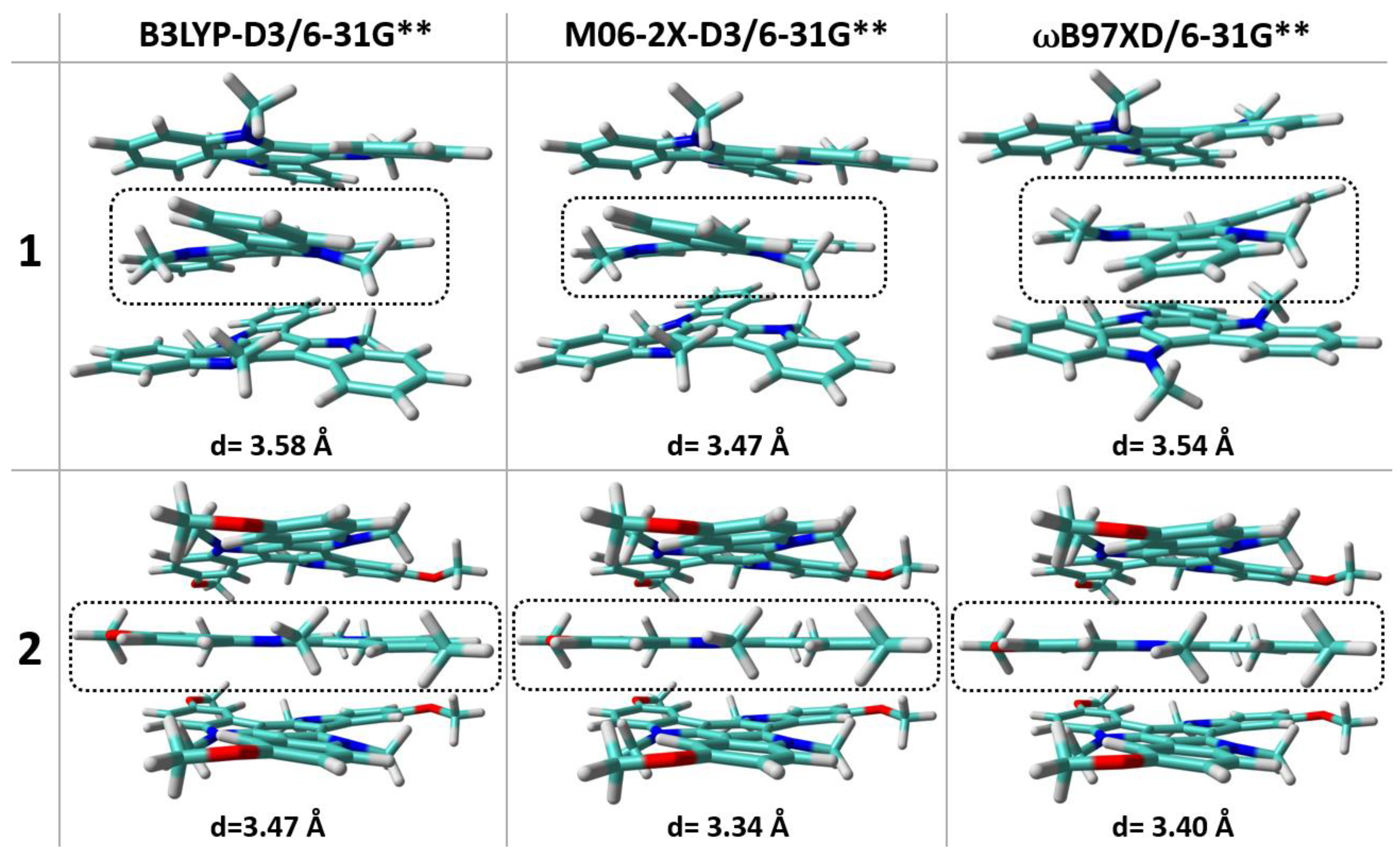
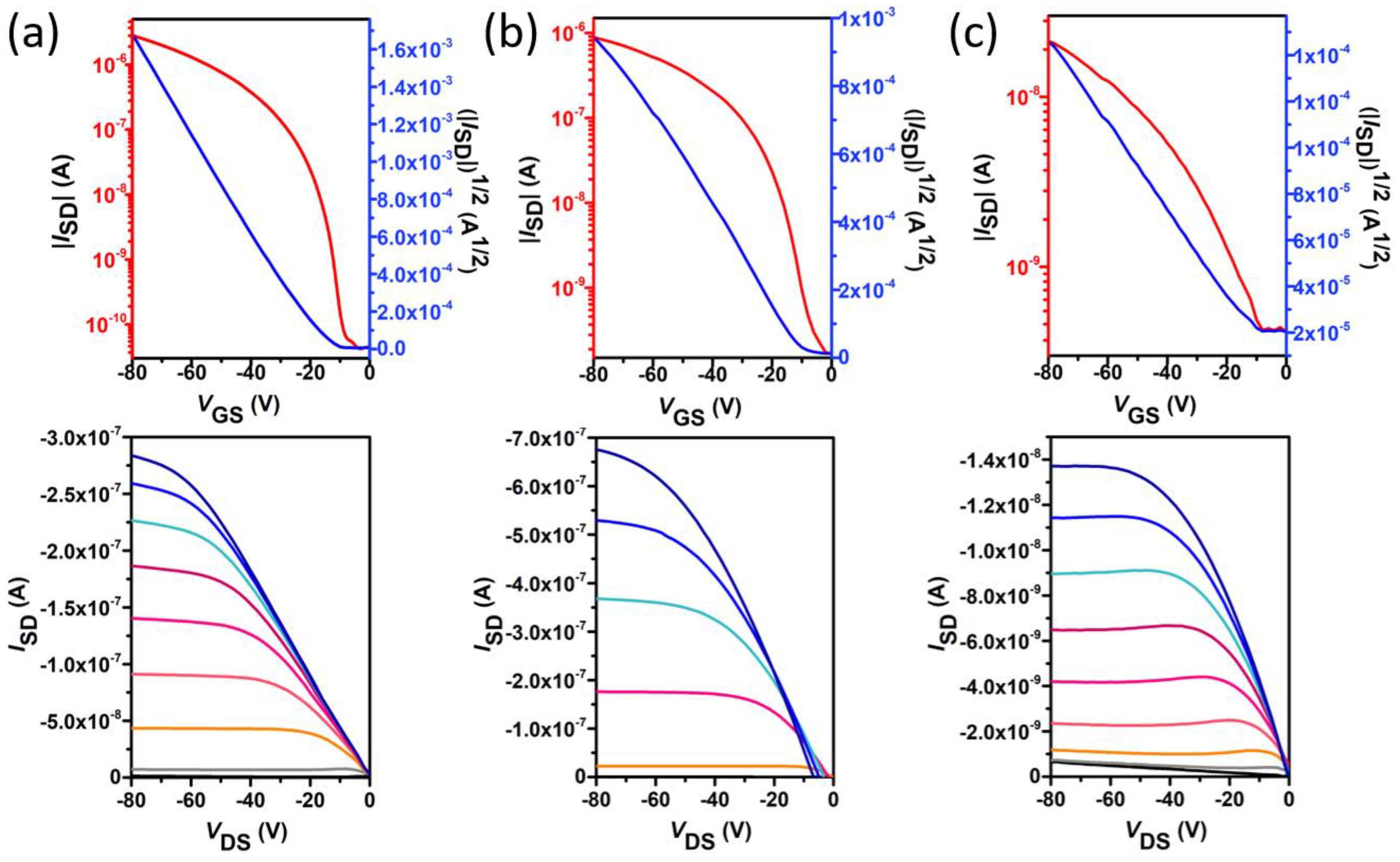
2.3. Electrical Characterization and Charge–Transport Parameters
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reig, M.; Bagdziunas, G.; Ramanavicius, A.; Puigdollers, J.; Velasco, D. Interface engineering and solid-state organization for triindole-based p-type organic thin-film transistors. Phys. Chem. Chem. Phys. 2018, 20, 17889–17898. [Google Scholar] [CrossRef] [PubMed]
- Reig, M.; Puigdollers, J.; Velasco, D. Molecular order of air-stable p-type organic thin-film transistors by tuning the extension of the π-conjugated core: The cases of indolo[3,2-b]carbazole and triindole semiconductors. J. Mater. Chem. C 2015, 3, 506–513. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, C.; Arrechea-Marcos, I.; Benito-Hernández, A.; Gutierrez-Puebla, E.; Monge, M.A.; López Navarrete, J.T.; Ruiz Delgado, M.C.; Ortiz, R.P.; Gómez-Lor, B. Solution-processed N-trialkylated triindoles for organic field effect transistors. J. Mater. Chem. C 2018, 6, 50–56. [Google Scholar] [CrossRef]
- Ruiz, C.; Pandey, U.K.; Termine, R.; García-Frutos, E.M.; López-Espejo, G.; Ortiz, R.P.; Huang, W.; Marks, T.J.; Facchetti, A.; Ruiz Delgado, M.C.; et al. Mobility versus Alignment of a Semiconducting π-Extended Discotic Liquid-Crystalline Triindole. ACS Appl. Mater. Interfaces 2016, 8, 26964–26971. [Google Scholar] [CrossRef]
- Qian, X.; Lu, L.; Zhu, Y.-Z.; Gao, H.-H.; Zheng, J.-Y. Triazatruxene-based organic dyes containing a rhodanine-3-acetic acid acceptor for dye-sensitized solar cells. Dye. Pigment. 2015, 113, 737–742. [Google Scholar] [CrossRef]
- Ghosh, N.N.; Habib, M.; Pramanik, A.; Sarkar, P.; Pal, S. Molecular engineering of anchoring groups for designing efficient triazatruxene-based organic dye-sensitized solar cells. New J. Chem. 2019, 43, 6480–6491. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, X.; Cai, B.; Wang, H.; Yu, Z.; Sun, L. Triazatruxene-based sensitizers for highly efficient solid-state dye-sensitized solar cells. Sol. Energy 2020, 212, 1–5. [Google Scholar] [CrossRef]
- Kil, D.R.; Lu, C.H.; Ji, J.-M.; Kim, C.H.; Kim, H.-K. Dopant-Free Triazatruxene-Based Hole Transporting Materials with Three Different End-Capped Acceptor Units for Perovskite Solar Cells. Nanomaterials 2020, 10, 936. [Google Scholar] [CrossRef]
- Rakstys, K.; Abate, A.; Dar, M.I.; Gao, P.; Jankauskas, V.; Jacopin, G.; Kamarauskas, E.; Kazim, S.; Ahmad, S.; Grätzel, M.; et al. Triazatruxene-Based Hole Transporting Materials for Highly Efficient Perovskite Solar Cells. J. Am. Chem. Soc. 2015, 137, 16172–16178. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, S.; Wu, X.; Xu, Y.; Li, H.; Liu, Y.; Tong, H.; Wang, L. Triazatruxene-based small molecules with thermally activated delayed fluorescence, aggregation-induced emission and mechanochromic luminescence properties for solution-processable nondoped OLEDs. J. Mater. Chem. C 2018, 6, 12503–12508. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Y.; Wu, S.; Zhao, Y.; Yu, T.; Su, W.; Ma, H.; Qian, L.; Wang, R. Benzo[d]imidazole-functionalized triazatruxenes as the emitting materials for solution-processed non-doped OLEDs. Dye. Pigment. 2021, 188, 109165. [Google Scholar] [CrossRef]
- dos Santos, P.L.; Ward, J.S.; Congrave, D.G.; Batsanov, A.S.; Eng, J.; Stacey, J.E.; Penfold, T.J.; Monkman, A.P.; Bryce, M.R. Triazatruxene: A Rigid Central Donor Unit for a D–A3 Thermally Activated Delayed Fluorescence Material Exhibiting Sub-Microsecond Reverse Intersystem Crossing and Unity Quantum Yield via Multiple Singlet–Triplet State Pairs. Adv. Sci. 2018, 5, 1700989. [Google Scholar] [CrossRef] [Green Version]
- Coya, C.; Ruiz, C.; Álvarez, Á.L.; Álvarez-García, S.; García-Frutos, E.M.; Gómez-Lor, B.; de Andrés, A. Star-shaped hexaaryltriindoles small molecules: Tuning molecular properties towards solution processed organic light emitting devices. Org. Electron. 2012, 13, 2138–2148. [Google Scholar] [CrossRef]
- Hu, Y.-C.; Lin, Z.-L.; Huang, T.-C.; Lee, J.-W.; Wei, W.-C.; Ko, T.-Y.; Lo, C.-Y.; Chen, D.-G.; Chou, P.-T.; Hung, W.-Y.; et al. New exciplex systems composed of triazatruxene donors and N-heteroarene-cored acceptors. Mater. Chem. Front. 2020, 4, 2029–2039. [Google Scholar] [CrossRef]
- Zhao, F.; Liu, C.; Sun, Y.; Li, Q.; Zhao, J.; Li, Z.; Zhang, B.; Lu, C.; Li, Q.; Qiao, S.; et al. Controlled self-assembly of Triazatruxene overlength microwires for optical waveguide. Org. Electron. 2019, 74, 276–281. [Google Scholar] [CrossRef]
- Gallego-Gómez, F.; García-Frutos, E.M.; Villalvilla, J.M.; Quintana, J.A.; Gutierrez-Puebla, E.; Monge, A.; Díaz-García, M.A.; Gómez-Lor, B. Very Large Photoconduction Enhancement Upon Self-Assembly of a New Triindole Derivative in Solution-Processed Films. Adv. Funct. Mater. 2011, 21, 738–745. [Google Scholar] [CrossRef]
- García-Frutos, E.M.; Pandey, U.K.; Termine, R.; Omenat, A.; Barberá, J.; Serrano, J.L.; Golemme, A.; Gómez-Lor, B. High Charge Mobility in Discotic Liquid-Crystalline Triindoles: Just a Core Business? Angew. Chem. Int. Ed. 2011, 50, 7399–7402. [Google Scholar] [CrossRef] [Green Version]
- Jing, J.; Heinrich, B.; Prel, A.; Steveler, E.; Han, T.; Bulut, I.; Méry, S.; Leroy, Y.; Leclerc, N.; Lévêque, P.; et al. Efficient 3D charge transport in planar triazatruxene-based dumbbell-shaped molecules forming a bridged columnar phase. J. Mater. Chem. A 2021, 9, 24315–24324. [Google Scholar] [CrossRef]
- Ye, Q.; Chang, J.; Shao, J.; Chi, C. Large core-expanded triazatruxene-based discotic liquid crystals: Synthesis, characterization and physical properties. J. Mater. Chem. 2012, 22, 13180–13186. [Google Scholar] [CrossRef]
- García-Frutos, E.M.; Omenat, A.; Barberá, J.; Serrano, J.L.; Gómez-Lor, B. Highly ordered π-extended discotic liquid-crystalline triindoles. J. Mater. Chem. 2011, 21, 6831–6836. [Google Scholar] [CrossRef]
- Benito-Hernández, A.; Pandey, U.K.; Cavero, E.; Termine, R.; García-Frutos, E.M.; Serrano, J.L.; Golemme, A.; Gómez-Lor, B. High Hole Mobility in Triindole-Based Columnar phases: Removing the Bottleneck of Homogeneous Macroscopic Orientation. Chem. Mater. 2013, 25, 117–121. [Google Scholar] [CrossRef]
- García-Frutos, E.M.; Gutierrez-Puebla, E.; Monge, M.A.; Ramírez, R.; Andrés, P.D.; Andrés, A.d.; Gómez-Lor, B. Crystal structure and charge-transport properties of N-trimethyltriindole: Novel p-type organic semiconductor single crystals. Org. Electron. 2009, 10, 643–652. [Google Scholar] [CrossRef]
- Toworakajohnkun, N.; Sukwattanasinitt, M.; Rashatasakhon, P. N-Bromosuccinimide Mediated Synthesis of Triazatruxenes from Indoles. Tetrahedron Lett. 2017, 58, 4149–4152. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, C.; García-Frutos, E.M.; da Silva Filho, D.A.; López Navarrete, J.T.; Ruiz Delgado, M.C.; Gómez-Lor, B. Symmetry Lowering in Triindoles: Impact on the Electronic and Photophysical Properties. J. Phys. Chem. C 2014, 118, 5470–5477. [Google Scholar] [CrossRef]
- Ruiz Delgado, M.C.; Casado, J.; Hernández, V.; López Navarrete, J.T.; Orduna, J.; Villacampa, B.; Alicante, R.; Raimundo, J.-M.; Blanchard, P.; Roncali, J. Electronic, Optical, and Vibrational Properties of Bridged Dithienylethylene-Based NLO Chromophores. J. Phys. Chem. C 2008, 112, 3109–3120. [Google Scholar] [CrossRef]
- Donohoo-Vallett, P.J.; Bragg, A.E. π-Delocalization and the Vibrational Spectroscopy of Conjugated Materials: Computational Insights on Raman Frequency Dispersion in Thiophene, Furan, and Pyrrole Oligomers. J. Phys. Chem. B 2015, 119, 3583–3594. [Google Scholar] [CrossRef]
- Castiglioni, C.; Lopez Navarrete, J.T.; Zerbi, G.; Gussoni, M. A simple interpretation of the vibrational spectra of undoped, doped and photoexcited polyacetylene: Amplitude mode theory in the GF formalism. Solid State Commun. 1988, 65, 625–630. [Google Scholar] [CrossRef]
- Hernandez, V.; Castiglioni, C.; Del Zoppo, M.; Zerbi, G. Confinement potential and π-electron delocalization in polyconjugated organic materials. Phys. Rev. B 1994, 50, 9815–9823. [Google Scholar] [CrossRef]
- Wilson, E.B.; Decius, J.C.; Cross, P.C. Molecular Vibrations: The Theory of Infrared and Raman Vibrational Spectra; McGraw-Hill: New York, NY, USA, 1955. [Google Scholar]
- García-Frutos, E.M.; Hennrich, G.; Gutierrez, E.; Monge, A.; Gómez-Lor, B. Self-Assembly of C3-Symmetrical Hexaaryltriindoles Driven by Solvophobic and CH−π Interactions. J. Org. Chem. 2010, 75, 1070–1076. [Google Scholar] [CrossRef]
- Sutton, C.; Marshall, M.S.; Sherrill, C.D.; Risko, C.; Brédas, J.-L. Rubrene: The Interplay between Intramolecular and Intermolecular Interactions Determines the Planarization of Its Tetracene Core in the Solid State. J. Am. Chem. Soc. 2015, 137, 8775–8782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apex 3; v 2019.1-0; Bruker AXS Inc.: Madison, WI, USA, 2016; Available online: https://www.brukersupport.com/ProductDetail/3177 (accessed on 1 February 2021).
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 09 Revision C 01; Gaussian Inc.: Wallingford, UK, 2009. [Google Scholar]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Gross, E.K.U.; Kohn, W. Time-Dependent Density-Functional Theory. Adv. Quant. Chem. 1990, 21, 255–291. [Google Scholar]
- Heinze, H.H.; Görling, A.; Rösch, N. An efficient method for calculating molecular excitation energies by time-dependent density-functional theory. J. Chem. Phys. 2000, 113, 2088–2099. [Google Scholar] [CrossRef]
- O’boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Brédas, J.-L.; Beljonne, D.; Coropceanu, V.; Cornil, J. Charge-Transfer and Energy-Transfer Processes in π-Conjugated Oligomers and Polymers: A Molecular Picture. Chem. Rev. 2004, 104, 4971–5004. [Google Scholar] [CrossRef]
- Senthilkumar, K.; Grozema, F.C.; Bickelhaupt, F.M.; Siebbeles, L.D.A. Charge transport in columnar stacked triphenylenes: Effects of conformational fluctuations on charge transfer integrals and site energies. J. Chem. Phys. 2003, 119, 9809–9817. [Google Scholar] [CrossRef] [Green Version]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 1 February 2021).
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | Eox (V) | HOMO (eV) 1 | LUMO (eV) 1 |
|---|---|---|---|---|
| 1 | H | 0.71 | −5.08 (−4.83) | −1.72 (−0.53) |
| 2 | OCH3 | 0.69 | −5.04 (−4.67) | −1.95 (−0.50) |
| 3 | COCH3 | 1.04 | −5.39 (−5.44) | −2.73 (−1.47) |
| Compound | Deposition Conditions | µh (cm2 V−1 s−1) | VT(V) | ION/IOFF |
|---|---|---|---|---|
| 1 | OTS, 60° | 2.2 × 10−2 (2.8 × 10−2) | −13 (−17) | 1 × 107 (3 × 107) |
| 2 | OTS, 90° | 1.4 × 10−3 (1.6 × 10−3) | −4 (−8) | 6 × 103 (1 × 104) |
| 3 | OTS, 120° | 3.1 × 10−5 (4.5 × 10−5) | −17 (−21) | 2 × 102 (3 × 102) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gámez-Valenzuela, S.; Benito-Hernández, A.; Echeverri, M.; Gutierrez-Puebla, E.; Ponce Ortiz, R.; Ruiz Delgado, M.C.; Gómez-Lor, B. Functionalized Crystalline N-Trimethyltriindoles: Counterintuitive Influence of Peripheral Substituents on Their Semiconducting Properties. Molecules 2022, 27, 1121. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27031121
Gámez-Valenzuela S, Benito-Hernández A, Echeverri M, Gutierrez-Puebla E, Ponce Ortiz R, Ruiz Delgado MC, Gómez-Lor B. Functionalized Crystalline N-Trimethyltriindoles: Counterintuitive Influence of Peripheral Substituents on Their Semiconducting Properties. Molecules. 2022; 27(3):1121. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27031121
Chicago/Turabian StyleGámez-Valenzuela, Sergio, Angela Benito-Hernández, Marcelo Echeverri, Enrique Gutierrez-Puebla, Rocío Ponce Ortiz, Maria Carmen Ruiz Delgado, and Berta Gómez-Lor. 2022. "Functionalized Crystalline N-Trimethyltriindoles: Counterintuitive Influence of Peripheral Substituents on Their Semiconducting Properties" Molecules 27, no. 3: 1121. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27031121