
The Biological Fate of Pharmaceutical Excipient β-Cyclodextrin: Pharmacokinetics, Tissue Distribution, Excretion, and Metabolism of β-Cyclodextrin in Rats

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Instruments and Conditions
2.2.1. Quantitative Analysis Conditions
2.2.2. Qualitative Analysis Conditions
2.3. Preparation of Standard and Quality Control Samples
2.4. Sample Preparation
2.4.1. Sample Preparation for Quantification Analysis
2.4.2. Sample Preparation for Qualitative Analysis
2.5. Animal Experiments
2.5.1. Pharmacokinetics Study
2.5.2. Tissue Distribution Study
2.5.3. Excretion Study
2.5.4. Metabolism Study
2.6. Method Validation
3. Results and Discussion
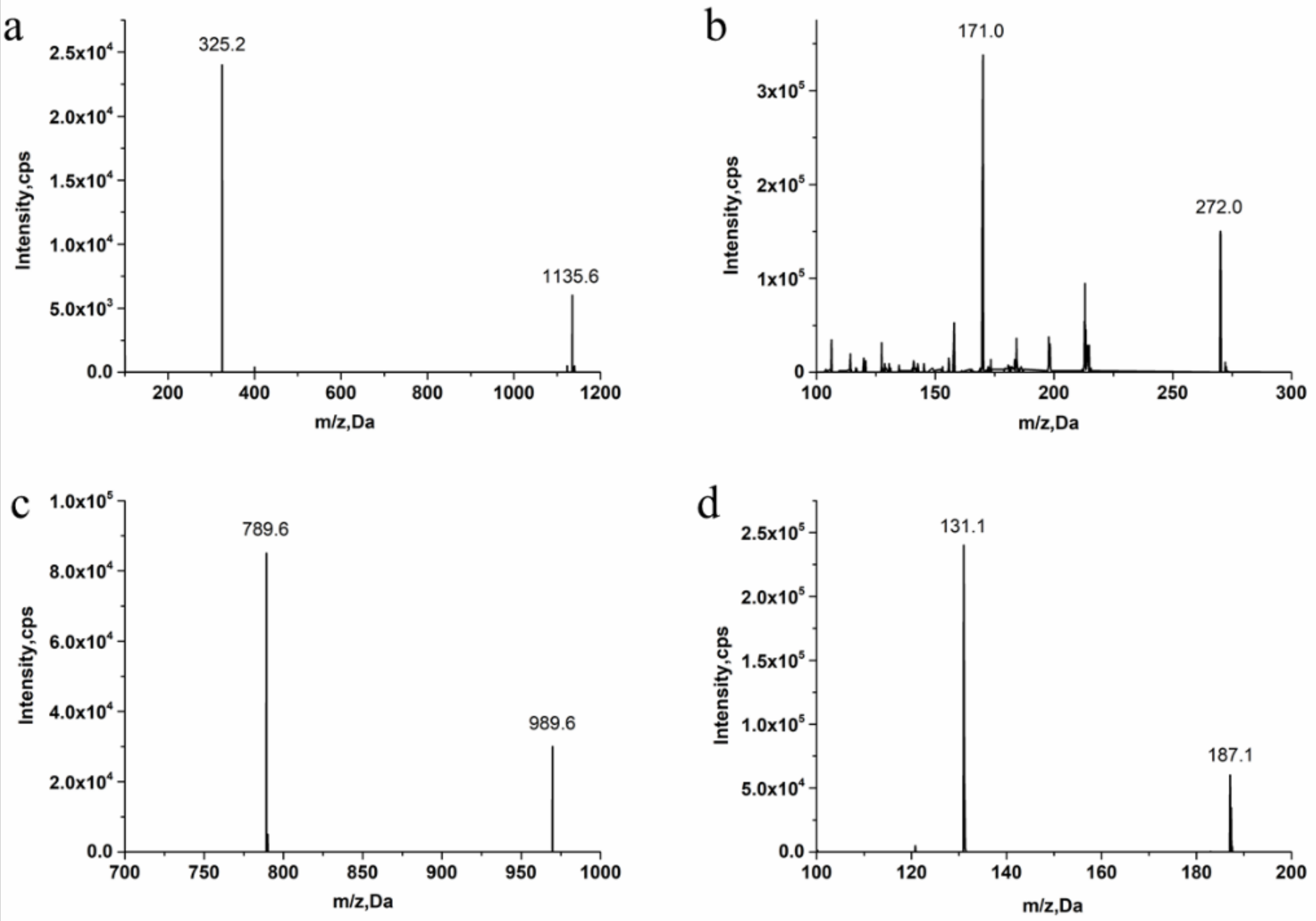
3.1. Method Development
3.2. Method Validation
3.2.1. Specificity
3.2.2. Linearity
3.2.3. Precision and Accuracy
3.2.4. Extraction Recovery and Matrix Effect
3.2.5. Stability
3.2.6. Dilution Reliability
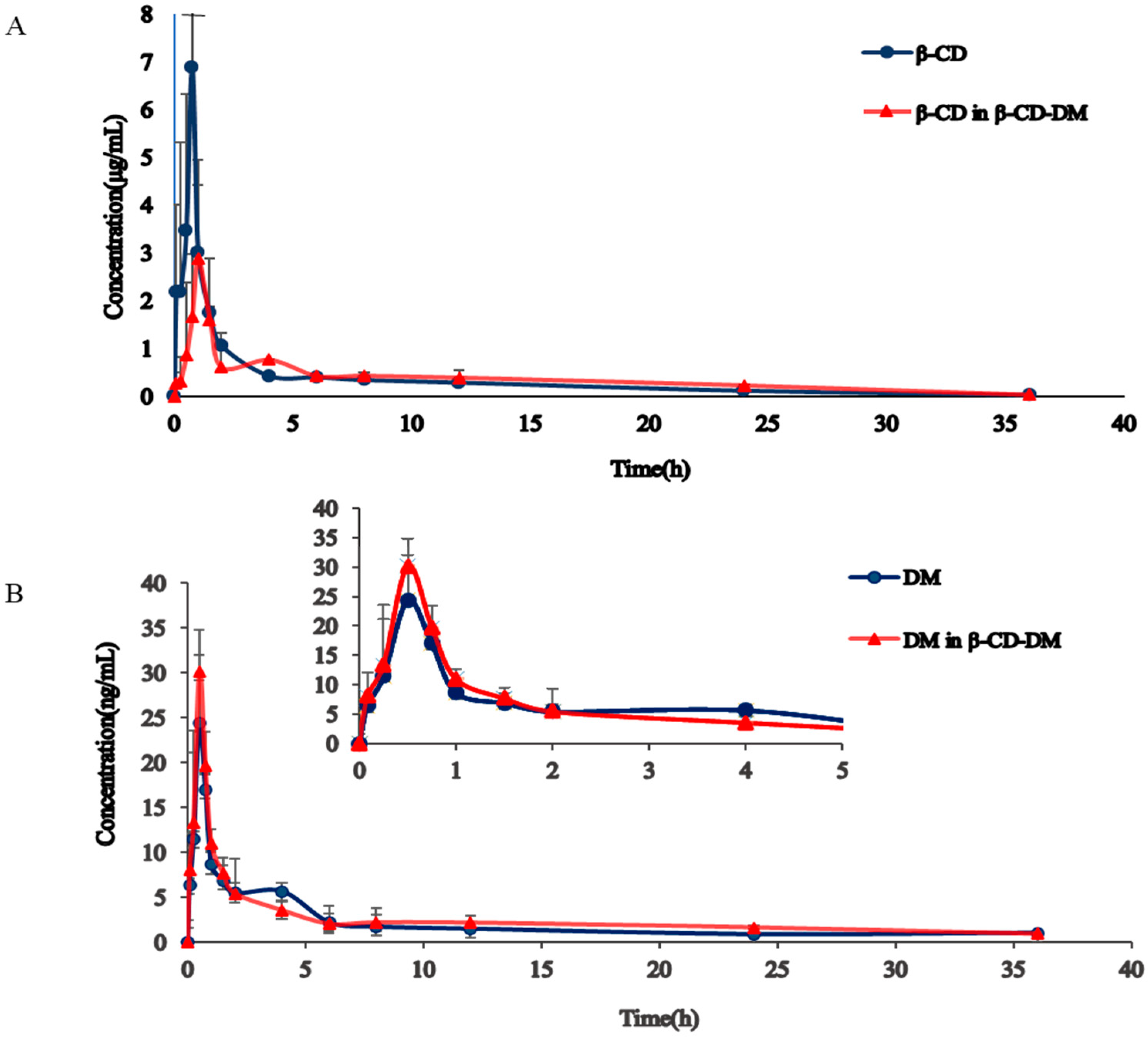
3.3. Pharmacokinetics Study
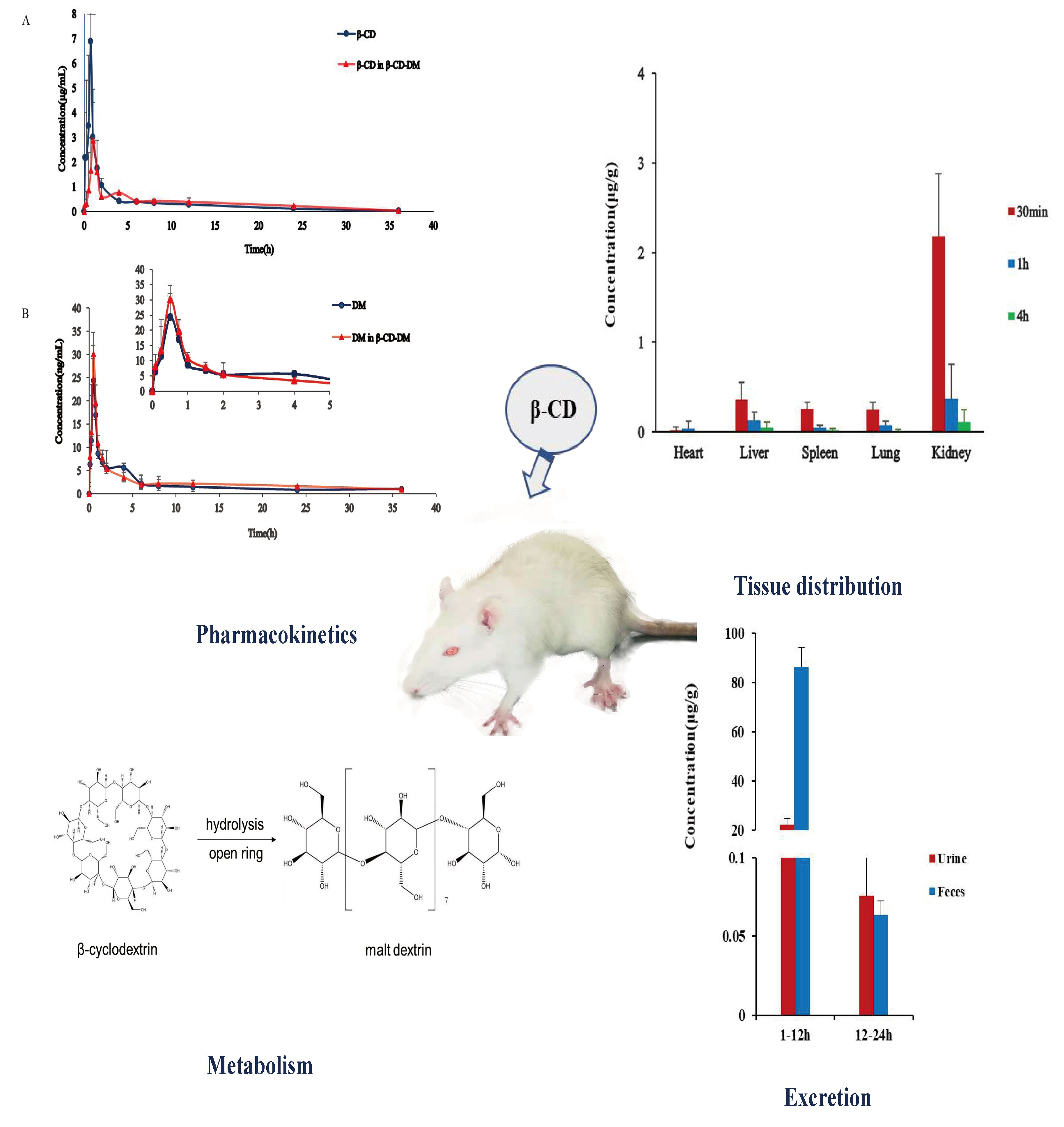
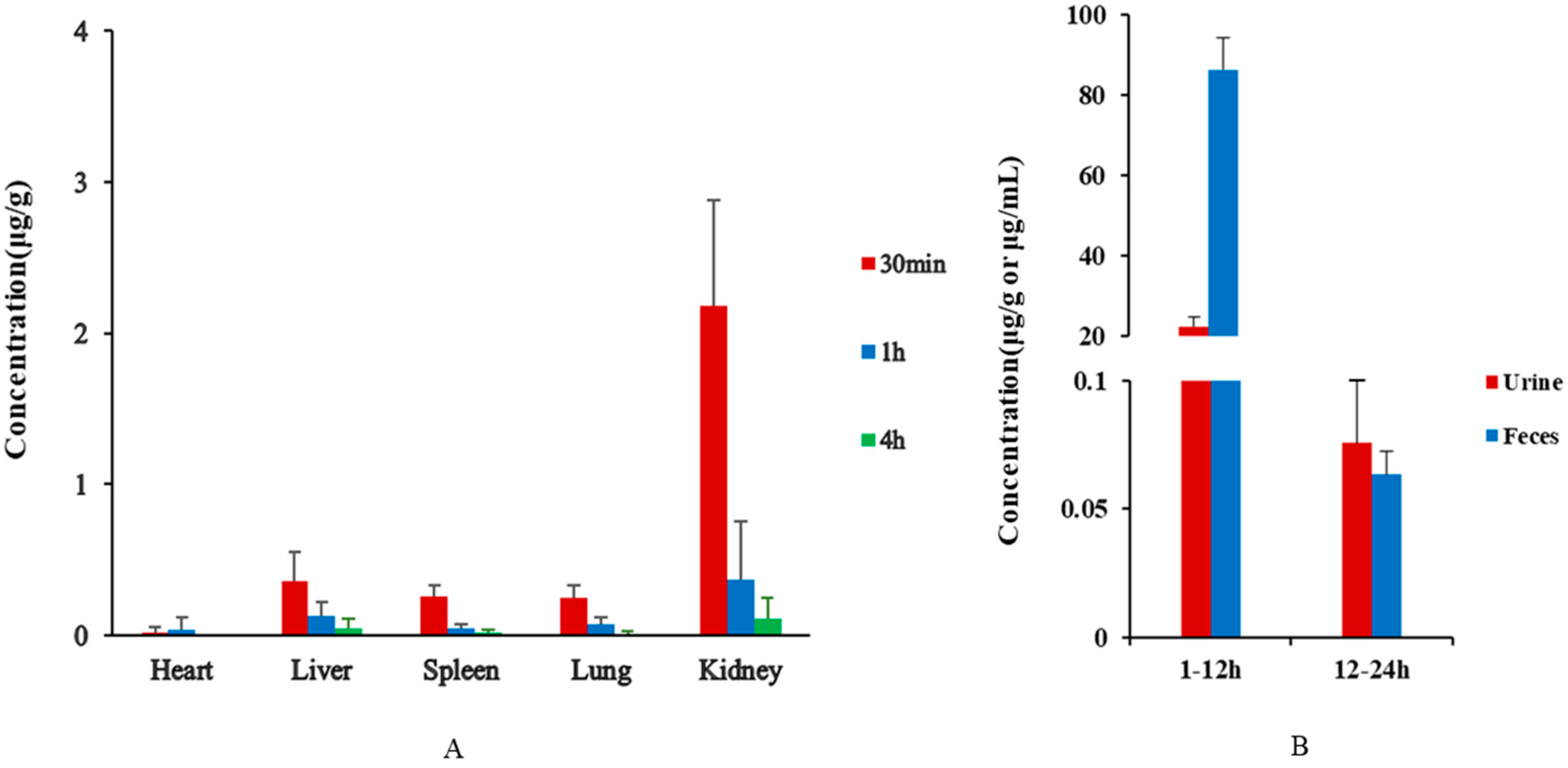
3.4. Tissue Distribution Study
3.5. Excretion Study
3.6. Metabolism Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
References
- Wang, D.; Cheow, W.S.; Amalina, N.; Faiezin, M.; Hadinoto, K. Selecting optimal pharmaceutical excipient formulation from life cycle assessment perspectives: A case study on ibuprofen tablet formulations. J. Clean. Prod. 2021, 292, 126074. [Google Scholar] [CrossRef]
- Kumar, M.; Bhatia, R.; Rawal, R.K. Applications of various analytical techniques in quality control of pharmaceutical excipients. J. Pharm. Biomed. Anal. 2018, 157, 122–136. [Google Scholar] [CrossRef] [PubMed]
- de Jong, H.J. The safety of pharmaceutical excipients. Therapie 1999, 54, 11–14. [Google Scholar] [PubMed]
- Elder, D.P.; Kuentz, M.; Holm, R. Pharmaceutical excipients-quality, regulatory and biopharmaceutical considerations. Eur. J. Pharm. Sci. 2016, 87, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Abdellah, A.; Noordin, M.I.; Ismail, W.A.W. Importance and globalization status of good manufacturing practice (GMP) requirements for pharmaceutical excipients. Saudi Pharm. J. 2015, 23, 9–13. [Google Scholar] [CrossRef] [Green Version]
- Barakat, N.S. Enhanced oral bioavailability of etodolac by self-emulsifying systems: In-vitro and in-vivo evaluation. J. Pharm. Pharmacol. 2010, 62, 173–180. [Google Scholar] [CrossRef]
- Li, W.; Cang, S.; Sun, Z.; Bi, K.; Li, Q.; Li, Z.; Liu, R. Development of an LC-MS/MS method for simultaneous quantitative analysis of macromolecular pharmaceutical adjuvant 2-hydroxypropyl-β-cyclodextrin and active pharmaceutical ingredients butylphthalide in rat plasma. J. Sep. Sci. 2021, 44, 2680–2692. [Google Scholar] [CrossRef]
- Szerman, N.; Schroh, I.; Rossi, A.L.; Rosso, A.M.; Krymkiewicz, N.; Ferrarotti, S.A. Cyclodextrin production by cyclodextrin glycosyltransferase from Bacillus circulans DF 9R. Bioresour. Technol. 2007, 98, 2886–2891. [Google Scholar] [CrossRef]
- Bhatt, P.; Lalani, R.; Vhora, I.; Patil, S.; Amrutiya, J.; Misra, A.; Mashru, R. Liposomes encapsulating native and cyclodextrin enclosed paclitaxel: Enhanced loading efficiency and its pharmacokinetic evaluation. Int. J. Pharm. 2018, 536, 95–107. [Google Scholar] [CrossRef]
- Frijlink, H.W.; Visser, J.; Hefting, N.R.; Oosting, R.; Meijer, D.K.; Lerk, C.F. The pharmacokinetics of beta-cyclodextrin and hydroxypropyl-beta-cyclodextrin in the rat. Pharm. Res. 1990, 7, 1248–1252. [Google Scholar] [CrossRef]
- Strege, M.A.; Huang, S.; Risley, D.S. Quantitative determination of beta-cyclodextrin in a powder insulin formulation for nasal delivery using hydrophilic interaction chromatography with evaporative light scattering detection. J. Liq. Chromatogr. Relat. Technol. 2019, 42, 74–78. [Google Scholar] [CrossRef]
- Fukuda, M.; Kubota, Y.; Ikuta, A.; Hasegawa, K.; Koizimi, K. Microanalyses of beta-cyclodextrin and glucosyl-beta-cyclodextrin in biological matrices by high-performance liquid chromatography with pulsed amperometric detection. Anal. Biochem. 1993, 212, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Hammes, W.; Bourscheidt, C.; Büchsler, U.; Stodt, G.; Bökens, H. Quantitative determination of α-cyclodextrin in human plasma by liquid chromatography/positive ion electrospray mass spectrometry. J. Mass Spectrom. 2000, 35, 378–384. [Google Scholar] [CrossRef]
- Xu, S.; Bian, R.; Chen, X. Pharmacological Experimental Methodology, 3rd ed.; People’s Medical Publishing House: Beijing, China, 2002; p. 1184. (In Chinese) [Google Scholar]
- Guler, L.P.; Yu, Y.Q.; Kenttamaa, H.I. An experimental and computational study of the gas-phase structures of five-carbon Monosaccharides. J. Phys. Chem. A 2002, 106, 6754–6764. [Google Scholar] [CrossRef]
- Vecsernyes, M.; Fenyvesi, F.; Bacskay, I.; Deli, M.A.; Szente, L.; Fenyvesi, E. Cyclodextrins, Blood-Brain Barrier, and Treatment of Neurological Diseases. Arch. Med. Res. 2014, 45, 711–729. [Google Scholar] [CrossRef]
- Giglio, V.; Viale, M.; Bertone, V.; Maric, I.; Vaccarone, R.; Vecchio, G. Cyclodextrin polymers as nanocarriers for sorafenib. Investig. New Drugs 2018, 36, 370–379. [Google Scholar] [CrossRef]
- Naqvi, R.F.; Khan, A.; Umer, M.F.; Malik, O.; Shahwani, N.A. Design and Optimization of Novel Taste-Masked Medicated Chocolates of Dextromethorphan with In vitro and In vivo Taste Evaluation. J. Pharm. Innov. 2020, (in press). [Google Scholar] [CrossRef]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef]
- Jambhekar, S.S.; Breen, P. Cyclodextrins in pharmaceutical formulations I: Structure and physicochemical properties, formation of complexes, and types of complex. Drug Discov. Today 2016, 21, 356–362. [Google Scholar] [CrossRef]
- Huang, T.; Zhao, Q.; Su, Y.; Ouyang, D. Investigation of molecular aggregation mechanism of glipizide/cyclodextrin complexation by combined experimental and molecular modeling approaches. Asian J. Pharm. Sci. 2019, 14, 609–620. [Google Scholar] [CrossRef]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Effects on drug permeation through biological membranes. J. Pharm. Pharmacol. 2011, 63, 1119–1135. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gao, J.; Ma, X.; Huang, G. Inclusion complex of tamibarotene with hydroxypropyl-β-cyclodextrin: Preparation, characterization, in-vitro and in-vivo evaluation. Asian J. Pharm. Sci. 2017, 12, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.Y.; Shin, D.C.; Nam, Y.S.; Cho, B.-K. Characterization, stability, and pharmacokinetics of sibutramine/β-cyclodextrin inclusion complex. J. Ind. Eng. Chem. 2012, 18, 1412–1417. [Google Scholar] [CrossRef]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Han, J.; Park, J.-H. Cyclodextrin polymer improves atherosclerosis therapy and reduces ototoxicity. J. Control. Release 2020, 319, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhang, Z.; Tong, J.; Sun, H.; Fawcett, J.P.; Gu, J. The biological fate of the polymer nanocarrier material monomethoxy poly(ethylene glycol)-block-poly(d,l-lactic acid) in rat. Acta Pharm. Sin. B 2021, 11, 1003–1009. [Google Scholar] [CrossRef]
- Li, P.; Song, J.; Ni, X.; Guo, Q.; Wen, H.; Zhou, Q.; Shen, Y.; Huang, Y.; Qiu, P.; Lin, S.; et al. Comparison in toxicity and solubilizing capacity of hydroxypropyl-beta-cyclodextrin with different degree of substitution. Int. J. Pharm. 2016, 513, 347–356. [Google Scholar] [CrossRef]
- Volkova, T.V.; Drozd, K.V.; Surov, A.O. Effect of polymers and cyclodextrins on solubility, permeability and distribution of enzalutamide and apalutamide antiandrogens. J. Mol. Liq. 2021, 322, 114937. [Google Scholar] [CrossRef]
- Rajewski, R.A.; Stella, V.J. Pharmaceutical applications of cyclodextrins. 2. In vivo drug delivery. J. Pharm. Sci. 1996, 85, 1142–1169. [Google Scholar] [CrossRef]
- Chaudhari, P.; Ghate, V.M.; Lewis, S.A. Supramolecular cyclodextrin complex: Diversity, safety, and applications in ocular therapeutics. Exp. Eye Res. 2019, 189, 107829. [Google Scholar] [CrossRef]
- Rojas, M.J.; Amaral-Fonseca, M.; Fernandez-Lafuente, R.; Giordano, R.D.L.C.; Tardioli, P.W. Recovery of starch from cassava bagasse for cyclodextrin production by sequential treatment with α-amylase and cyclodextrin glycosyltransferase. Biocatal. Agric. Biotechnol. 2019, 22, 101411. [Google Scholar] [CrossRef]
- Abdalla, M.; Jiang, B.; Hassanin, H.A.M.; Zheng, L.; Chen, J. One-pot production of maltoheptaose (DP7) from starch by sequential addition of cyclodextrin glucotransferase and cyclomaltodextrinase. Enzym. Microb. Technol. 2021, 149, 109847. [Google Scholar] [CrossRef] [PubMed]
- Manas, N.H.A.; Pachelles, S.; Mahadi, N.M.; Illias, R.M. The Characterisation of an Alkali-Stable Maltogenic Amylase from Bacillus lehensis G1 and Improved Malto-Oligosaccharide Production by Hydrolysis Suppression. PLoS ONE 2014, 9, e106481. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Structure | Q1Mass (Da) | Q3Mass (Da) | DP (V) | EP (V) | CE (V) | CXP (V) |
|---|---|---|---|---|---|---|---|
| β-CD |  | 1135.6 | 325.2 | 142.7 | 8.4 | 44.8 | 16.4 |
| DM |  | 272.0 | 171.0 | 94.1 | 7.3 | 52.5 | 11.0 |
| ginsenoside Re (IS1) |  | 969.6 | 789.6 | 207.1 | 10 | 61.7 | 13 |
| Psoralen (IS2) |  | 187.1 | 131.1 | 80 | 11 | 35 | 10 |
| Sample | Analyte | Liner Range (μg/mL) | Slope | Intercept | Regression Coefficient |
|---|---|---|---|---|---|
| Plasma | β-CD a | 5.0000 × 10−2–1.0000 × 101 | 4.1848 × 10−2 | 1.6829 × 10−1 | 0.9977 |
| DM b | 2.5000 × 10−5–1.0000 × 10−1 | 1.6445 × 10−2 | 3.4248 × 10−2 | 0.9970 | |
| Tissue | β-CD a | 5.0000 × 10−3–5.0000 | 5.8375 × 10−2 | 1.2174 × 10−2 | 0.9952 |
| Urine | β-CD a | 5.0000 × 10−2–1.0000 × 101 | 6.0808 × 10−2 | 2.6697 × 10−1 | 0.9991 |
| Feces | β-CD a | 5.0000 × 10−2–1.0000 × 101 | 5.7079 × 10−2 | 3.0097 × 10−1 | 0.9971 |
| Sample | Analyte | Concentration | Concentration (μg/mL) | Inter-Day (RSD,%) | Intra-Day (RSD,%) | Accuracy (RE,%) |
|---|---|---|---|---|---|---|
| Plasma | β-CD | LLOQ | 0.05 | 12.96 | 7.76 | −1.70 |
| QC1 | 0.15 | 9.70 | 6.66 | −0.02 | ||
| QC2 | 2.50 | 9.18 | 2.51 | 3.77 | ||
| QC3 | 8.00 | 6.10 | 6.18 | 3.82 | ||
| DM | LLOQ | 2.50 × 10−5 | 11.60 | 4.53 | -1.50 | |
| QC1 | 7.50 × 10−5 | 7.11 | 7.11 | −3.30 | ||
| QC2 | 2.5 × 10−3 | 9.60 | 2.12 | 6.28 | ||
| QC3 | 8.0 × 10−2 | 11.19 | 6.10 | −2.42 | ||
| Tissue | β−CD | LLOQ | 0.05 | 7.12 | 4.33 | −0.32 |
| QC1 | 0.15 | 13.62 | 4.72 | −0.26 | ||
| QC2 | 2.50 | 2.73 | 2.53 | 0.70 | ||
| QC3 | 8.00 | 8.28 | 3.03 | −1.22 | ||
| Urine | β−CD | LLOQ | 0.05 | 14.40 | 6.70 | −6.86 |
| QC1 | 0.15 | 6.97 | 4.22 | −2.55 | ||
| QC2 | 2.50 | 9.70 | 7.07 | 0.71 | ||
| QC3 | 8.00 | 11.78 | 4.13 | 0.67 | ||
| Feces | β−CD | LLOQ | 0.05 | 5.34 | 7.76 | −1.79 |
| QC1 | 0.15 | 5.66 | 5.26 | −5.23 | ||
| QC2 | 2.50 | 4.63 | 6.35 | 5.24 | ||
| QC3 | 8.00 | 14.55 | 2.74 | 6.89 |
| Analyte | Groups | AUC0–t (ng/mL/h) | AUC0–∞ (ng/mL/h/) | Cmax(ng/mL/h) | t1/2(h) | CLZ (L/h/Kg) | VZ (L/Kg) |
|---|---|---|---|---|---|---|---|
| β-CD | β-CD | 1.33 × 104 ± 6.51 × 103 | 1.38 × 104 ± 6.38 × 103 | 7.30 × 103 ± 2.31 × 103 | 8.33 ± 2.46 | 3.27 × 101 ± 1.02 × 101 | 4.00 × 102 ± 1.76 × 102 |
| β-CD-DM | 1.32 × 104 ± 2.80 × 103 | 1.37 × 104 ± 2.90 × 103 | 3.81 × 103 ± 6.99 × 102 ** | 8.21 ± 1.47 | 3.03 × 101 ± 6.18 | 3.54 × 102 ± 7.16 × 101 | |
| DM | DM | 7.73 × 101 ± 1.64 × 101 | 9.28 × 101 ± 1.11 × 101 | 2.91 × 101 ± 3.17 | 1.68 × 101 ± 8.56 | 6.55 × 101 ± 8.60 | 1.64 × 103 ± 9.96 × 102 |
| β-CD-DM | 9.22 × 101 ± 3.14 × 101 | 1.47 × 102 ± 4.93 × 101 * | 3.27 × 101 ± 4.67 | 3.04 × 101 ± 7.48 * | 4.41 × 101 ± 1.25 × 101 ** | 1.90 × 103 ± 6.43 × 102 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, K.; Jiang, K.; Wang, Y.; Zhao, Z.; Cang, S.; Bi, K.; Li, Q.; Liu, R. The Biological Fate of Pharmaceutical Excipient β-Cyclodextrin: Pharmacokinetics, Tissue Distribution, Excretion, and Metabolism of β-Cyclodextrin in Rats. Molecules 2022, 27, 1138. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27031138
Mu K, Jiang K, Wang Y, Zhao Z, Cang S, Bi K, Li Q, Liu R. The Biological Fate of Pharmaceutical Excipient β-Cyclodextrin: Pharmacokinetics, Tissue Distribution, Excretion, and Metabolism of β-Cyclodextrin in Rats. Molecules. 2022; 27(3):1138. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27031138
Chicago/Turabian StyleMu, Kunqian, Kaiwen Jiang, Yue Wang, Zihan Zhao, Song Cang, Kaishun Bi, Qing Li, and Ran Liu. 2022. "The Biological Fate of Pharmaceutical Excipient β-Cyclodextrin: Pharmacokinetics, Tissue Distribution, Excretion, and Metabolism of β-Cyclodextrin in Rats" Molecules 27, no. 3: 1138. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27031138