
Suppression of Charge Recombination by Auxiliary Atoms in Photoinduced Charge Separation Dynamics with Mn Oxides: A Theoretical Study


Abstract
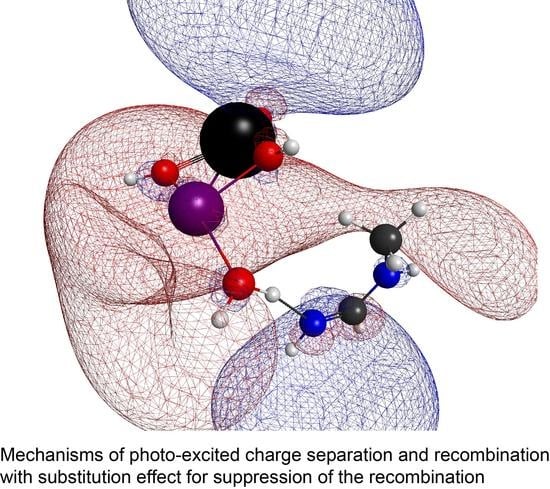
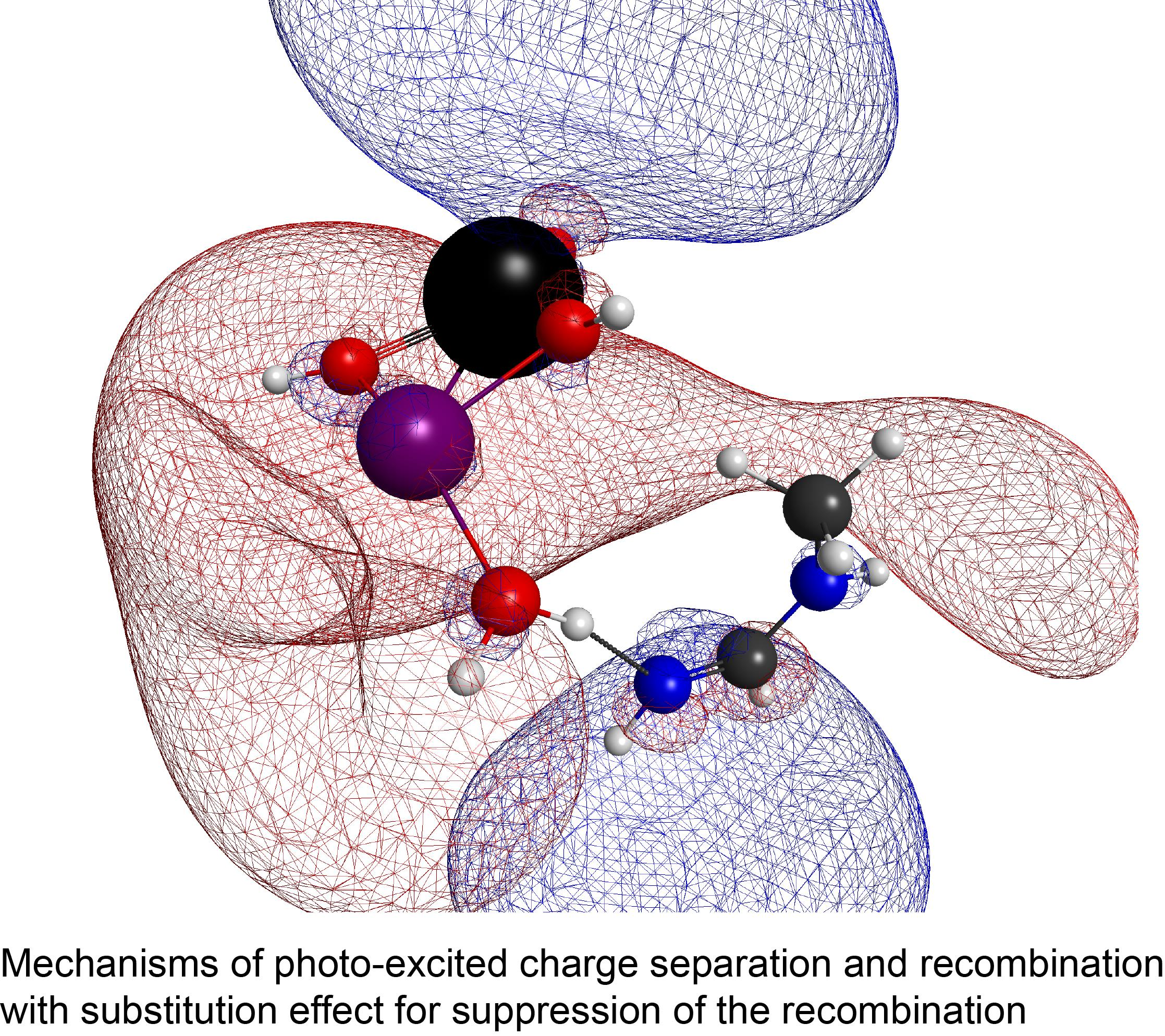
:
1. Introduction
2. Dynamics of Charge Separation
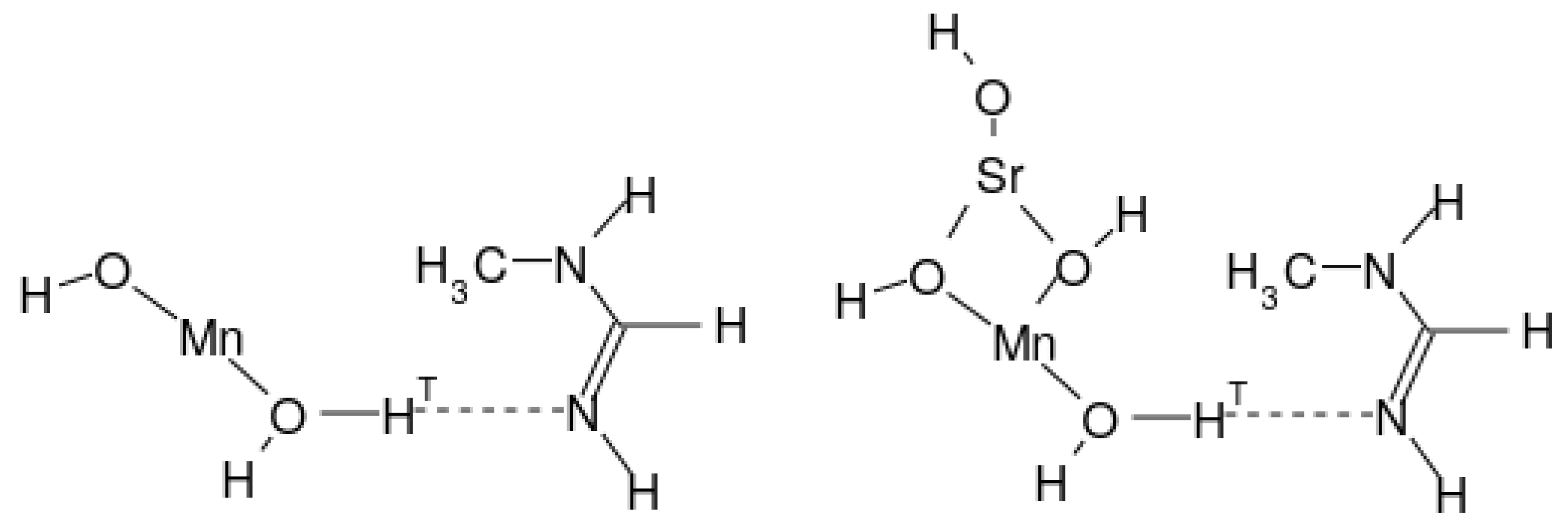
2.1. Molecular Systems
2.2. Global Features of the Potential Energy Surfaces and Excited State Dynamics
2.3. Computational Background
2.3.1. Theory of Nonadiabatic Electron Wave-Packet Dynamics
2.3.2. Computational Details
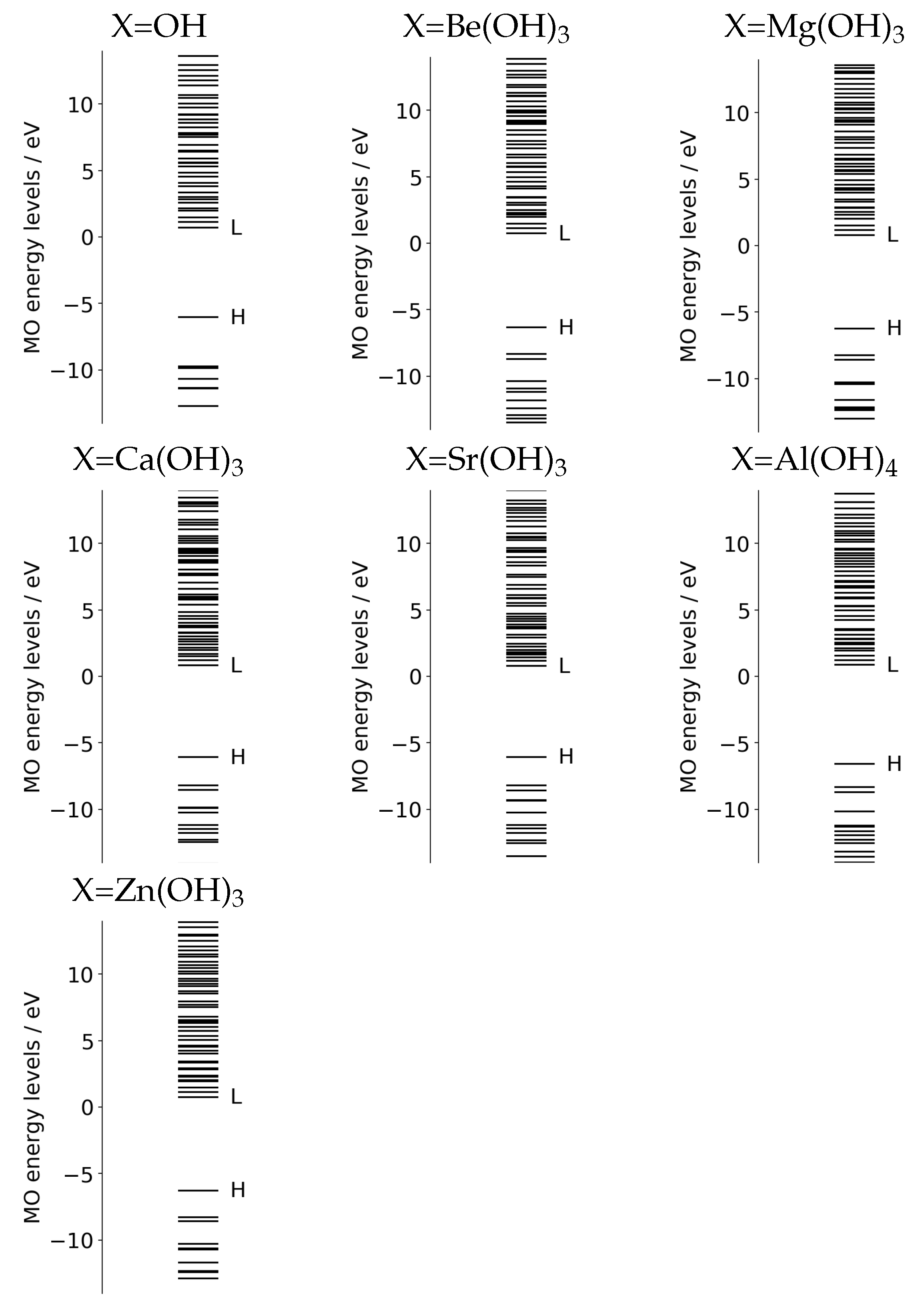
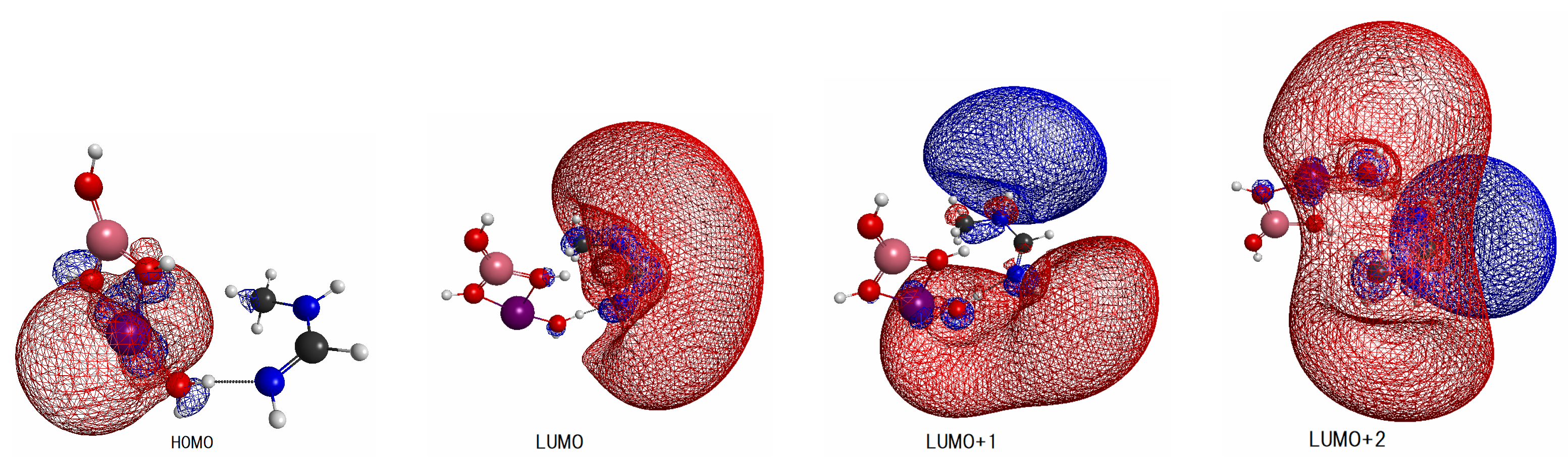
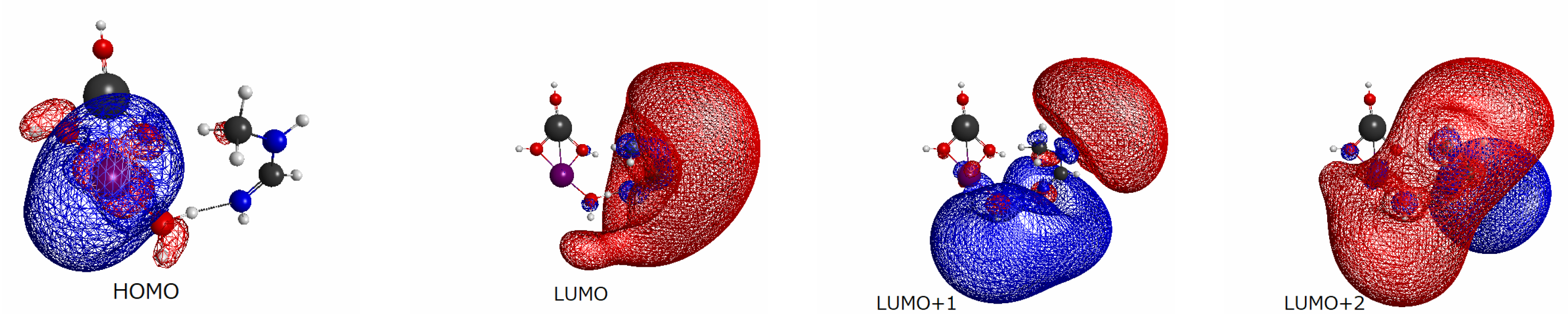
2.3.3. Basic Molecular Orbitals
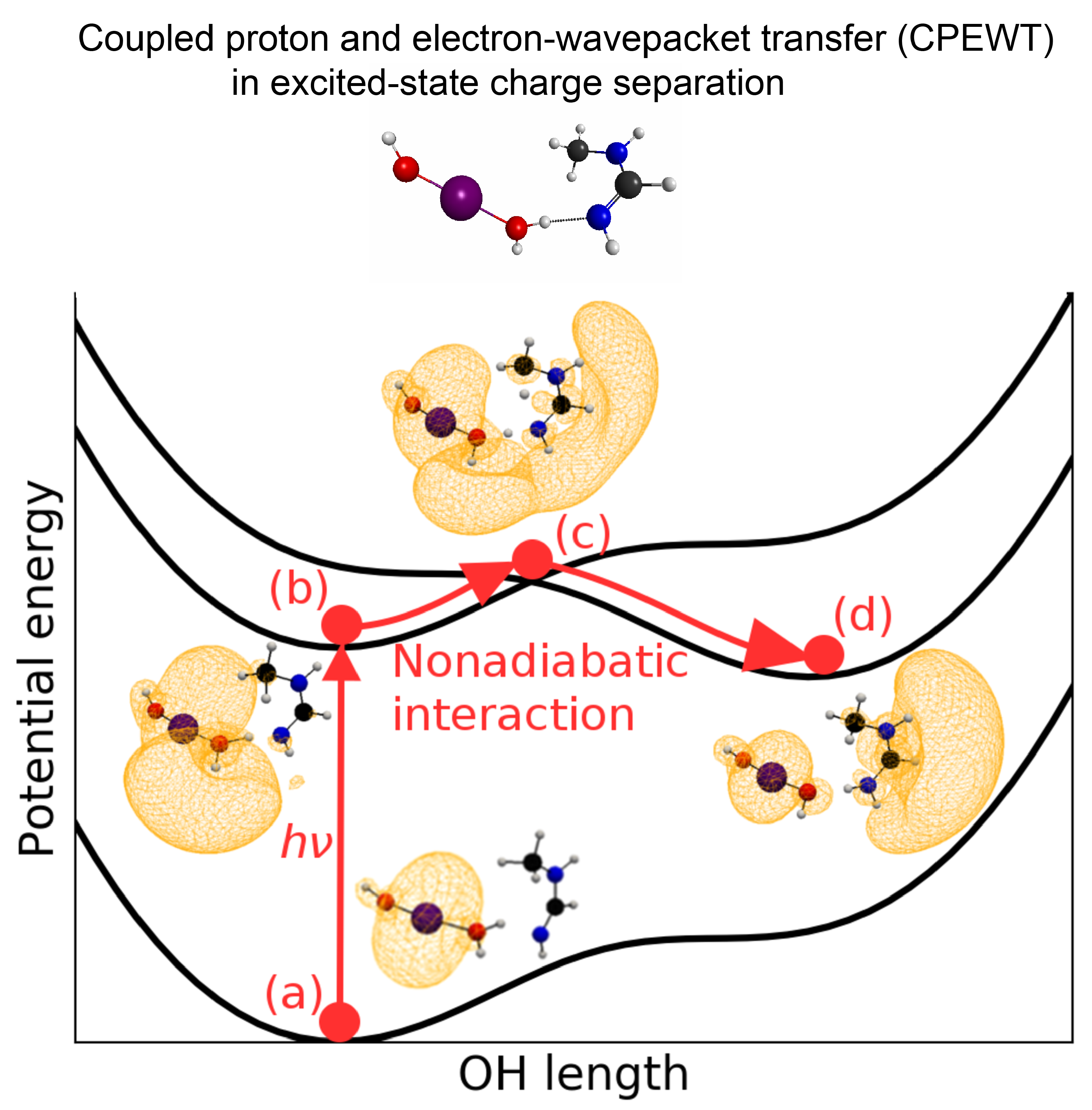
2.4. Coupled Proton and Electron Wave-Packet Dynamics (CPEWT) in Excited States
2.4.1. Running Nonadiabatic Electron Wave-Packet Dynamics
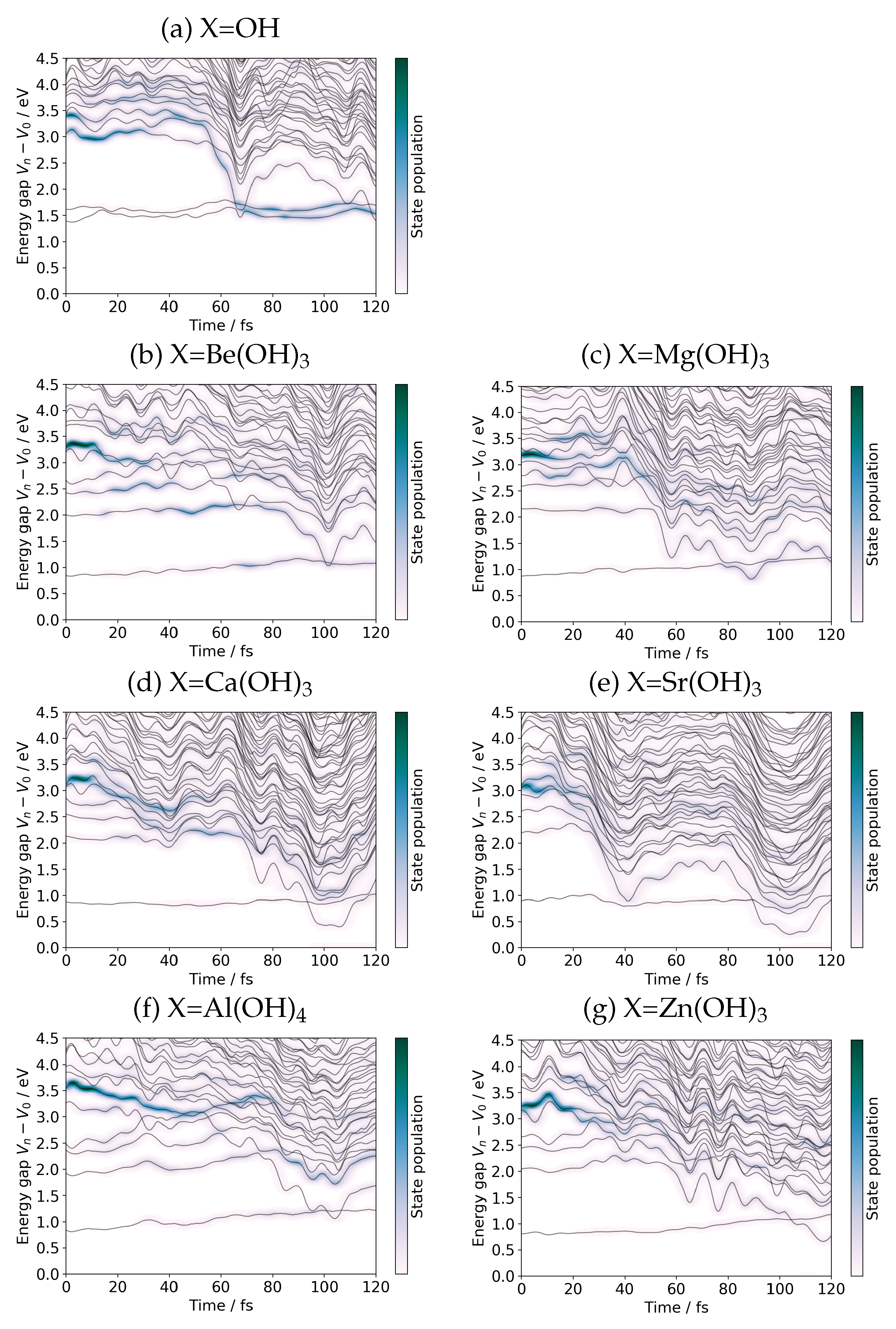
2.4.2. Real-Time Tracking of the Dynamics
3. Charge Recombination
3.1. Mechanism of Charge Recombination
3.2. Suppression of the Charge Recombination
3.3. Competence of Suppressing the Charge Recombination
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wydrzynski, T.; Satoh, K. (Eds.) Photosystem 2; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Siegbahn, P.E. Structures and Energetics for O2 Formation in Photosystem II. Acc. Chem. Res. 2009, 42, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef]
- Suga, M.; Akita, F.; Hirata, K.; Ueno, G.; Murakami, H.; Nakajima, Y.; Shimizu, T.; Yamashita, K.; Yamamoto, M.; Ago, H.; et al. Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature 2015, 517, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Suga, M.; Akita, F.; Sugahara, M.; Kubo, M.; Nakajima, Y.; Nakane, T.; Yamashita, K.; Umena, Y.; Nakabayashi, M.; Yamane, T.; et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 2017, 543, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Young, I.D.; Ibrahim, M.; Chatterjee, R.; Gul, S.; Fuller, F.D.; Koroidov, S.; Brewster, A.S.; Tran, R.; Alonso-Mori, R.; Kroll, T.; et al. Structure of photosystem II and substrate binding at room temperature. Nature 2016, 540, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, J.; Chatterjee, R.; Young, I.D.; Fuller, F.D.; Lassalle, L.; Ibrahim, M.; Gul, S.; Fransson, T.; Brewster, A.S.; Alonso Mori, R.; et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 2018, 563, 421–425. [Google Scholar] [CrossRef]
- Yano, J.; Yachandra, V. Mn4Ca Cluster in Photosynthesis: Where and How Water is Oxidized to Dioxygen. Chem. Rev. 2014, 114, 4175–4205. [Google Scholar] [CrossRef]
- Askerka, M.; Brudvig, G.W.; Batista, V.S. The O2-Evolving Complex of Photosystem II: Recent Insights from Quantum Mechanics/Molecular Mechanics (QM/MM), Extended X-ray Absorption Fine Structure (EXAFS), and Femtosecond X-ray Crystallography Data. Acc. Chem. Res. 2016, 50, 41–48. [Google Scholar] [CrossRef]
- Mauthe, S.; Fleischer, I.; Bernhardt, T.M.; Lang, S.M.; Barnett, R.N.; Landman, U. A Gas-Phase CanMn4-nO4+ Cluster Model for the Oxygen-Evolving Complex of Photosystem II. Angew. Chem. Int. Ed. 2019, 131, 8592–8597. [Google Scholar] [CrossRef]
- Zhang, Y.; Masuzaki, D.; Mafuné, F. Hydrophilicity and oxophilicity of the isolated CaMn4O5 cationic cluster modeling inorganic core of the oxygen-evolving complex. Chem. Commun. 2019, 55, 14327–14330. [Google Scholar] [CrossRef]
- Liu, X.; Sobolewski, A.L.; Borrelli, R.; Domcke, W. Computational investigation of the photoinduced homolytic dissociation of water in the pyridine-water complex. Phys. Chem. Chem. Phys. 2013, 15, 5957–5966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, W.-J.; Tan, L.-L.; Ng, Y.H.; Yong, S.-T.; Chai, S.-P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef] [PubMed]
- Welsch, R.; Driscoll, E.; Dawlaty, J.M.; Miller, T.F., III. Molecular Seesaw: How Increased Hydrogen Bonding Can Hinder Excited-State Proton Transfer. J. Phys. Chem. Lett. 2016, 7, 3616–3620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrmaier, J.; Janicki, M.J.; Sobolewski, A.L.; Domcke, W. Mechanism of photocatalytic water splitting with triazine-based carbon nitrides: Insights from ab initio calculations for the triazine–water complex. Phys. Chem. Chem. Phys. 2018, 20, 14420–14430. [Google Scholar] [CrossRef] [PubMed]
- Rabe, E.J.; Corp, K.L.; Sobolewski, A.L.; Domcke, W.; Schlenker, C.W. Proton-Coupled Electron Transfer from Water to a Model Heptazine-Based Molecular Photocatalyst. J. Phys. Chem. Lett. 2018, 9, 6257–6261. [Google Scholar] [CrossRef]
- Pang, X.; Jiang, C.; Xie, W.; Domcke, W. Photoinduced electron-driven proton transfer from water to an N-heterocyclic chromophore: Nonadiabatic dynamics studies for pyridine–water clusters. Phys. Chem. Chem. Phys. 2019, 21, 14073–14079. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, K.; Takatsuka, K. Early-stage dynamics in coupled proton–electron transfer from π-π* state of phenol to solvent ammonia clusters: An electron dynamics study. J. Phys. Chem. A 2012, 116, 11167–11179. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takatsuka, K. An Electron Dynamics Mechanism of Charge Separation in the Initial-Stage Dynamics of Photoinduced Water Splitting in X_Mn_Water (X=OH, OCaH) and Electron–Proton Acceptors. ChemPhysChem 2015, 16, 2534–2537. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takatsuka, K. Dynamical mechanism of charge separation by photoexcited generation of proton–electron pairs in organic molecular systems. A nonadiabatic electron wave-packet study. Chem. Phys. 2016, 475, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Ushiyama, H.; Takatsuka, K. Mechanism of the elementary processes of electron wave-packet dynamics coupled with proton and hydrogen-atom migration in H2O + H3O+. Angew. Chem. Int. Ed. 2007, 46, 587–590. [Google Scholar] [CrossRef]
- Nagashima, K.; Takatsuka, K. Electron-wave-packet reaction dynamics in proton transfer of formamide. J. Phys. Chem. A 2009, 113, 15240–15249. [Google Scholar] [CrossRef] [PubMed]
- Takatsuka, K. Electron dynamics in molecular elementary processes and chemical reactions. Bull. Chem. Soc. Jpn. 2021, 94, 1421–1477. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takatsuka, K. Charge separation and successive reconfigurations of electrons and protons driving water-splitting catalytic cycle with tetranuclear Mn oxo complex. On the mechanism of water splitting in PSII. Phys. Chem. Chem. Phys. 2020, 22, 7912–7934. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Takatsuka, K. Collision induced charge separation in ground-state water splitting dynamics. Phys. Chem. Chem. Phys. 2018, 20, 12229–12240. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.W.; Hayashi, S.; Lundberg, M.; Nakatsu, T.; Kato, H.; Morokuma, K. Mechanism of Efficient Firefly Bioluminescence via Adiabatic Transition State and Seam of Sloped Conical Intersection. J. Am. Chem. Soc. 2008, 130, 12880–12881. [Google Scholar] [CrossRef]
- Ghanotakis, D.F.; Babcock, G.T.; Yocum, C.F. Calcium reconstitutes high rates of oxygen evolution in polypeptide depleted Photosystem II preparations. FEBS Lett. 1984, 167, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Ono, T.A.; Inoue, Y. Discrete extraction of the Ca atom functional for O2 evolution in higher plant photosystem II by a simple low pH treatment. FEBS Lett. 1988, 227, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Boussac, A.; Zimmermann, J.L.; Rutherford, A.W.; Lavergne, J. Histidine oxidation in the oxygen-evolving photosystem-II enzyme. Nature 1990, 347, 303–306. [Google Scholar] [CrossRef]
- Boussac, A.; Rappaport, F.; Carrier, P.; Verbavatz, J.M.; Gobin, R.; Kirilovsky, D.; Rutherford, A.W.; Sugiura, M. Biosynthetic Ca2+/Sr2+ exchange in the photosystem II oxygen-evolving enzyme of Thermosynechococcus elongatus. J. Biol. Chem. 2004, 279, 22809–22819. [Google Scholar] [CrossRef] [Green Version]
- Baer, M. Beyond Born-Oppenheimer; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Nakamura, H. Nonadiabatic Transition: Concepts, Basic Theories and Applications; World Scientific: Singapore, 2012. [Google Scholar]
- Domcke, W.; Yarkony, D.; Köppel, H. Conical Intersections; Advanced Series in Physical Chemistry; World Scientific: Singapore, 2004; Volume 15. [Google Scholar]
- Jasper, A.; Kendrick, B.K.; Mead, C.A.; Truhlar, D.G. Modern Trends in Chemical Reaction Dynamics Part I Chapter 8; Yang, A., Liu, K., Eds.; World Scientific: Singapore, 2004. [Google Scholar]
- Mead, C.A.; Truhlar, D.G. On the determination of Born–Oppenheimer nuclear motion wave functions includingcomplications due to conical intersections and identical nuclei. J. Chem. Phys. 1979, 70, 2284–2296. [Google Scholar] [CrossRef]
- Shu, Y.; Varga, Z.; Sampaio de Oliveira-Filho, A.G.; Truhlar, D.G. Permutationally Restrained Diabatization by Machine Intelligence. J. Chem. Theory Comput. 2021, 17, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: Oxford, UK, 1954. [Google Scholar]
- Takatsuka, K.; Yonehara, T. Nonadiabatic chemical dynamics in intermediate and intense laser fields. Adv. Chem. Phys. 2009, 144, 93–156. [Google Scholar]
- Takatsuka, K.; Yonehara, T. Exploring dynamical electron theory beyond the Born-Oppenheimer framework: From chemical reactivity to non-adiabatically coupled electronic and nuclear wave-packets on-the-fly under laser field. Phys. Chem. Chem. Phys. 2011, 13, 4987–5016. [Google Scholar] [CrossRef] [PubMed]
- Yonehara, T.; Hanasaki, K.; Takatsuka, K. Fundamental Approaches to Nonadiabaticity: Toward a Chemical Theory beyond the Born–Oppenheimer Paradigm. Chem. Rev. 2012, 112, 499–542. [Google Scholar] [CrossRef] [PubMed]
- Takatsuka, K.; Yonehara, T.; Hanasaki, K.; Arasaki, Y. Chemical Theory beyond the Born-Oppenheimer Paradigm: Nonadiabatic Electronic and Nuclear Dynamics in Chemical Reactions; World Scientific: Singapore, 2015. [Google Scholar]
- Takatsuka, K. Time-dependent variational dynamics for nonadiabatically coupled nuclear and electronic quantum wave-packets in molecules. Eur. Phys. J. D 2021, 75, 252. [Google Scholar] [CrossRef]
- Takatsuka, K.; Fueno, T.; Yamaguchi, K. Distribution of odd electrons in ground-state molecules. Theor. Chim. Acta 1978, 48, 175–183. [Google Scholar] [CrossRef]
- Staroverov, V.N.; Davidson, E.R. Distribution of effectively unpaired electrons. Chem. Phys. Lett. 2000, 330, 161–168. [Google Scholar] [CrossRef]
- Yonehara, T.; Takatsuka, K. Phase-space averaging and natural branching of nuclear paths for nonadiabatic electron wave-packet dynamics. J. Chem. Phys. 2008, 129, 134109. [Google Scholar] [CrossRef]
- Takatsuka, K. Generalization of Classical Mechanics for Nuclear Motions on Nonadiabatically Coupled Potential Energy Surfaces in Chemical Reactions. J. Phys. Chem. A 2007, 111, 10196–10204. [Google Scholar] [CrossRef]
- Meyer, H.-D.; Miller, W.H. A classical analog for electronic degrees of freedom in nonadiabatic collision processes. J. Chem. Phys. 1979, 70, 3214–3223. [Google Scholar] [CrossRef]
- Micha, D.A. A self-consistent eikonal treatment of electronic transitions in molecular collisions. J. Chem. Phys. 1983, 78, 7138–7145. [Google Scholar] [CrossRef]
- García-Vela, A.; Gerber, R.; Imre, D. Mixed quantum wave-packet/classical trajectory treatment of the photodissociation process ArHCl → Ar+ H + Cl. J. Chem. Phys. 1992, 97, 7242–7250. [Google Scholar] [CrossRef]
- Miller, W.H. Spiers memorial lecture quantum and semiclassical theory of chemical reaction rates. Faraday Discuss. 1998, 110, 1–21. [Google Scholar] [CrossRef]
- Hack, M.D.; Jasper, A.W.; Volobuev, Y.L.; Schwenke, D.W.; Truhlar, D.G. Do semiclassical trajectory theories provide an accurate picture of radiationless decay for systems with accessible surface crossings? J. Phys. Chem. A 2000, 104, 217–232. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S.; Michael, W.S. Chapter Advances in electronic structure theory: GAMESS a decade later. In Theory and Applications of Computational Chemistry: The First Forty Years; Elsevier: Amsteldam, The Netherlands, 2005; pp. 1167–1189. [Google Scholar]
- Stevens, W.J.; Krauss, M.; Basch, H.; Jasien, P.G. Relativistic compact effective potentials and efficient, shared-exponent basis sets for the third-, fourth-, and fifth-row atoms. Can. J. Chem. 1992, 70, 612–630. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takatsuka, K. On the Elementary Chemical Mechanisms of Directional Proton Transfers: A Nonadiabatic Electron Wave-Packet Dynamics Study. J. Phys. Chem. A 2019, 123, 4125–4138. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takatsuka, K. Photoinduced charge separation catalyzed by Mn-oxides onto a Y-shaped branching acceptor efficiently preventing charge recombination. ChemPhysChem 2017, 18, 537–548. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takatsuka, K. On photocatalytic cycle of water splitting with small manganese oxides and the roles of water cluster as a direct resource of oxygen molecules. Phys. Chem. Chem. Phys. 2018, 20, 6708–6725. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Takatsuka, K. Binuclear Mn oxo complex as a self-contained photocatalyst in water-splitting cycle: Role of additional Mn oxides as a buffer of electrons and protons. J. Chem. Phys. 2020, 152, 024115. [Google Scholar] [CrossRef]
- Migliore, A.; Polizzi, N.F.; Therien, M.J.; Beratan, D.N. Biochemistry and theory of proton-coupled electron transfer. Chem. Rev. 2014, 114, 3381–3465. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
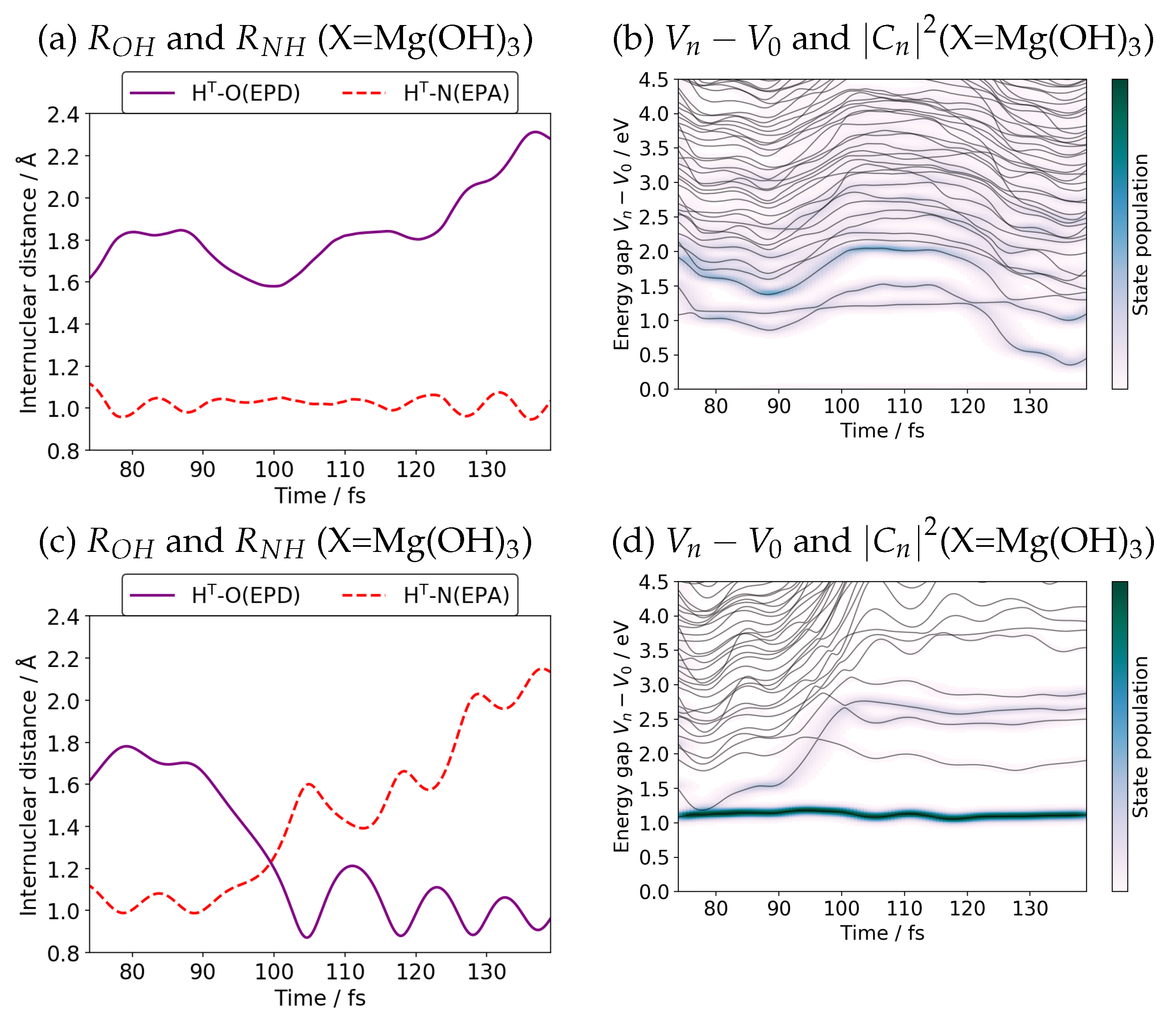{kind=link}
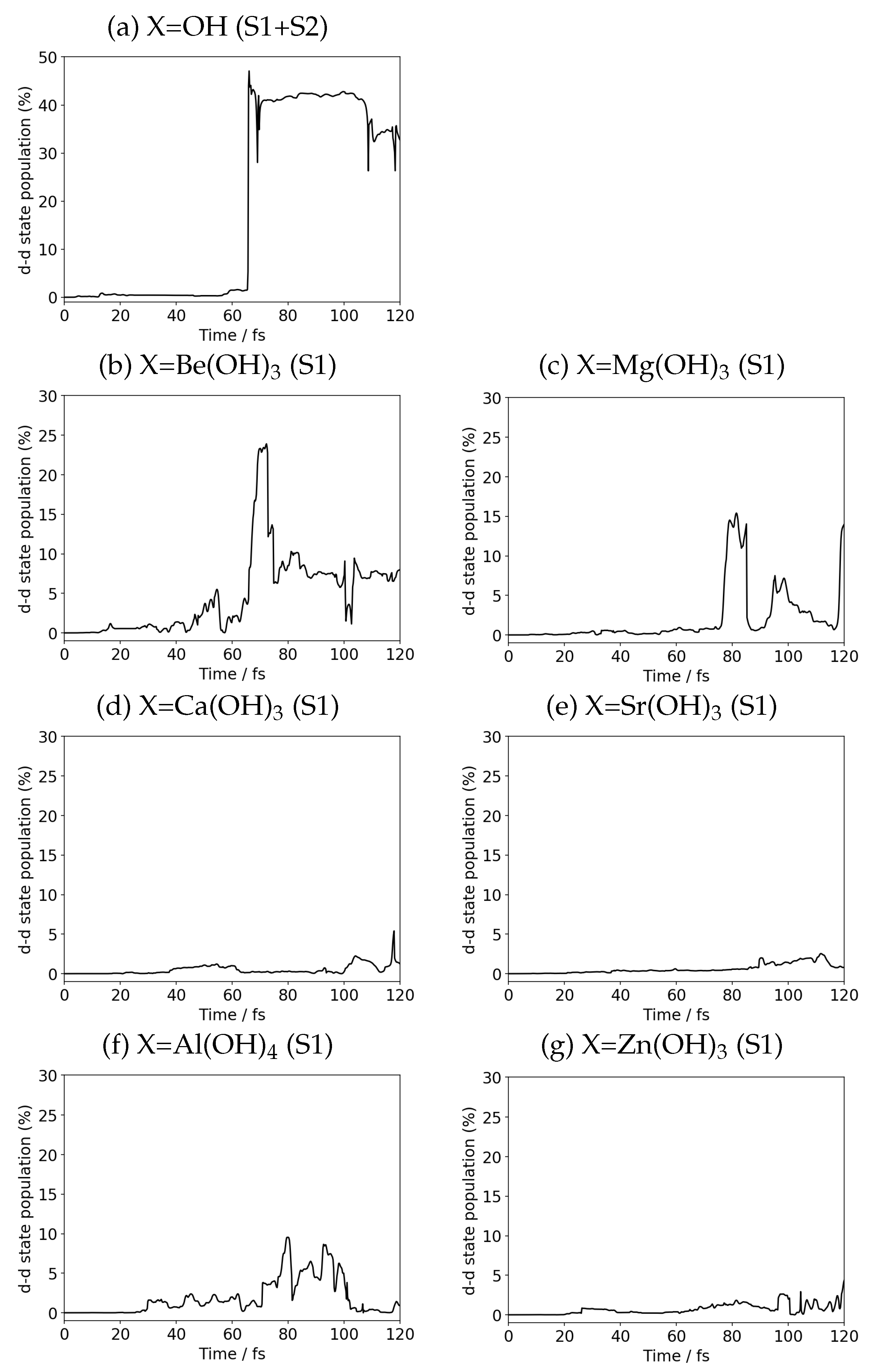{kind=link}
| X | Population (%·Time) |
|---|---|
| OH | 22.07 |
| Be(OH) | 5.62 |
| Mg(OH) | 2.44 |
| Ca(OH) | 0.59 |
| Sr(OH) | 0.73 |
| Al(OH) | 2.26 |
| Zn(OH) | 0.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohnishi, Y.; Yamamoto, K.; Takatsuka, K. Suppression of Charge Recombination by Auxiliary Atoms in Photoinduced Charge Separation Dynamics with Mn Oxides: A Theoretical Study. Molecules 2022, 27, 755. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030755
Ohnishi Y, Yamamoto K, Takatsuka K. Suppression of Charge Recombination by Auxiliary Atoms in Photoinduced Charge Separation Dynamics with Mn Oxides: A Theoretical Study. Molecules. 2022; 27(3):755. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030755
Chicago/Turabian StyleOhnishi, Yu, Kentaro Yamamoto, and Kazuo Takatsuka. 2022. "Suppression of Charge Recombination by Auxiliary Atoms in Photoinduced Charge Separation Dynamics with Mn Oxides: A Theoretical Study" Molecules 27, no. 3: 755. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030755