
Potential Immunogenic Activity of Computationally Designed mRNA- and Peptide-Based Prophylactic Vaccines against MERS, SARS-CoV, and SARS-CoV-2: A Reverse Vaccinology Approach

 ,
,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Sequences Retrieval
2.2. Prioritization of Putative Vaccine Epitopes
2.2.1. T-Cell Epitopes
2.2.2. HTL Epitopes
2.2.3. B-Cell Epitopes
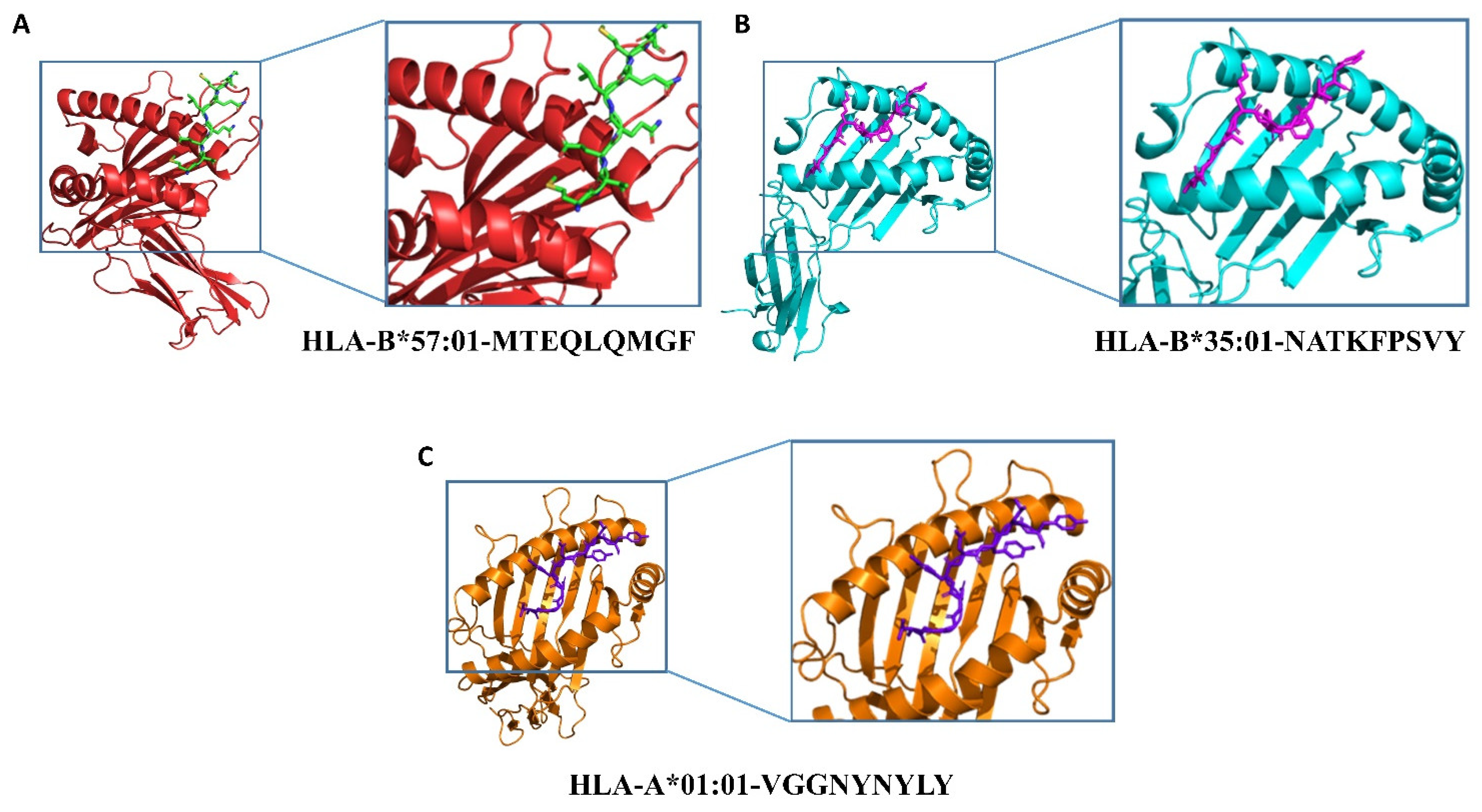
2.3. Epitopes–HLAs Molecular Docking
2.4. Prophylactic MEVC Designs
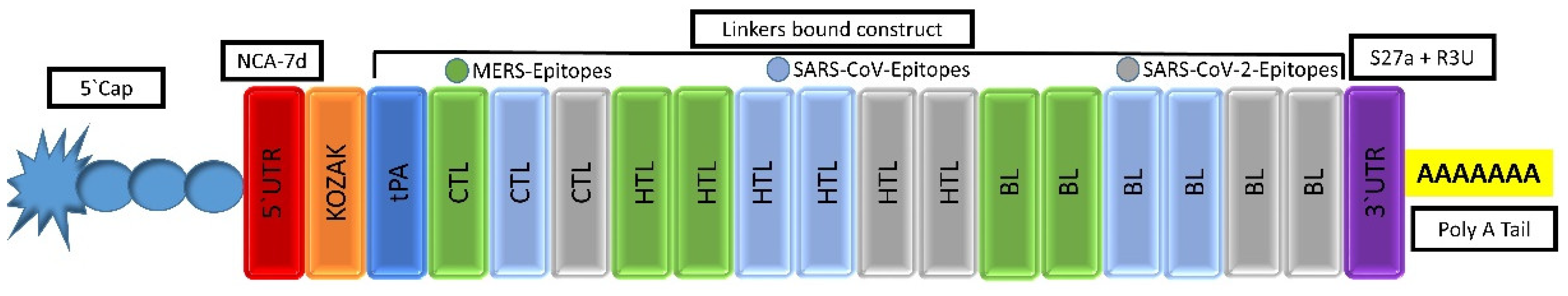
2.4.1. mRNA-Based Vaccine Construction
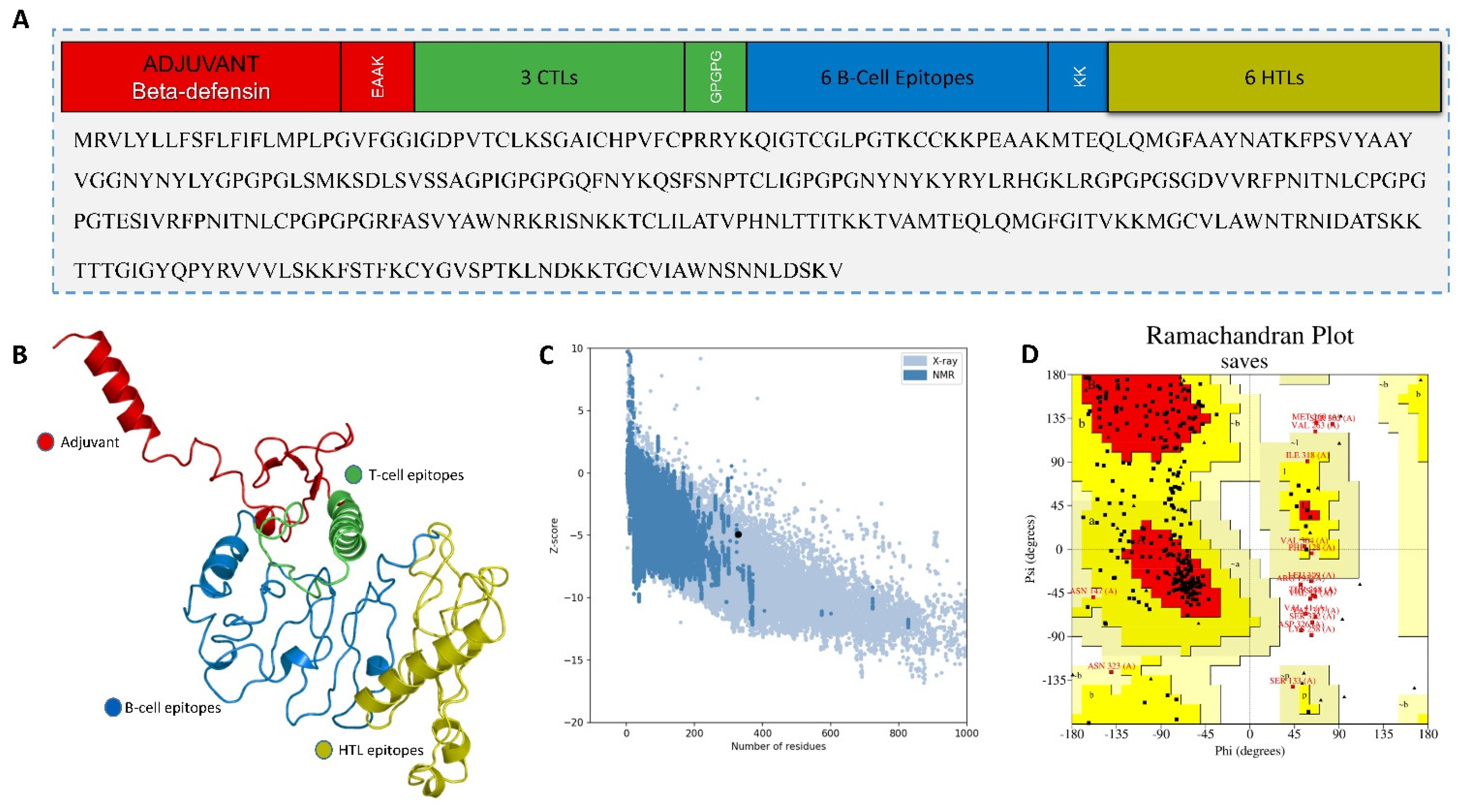
2.4.2. Multiepitope-Based Vaccine Construct
2.5. In Silico Cloning of the MEVC
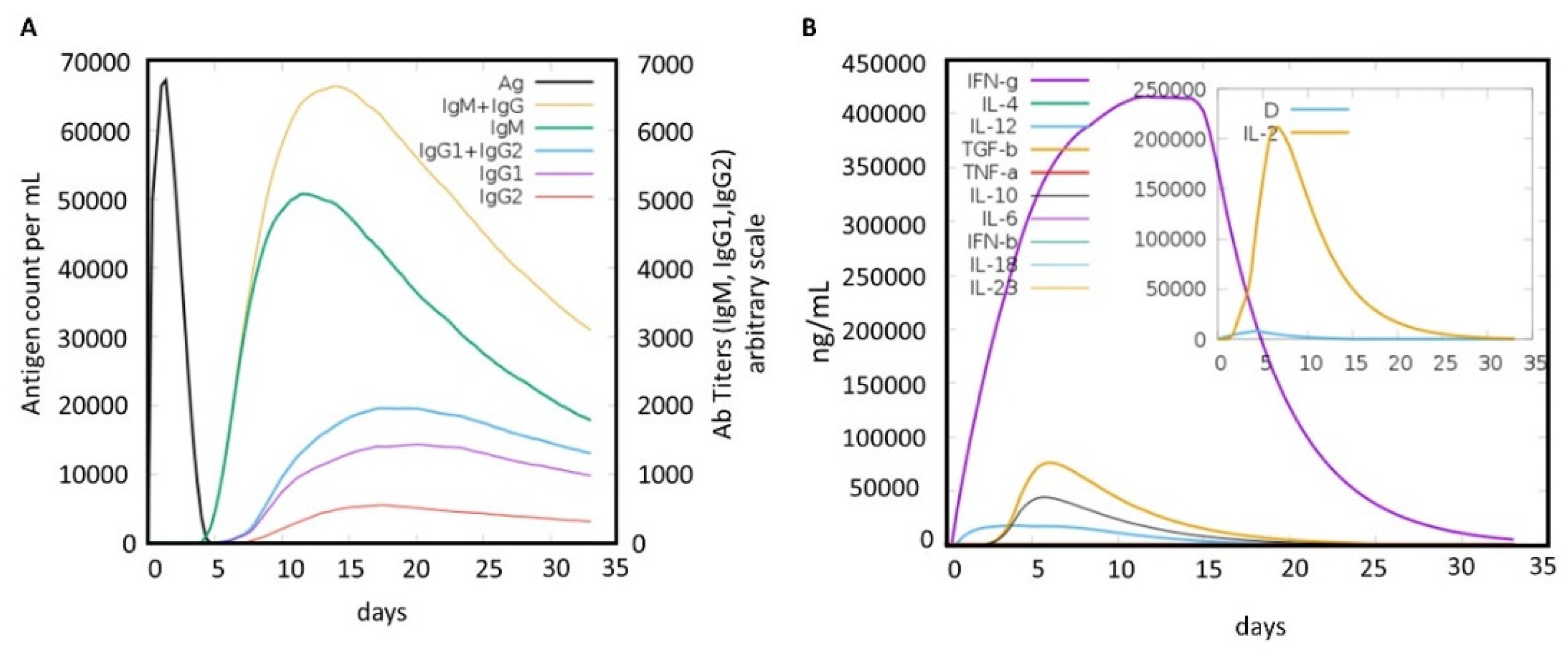
2.6. Immune Simulation of the MEVC
2.7. Molecular Docking of MEVC with Human TLR4
3. Materials and Methods
3.1. RBD Sequence Retrieval
3.2. Putative Vaccine Epitope Screening
3.2.1. Prediction of CTL Epitopes
3.2.2. Prediction of HTL Epitopes
3.2.3. B-Cell Epitope Prediction
3.3. Immunogenic and MHC-I Binding Potential Evaluation
3.3.1. Antigenicity Profiling
3.3.2. Allergenicity Profiling
3.3.3. MHC-I Binders Profiling
3.4. Molecular Docking of CTL and HTL Epitopes and HLAs
3.5. Vaccine Construction
3.5.1. mRNA Vaccine Sequence Construction
3.5.2. Multiepitope Vaccine Construction, Structural Modeling, and Validation
3.6. Molecular Docking Analysis
3.7. In Silico Cloning and Codon Optimization
3.8. Immune Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weiss, S.R. Forty years with coronaviruses. J. Exp. Med. 2020, 217, e20200537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, C.M.; Frieman, M.B. Coronaviruses: Important emerging human pathogens. J. Virol. 2014, 88, 5209–5212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meo, S.; Alhowikan, A.; Al-Khlaiwi, T.; Meo, I.; Halepoto, D.; Iqbal, M.; Usmani, A.; Hajjar, W.; Ahmed, N. Novel coronavirus 2019-nCoV: Prevalence, biological and clinical characteristics comparison with SARS-CoV and MERS-CoV. Eur. Rev. Med. Pharm. Sci. 2020, 24, 2012–2019. [Google Scholar]
- Khan, A.; Waris, H.; Rafique, M.; Suleman, M.; Mohammad, A.; Ali, S.S.; Khan, T.; Waheed, Y.; Liao, C.; Wei, D.-Q. The Omicron (B. 1.1. 529) variant of SARS-CoV-2 binds to the hACE2 receptor more strongly and escapes the antibody response: Insights from structural and simulation data. Int. J. Biol. Macromol. 2022, 200, 438–448. [Google Scholar] [CrossRef]
- Vos, L.; Bruyndonckx, R.; Zuithoff, N.; Little, P.; Oosterheert, J.; Broekhuizen, B.; Lammens, C.; Loens, K.; Viveen, M.; Butler, C. Lower respiratory tract infection in the community: Associations between viral aetiology and illness course. Clin. Microbiol. Infect. 2021, 27, 96–104. [Google Scholar] [CrossRef]
- Zhong, N.; Zheng, B.; Li, Y.; Poon, L.; Xie, Z.; Chan, K.; Li, P.; Tan, S.; Chang, Q.; Xie, J. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 2003, 362, 1353–1358. [Google Scholar] [CrossRef] [Green Version]
- Azhar, E.I.; Hui, D.S.; Memish, Z.A.; Drosten, C.; Zumla, A. The middle east respiratory syndrome (MERS). Infect. Dis. Clin. 2019, 33, 891–905. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Saberi, A.; Gulyaeva, A.A.; Brubacher, J.L.; Newmark, P.A.; Gorbalenya, A.E. A planarian nidovirus expands the limits of RNA genome size. PLoS Pathog. 2018, 14, e1007314. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Al-Qahtani, A.A. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): Emergence, history, basic and clinical aspects. Saudi J. Biol. Sci. 2020, 27, 2531–2538. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Leibowitz, J.L. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015, 206, 120–133. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [Green Version]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [Green Version]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Llanes, A.; Restrepo, C.M.; Caballero, Z.; Rajeev, S.; Kennedy, M.A.; Lleonart, R. Betacoronavirus genomes: How genomic information has been used to deal with past outbreaks and the COVID-19 pandemic. Int. J. Mol. Sci. 2020, 21, 4546. [Google Scholar] [CrossRef]
- Borbone, N.; Piccialli, G.; Roviello, G.N.; Oliviero, G. Nucleoside analogs and nucleoside precursors as drugs in the fight against SARS-CoV-2 and other coronaviruses. Molecules 2021, 26, 986. [Google Scholar] [CrossRef] [PubMed]
- George, P.J.; Tai, W.; Du, L.; Lustigman, S. The potency of an anti-MERS coronavirus subunit vaccine depends on a unique combinatorial adjuvant formulation. Vaccines 2020, 8, 251. [Google Scholar] [CrossRef] [PubMed]
- Peeri, N.C.; Shrestha, N.; Rahman, M.S.; Zaki, R.; Tan, Z.; Bibi, S.; Baghbanzadeh, M.; Aghamohammadi, N.; Zhang, W.; Haque, U. The SARS, MERS and novel coronavirus (COVID-19) epidemics, the newest and biggest global health threats: What lessons have we learned? Int. J. Epidemiol. 2020, 49, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Khan, S.; Saleem, S.; Nizam-Uddin, N.; Mohammad, A.; Khan, T.; Ahmad, S.; Arshad, M.; Ali, S.S.; Suleman, M. Immunogenomics guided design of immunomodulatory multi-epitope subunit vaccine against the SARS-CoV-2 new variants, and its validation through in silico cloning and immune simulation. Comput. Biol. Med. 2021, 133, 104420. [Google Scholar] [CrossRef]
- Chowdhury, K.H.; Chowdhury, M.; Mahmud, S.; Tareq, A.M.; Hanif, N.B.; Banu, N.; Reza, A.; Emran, T.B.; Simal-Gandara, J. Drug repurposing approach against novel coronavirus disease (COVID-19) through virtual screening targeting SARS-CoV-2 main protease. Biology 2021, 10, 2. [Google Scholar] [CrossRef]
- Khan, T.; Khan, A.; Wei, D.-Q. MMV-db: Vaccinomics and RNA-based therapeutics database for infectious hemorrhagic fever-causing mammarenaviruses. Database 2021, 2021, baab063. [Google Scholar] [CrossRef]
- Khan, T.; Khan, A.; Nasir, S.N.; Ahmad, S.; Ali, S.S.; Wei, D.-Q. CytomegaloVirusDb: Multi-Omics knowledge database for Cytomegaloviruses. Comput. Biol. Med. 2021, 135, 104563. [Google Scholar] [CrossRef]
- Alexander, J.; Fikes, J.; Hoffman, S.; Franke, E.; Sacci, J.; Appella, E.; Chisari, F.V.; Guidotti, L.G.; Chesnut, R.W.; Livingston, B. The optimization of helper T lymphocyte (HTL) function in vaccine development. Immunol. Res. 1998, 18, 79–92. [Google Scholar] [CrossRef]
- Khan, A.; Khan, S.; Ahmad, S.; Anwar, Z.; Hussain, Z.; Safdar, M.; Rizwan, M.; Waseem, M.; Hussain, A.; Akhlaq, M. HantavirusesDB: Vaccinomics and RNA-based therapeutics database for the potentially emerging human respiratory pandemic agents. Microb. Pathog. 2021, 160, 105161. [Google Scholar] [CrossRef]
- Humayun, F.; Cai, Y.; Khan, A.; Farhan, S.A.; Khan, F.; Rana, U.I.; binte Qamar, A.; Fawad, N.; Shamas, S. Structure-guided design of multi-epitopes vaccine against variants of concern (VOCs) of SARS-CoV-2 and validation through In silico cloning and immune simulations. Comput. Biol. Med. 2022, 140, 105122. [Google Scholar] [CrossRef]
- Gomes, L.R.; Durans, A.M.; Napoleão-Pêgo, P.; Waterman, J.A.; Freitas, M.S.; De Sá, N.B.; Pereira, L.V.; Furtado, J.S.; Aquino, R.G.; Machado, M.C. Multiepitope Proteins for the Differential Detection of IgG Antibodies against RBD of the Spike Protein and Non-RBD Regions of SARS-CoV-2. Vaccines 2021, 9, 986. [Google Scholar] [CrossRef] [PubMed]
- Das, B.K.; Chakraborty, D. Epitope-Based Potential Vaccine Candidate for Humoral and Cell-Mediated Immunity to Combat Severe Acute Respiratory Syndrome Coronavirus 2 Pandemic. J. Phys. Chem. Lett. 2020, 11, 9920–9930. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Zhang, G.; Wang, Y.; Zhang, Z.; Hu, H.; Shen, S.; Wu, J.; Li, B.; Li, X.; Fang, Y. Structural basis for SARS-CoV-2 neutralizing antibodies with novel binding epitopes. PLoS Biol. 2021, 19, e3001209. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Hasan, A.; Babadaei, M.M.N.; Bloukh, S.H.; Chowdhury, M.E.; Sharifi, M.; Haghighat, S.; Falahati, M. Targeting SARS-CoV2 spike protein receptor binding domain by therapeutic antibodies. Biomed. Pharmacother. 2020, 130, 110559. [Google Scholar] [CrossRef] [PubMed]
- Poland, G.; Ovsyannikova, I.; Jacobson, R.; Smith, D. Heterogeneity in vaccine immune response: The role of immunogenetics and the emerging field of vaccinomics. Clin. Pharmacol. Ther. 2007, 82, 653–664. [Google Scholar] [CrossRef]
- Shamriz, S.; Ofoghi, H.; Moazami, N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput. Biol. Med. 2016, 76, 24–29. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Stranzl, T.; Larsen, M.V.; Lundegaard, C.; Nielsen, M. NetCTLpan: Pan-specific MHC class I pathway epitope predictions. Immunogenetics 2010, 62, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.P.; Mishra, B.N. Major histocompatibility complex linked databases and prediction tools for designing vaccines. Hum. Immunol. 2016, 77, 295–306. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Modeling 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein–protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef]
- Kozak, M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 1986, 44, 283–292. [Google Scholar] [CrossRef]
- Liu, Q. Comparative analysis of base biases around the stop codons in six eukaryotes. Biosystems 2005, 81, 281–289. [Google Scholar] [CrossRef]
- Chen, X.; Zaro, J.L.; Shen, W.-C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef] [Green Version]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019, 9, 4409. [Google Scholar] [CrossRef] [Green Version]
- Schönefuß, A.; Wendt, W.; Schattling, B.; Schulten, R.; Hoffmann, K.; Stuecker, M.; Tigges, C.; Lübbert, H.; Stichel, C. Upregulation of cathepsin S in psoriatic keratinocytes. Exp. Dermatol. 2010, 19, e80–e88. [Google Scholar] [CrossRef] [PubMed]
- Riese, R.J.; Mitchell, R.N.; Villadangos, J.A.; Shi, G.-P.; Palmer, J.T.; Karp, E.R.; De Sanctis, G.T.; Ploegh, H.L.; Chapman, H.A. Cathepsin S activity regulates antigen presentation and immunity. J. Clin. Investig. 1998, 101, 2351–2363. [Google Scholar] [CrossRef] [PubMed]
- Michel-Todó, L.; Reche, P.A.; Bigey, P.; Pinazo, M.-J.; Gascón, J.; Alonso-Padilla, J. In silico Design of an Epitope-Based Vaccine Ensemble for Chagas Disease. Front. Immunol. 2019, 10, 2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahammad, I.; Lira, S.S. Designing a novel mRNA vaccine against SARS-CoV-2: An immunoinformatics approach. Int. J. Biol. Macromol. 2020, 162, 820–837. [Google Scholar] [CrossRef] [PubMed]
- Gallie, D.R. The cap and poly (A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 1991, 5, 2108–2116. [Google Scholar] [CrossRef] [Green Version]
- Munroe, D.; Jacobson, A. mRNA poly (A) tail, a 3′enhancer of translational initiation. Mol. Cell. Biol. 1990, 10, 3441–3455. [Google Scholar]
- Zhao, Y.; Moon, E.; Carpenito, C.; Paulos, C.M.; Liu, X.; Brennan, A.L.; Chew, A.; Carroll, R.G.; Scholler, J.; Levine, B.L. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010, 70, 9053–9061. [Google Scholar] [CrossRef] [Green Version]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.Z.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- Bernstein, P.; Ross, J. Poly (A), poly (A) binding protein and the regulation of mRNA stability. Trends Biochem. Sci. 1989, 14, 373–377. [Google Scholar] [CrossRef]
- Wang, Z.; Day, N.; Trifillis, P.; Kiledjian, M. An mRNA stability complex functions with poly (A)-binding protein to stabilize mRNA in vitro. Mol. Cell. Biol. 1999, 19, 4552–4560. [Google Scholar] [CrossRef] [Green Version]
- Pourseif, M.M.; Parvizpour, S.; Jafari, B.; Dehghani, J.; Naghili, B.; Omidi, Y. A domain-based vaccine construct against SARS-CoV-2, the causative agent of COVID-19 pandemic: Development of self-amplifying mRNA and peptide vaccines. BioImpacts 2021, 11, 65. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castiglione, F.; Mantile, F.; De Berardinis, P.; Prisco, A. How the interval between prime and boost injection affects the immune response in a computational model of the immune system. Comput. Math. Methods Med. 2012, 2012, 842329. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
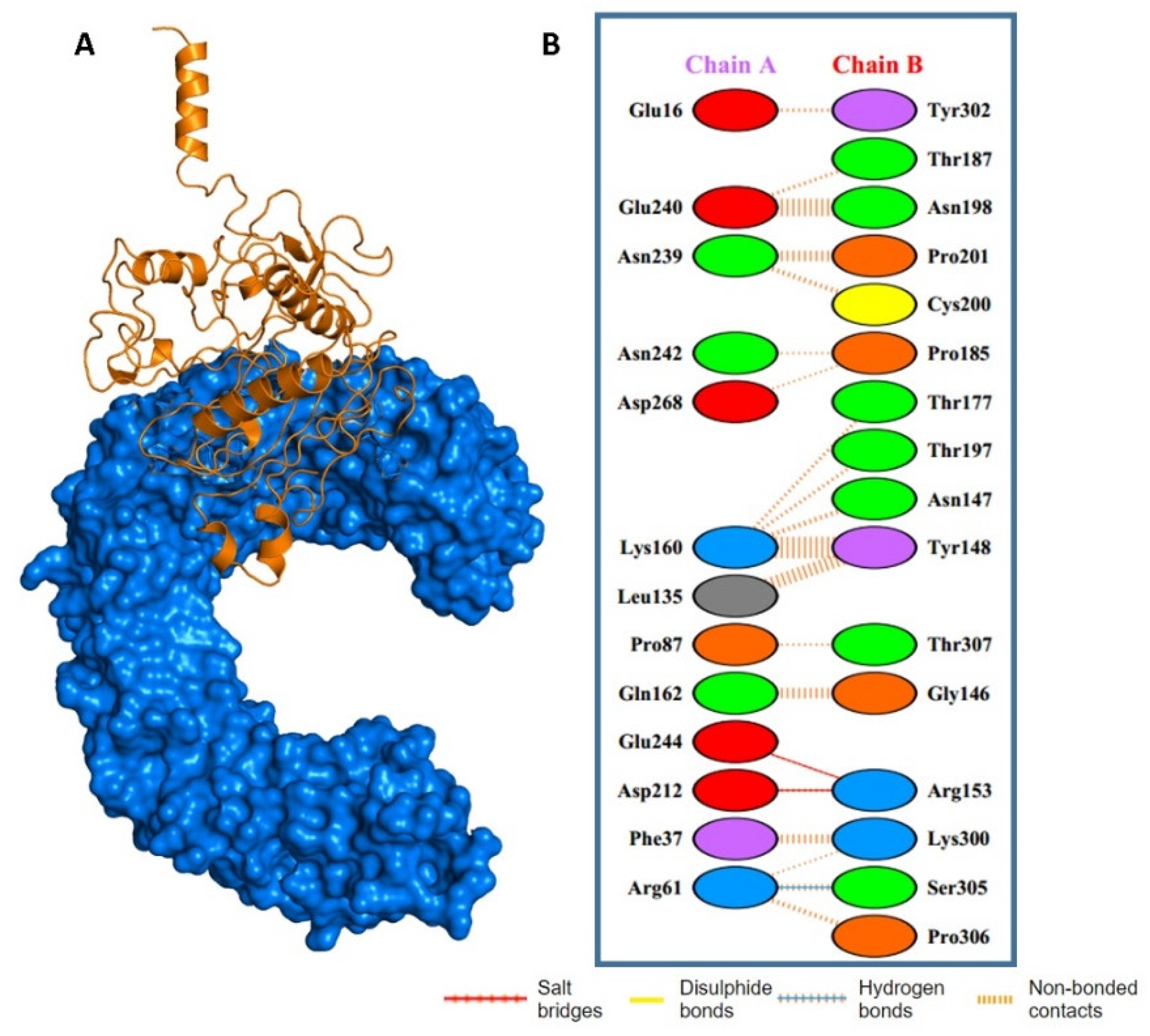{kind=link}
| Accession ID | Domain Name | Protein Name | Organism | Position of RBD in the Spike (GP) |
|---|---|---|---|---|
| K9N5Q8 | BetaCoV S1-CTD | Spike glycoprotein | MERS | 381–587 |
| P59594 | RBD | Spike glycoprotein | SARS-CoV | 306–527 |
| P0DTC2 | RBD | Spike glycoprotein | SARS-CoV-2 | 319–541 |
| Target Species | Number of Predicted T-Cell Epitopes | Immunogenic T-Cell Epitopes | Position in the RBD |
|---|---|---|---|
| MERS | 11 | MTEQLQMGF | 183–191 |
| SARS-CoV | 9 | NATKFPSVY | 25–33 |
| SARS-CoV-2 | 9 | VGGNYNYLY | 127–135 |
| Target Species | Number of Predicted HTL Epitopes | Immunogenic HTL Epitopes | Position in the RBD |
|---|---|---|---|
| MERS | 1351 | LSMKSDLSVSSAGPI QFNYKQSFSNPTCLI | 70–84 86–100 |
| SARS-CoV | 1456 | NYNYKYRYLRHGKLR SGDVVRFPNITNLCP | 130–144 5–19 |
| SARS-CoV-2 | 1463 | TESIVRFPNITNLCP RFASVYAWNRKRISN | 5–19 28–42 |
| Target Species | Number of Predicted B-Cell Epitopes | Immunogenic B-Cell Epitopes | Position in the RBD |
|---|---|---|---|
| MERS | 18 | TCLILATVPHNLTTIT TVAMTEQLQMGFGITV | 97–112 180–195 |
| SARS-CoV | 24 | MGCVLAWNTRNIDATS TTTGIGYQPYRVVVLS | 112–127 180–195 |
| SARS-CoV-2 | 23 | FSTFKCYGVSPTKLND TGCVIAWNSNNLDSKV | 56–71 112–127 |
| Species | Type of Peptide | Peptide Sequence | Position in the RBD | HLA | Binding Energies |
|---|---|---|---|---|---|
| MERS | CTL | MTEQLQMGF | 183–191 | HLA-B*57:01 | −27.86 kcal/mol |
| SARS-CoV | CTL | NATKFPSVY | 25–33 | HLA-B*35:01 | −20.65 kcal/mol |
| SARS-CoV-2 | CTL | VGGNYNYLY | 127–135 | HLA-A*01:01 | −21.87 kcal/mol |
| MERS | HTL | LSMKSDLSVSSAGPI | 70–84 | HLA-B*58:01 | −37.1 kcal/mol |
| MERS | HTL | QFNYKQSFSNPTCLI | 86–100 | HLA-B*15:01 | −31.49 kcal/mol |
| SARS-CoV | HTL | NYNYKYRYLRHGKLR | 130–144 | HLA-A*24:02 | −33.44 kcal/mol |
| SARS-CoV | HTL | SGDVVRFPNITNLCP | 5–19 | HLA-A*02:06 | −35.7 kcal/mol |
| SARS-CoV-2 | HTL | TESIVRFPNITNLCP | 5–19 | HLA-B*51:01 | −29.69 kcal/mol |
| SARS-CoV-2 | HTL | RFASVYAWNRKRISN | 28–42 | HLA-A*68:01 | −46.27 kcal/mol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, T.; Khan, A.; Ansari, J.K.; Najmi, M.H.; Wei, D.-Q.; Muhammad, K.; Waheed, Y. Potential Immunogenic Activity of Computationally Designed mRNA- and Peptide-Based Prophylactic Vaccines against MERS, SARS-CoV, and SARS-CoV-2: A Reverse Vaccinology Approach. Molecules 2022, 27, 2375. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072375
Khan T, Khan A, Ansari JK, Najmi MH, Wei D-Q, Muhammad K, Waheed Y. Potential Immunogenic Activity of Computationally Designed mRNA- and Peptide-Based Prophylactic Vaccines against MERS, SARS-CoV, and SARS-CoV-2: A Reverse Vaccinology Approach. Molecules. 2022; 27(7):2375. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072375
Chicago/Turabian StyleKhan, Taimoor, Abbas Khan, Jawad Khaliq Ansari, Muzammil Hasan Najmi, Dong-Qing Wei, Khalid Muhammad, and Yasir Waheed. 2022. "Potential Immunogenic Activity of Computationally Designed mRNA- and Peptide-Based Prophylactic Vaccines against MERS, SARS-CoV, and SARS-CoV-2: A Reverse Vaccinology Approach" Molecules 27, no. 7: 2375. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072375