
Explorations on Thermodynamic and Kinetic Performances of Various Cationic Exchange Durations for Synthetic Clinoptilolite


Abstract
:1. Introduction
2. Results and Discussion
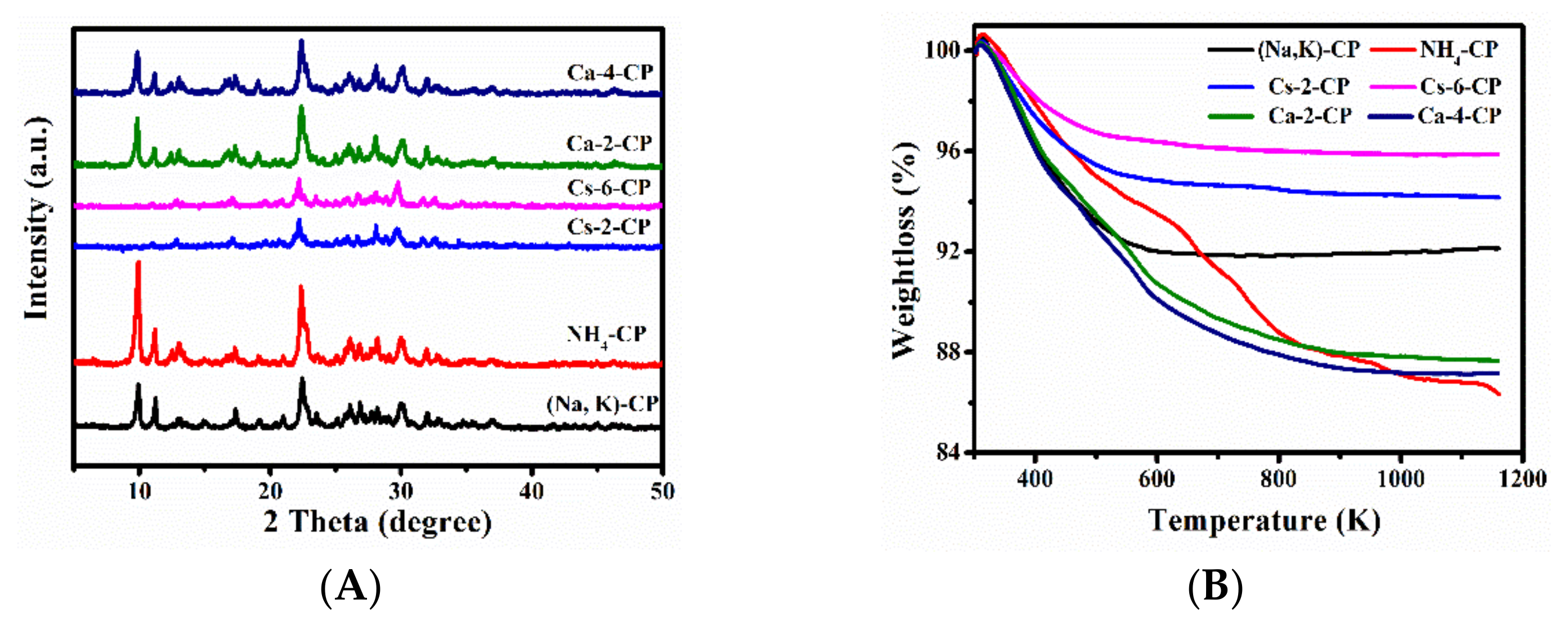
2.1. Physical-Chemical Properties of before and after Ion-Exchanged CPs
2.2. Physical-Chemical Properties of before and after Ion-Exchanged CPs
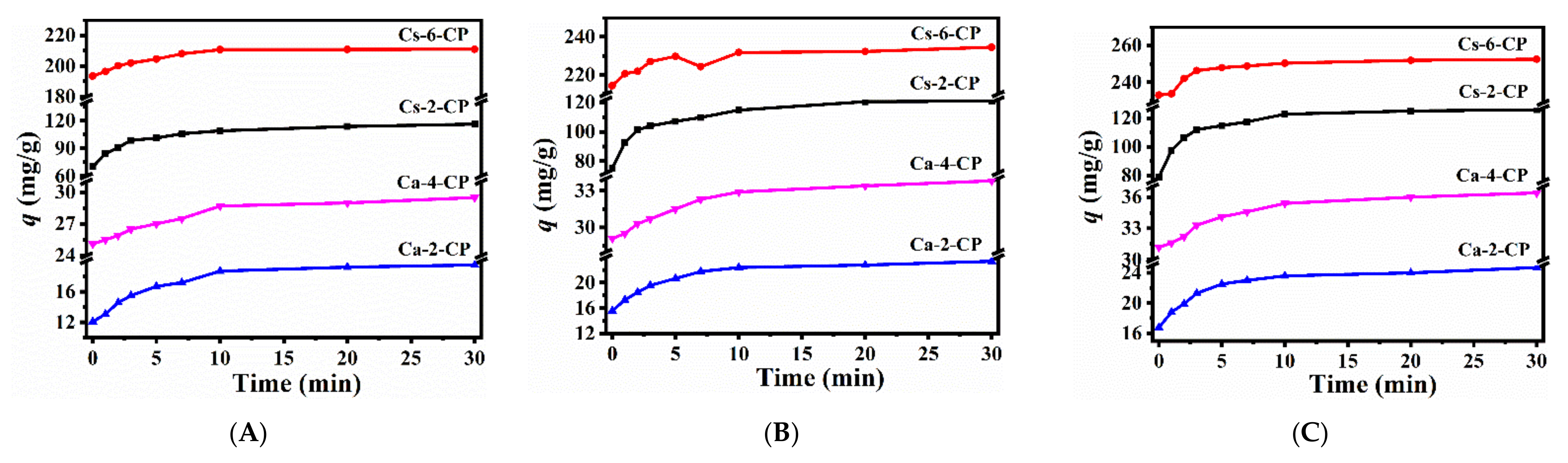
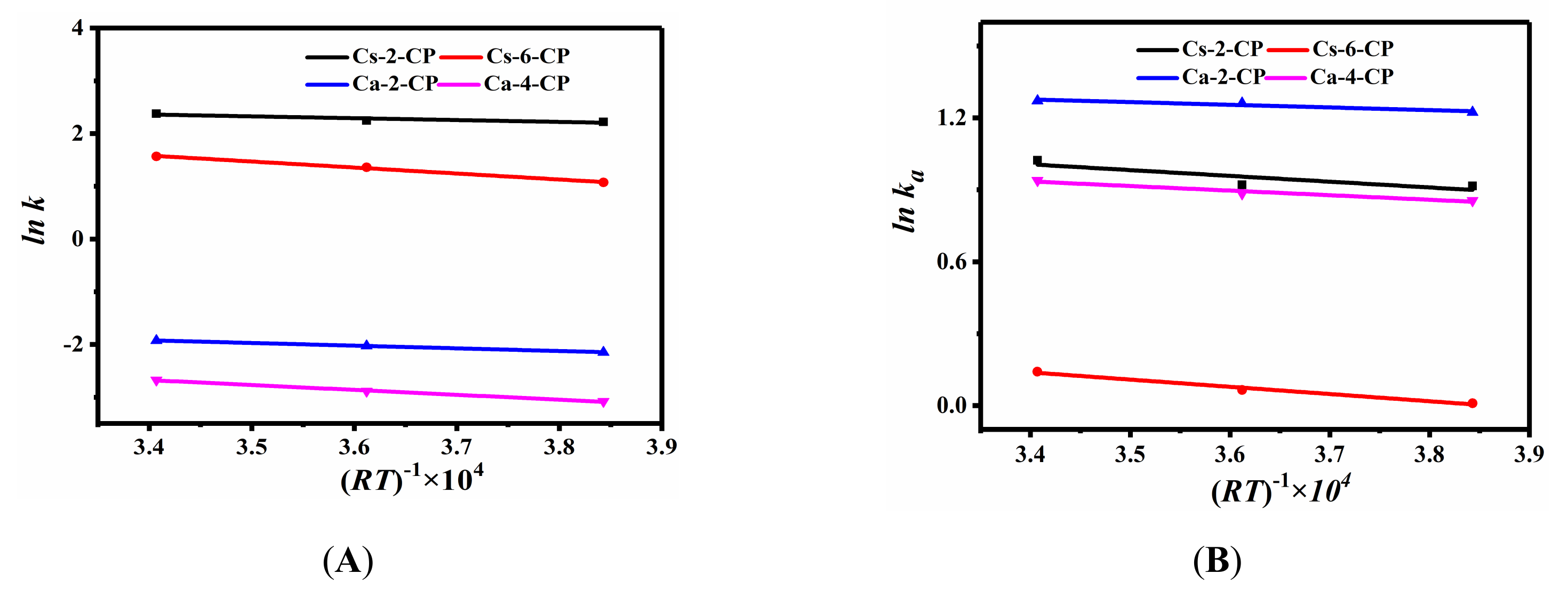
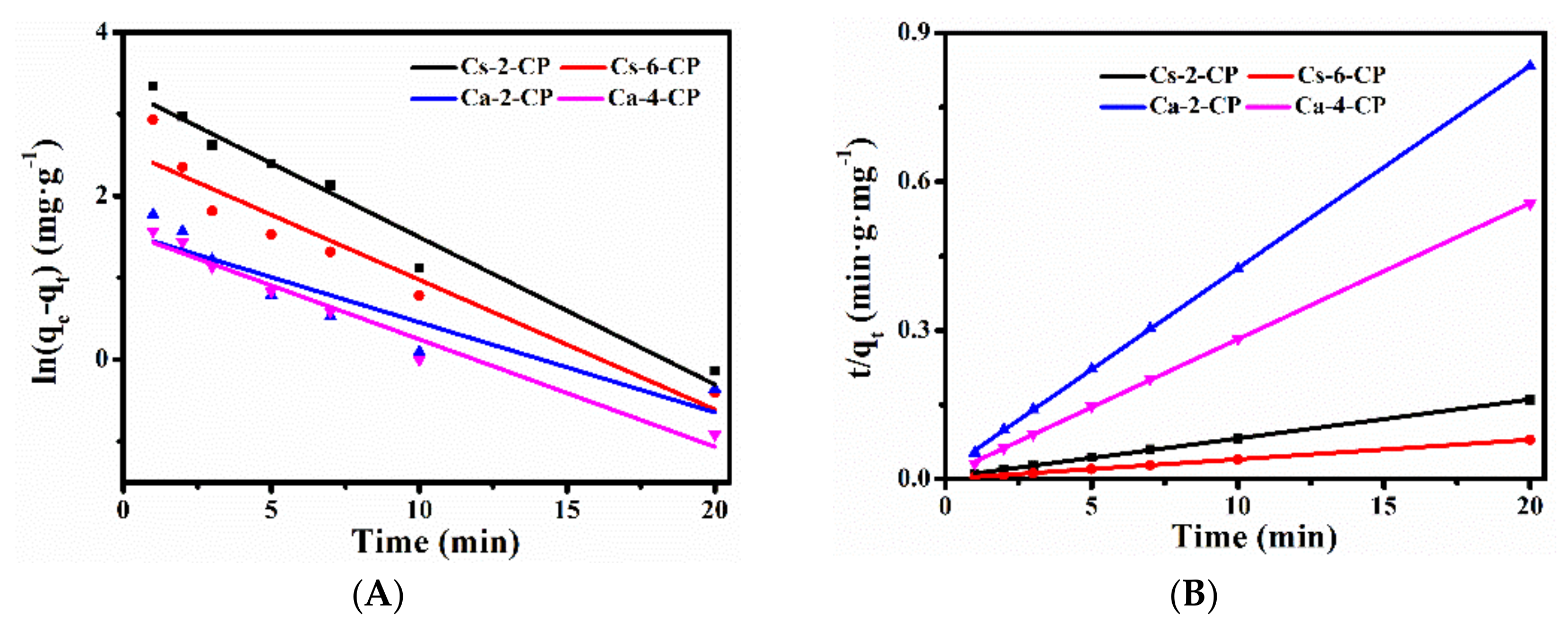
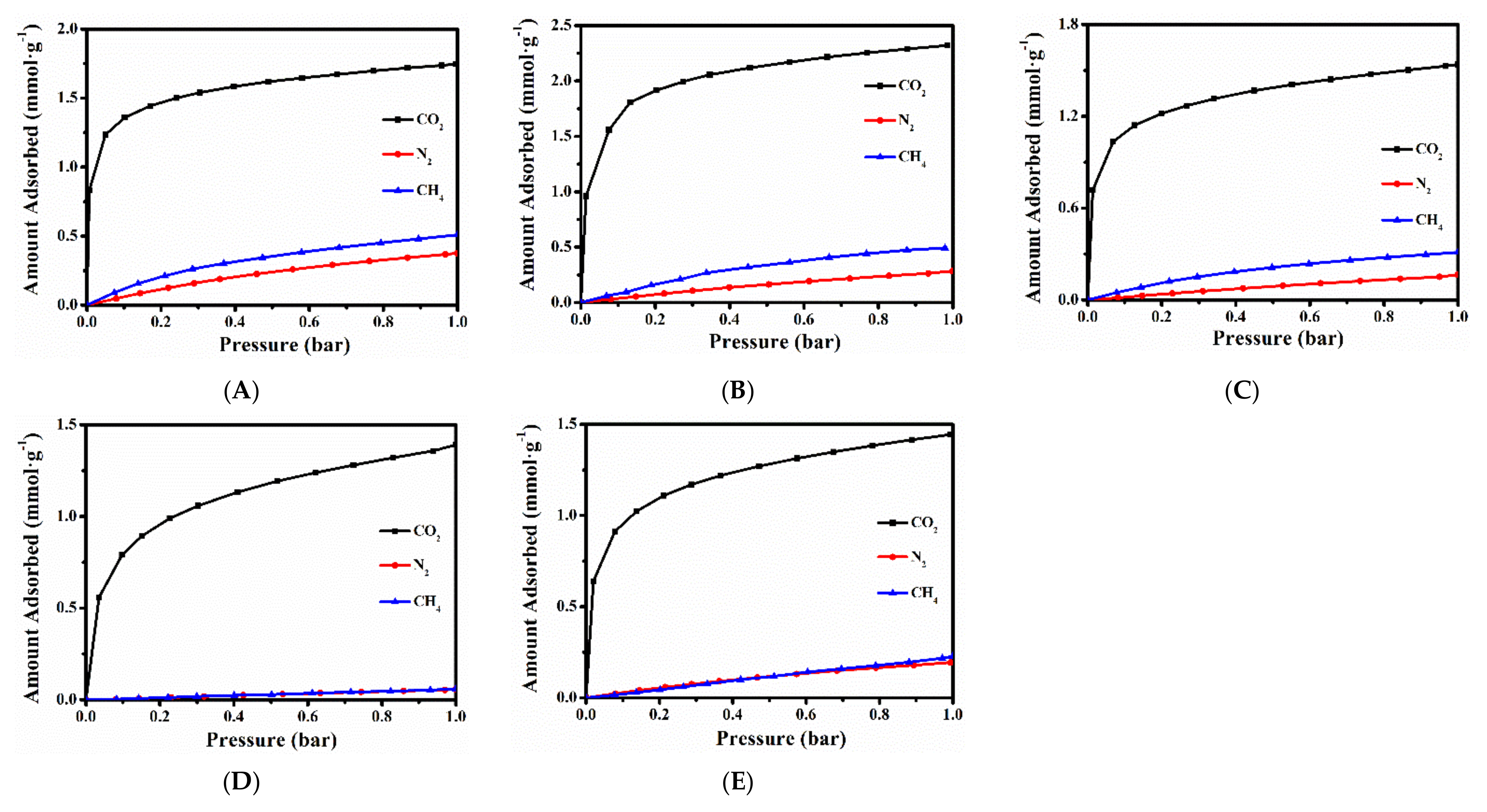
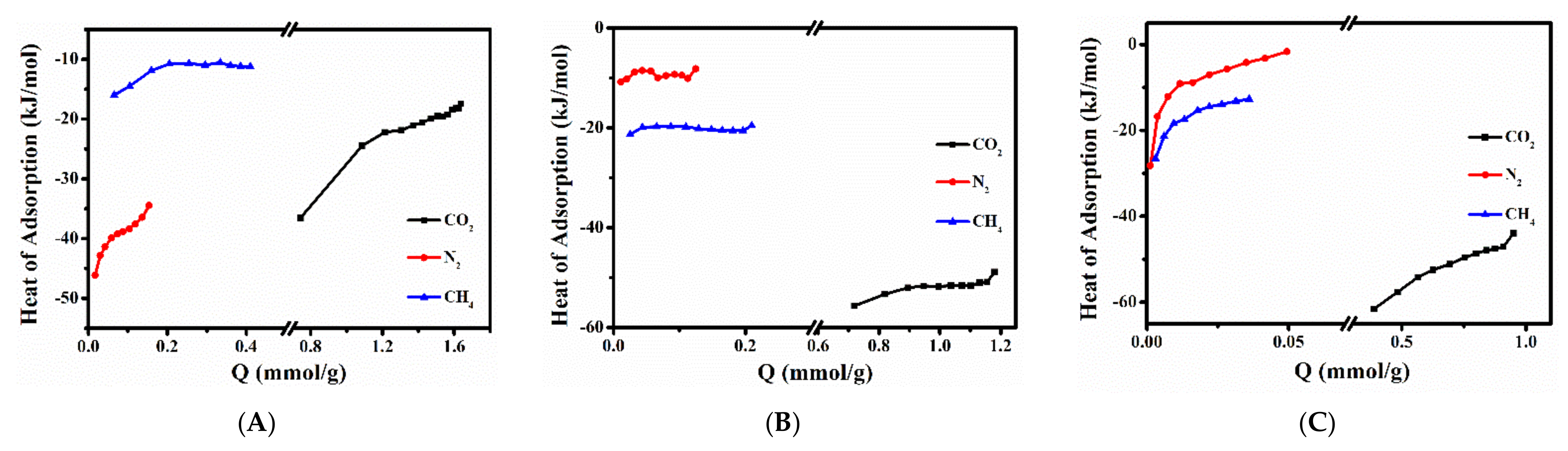
2.3. CH4, N2 and CO2 Adsorption Performances of Various Ion-Exchanged CPs
3. Materials and Methods
3.1. Materials
3.2. Synthesis of CP and Procedure of Ion Exchange
3.3. Characterizations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Williams, C.D. Synthesis of pure clinoptilolite without the use of seed crystals. Chem. Commun. 1997, 21, 2113–2114. [Google Scholar] [CrossRef]
- Wilson, S.T.; Lok, B.M.; Messina, C.A.; Cannan, T.R.; Flanigen, E.M. Aluminophosphate molecular sieves: A new class of mi-croporous crystalline inorganic solids. J. Am. Chem. Soc. 1982, 104, 1146–1147. [Google Scholar] [CrossRef]
- Gottardi, G.; Galli, E. Natural Zeolites; Springer: Berlin, Germany, 1985. [Google Scholar]
- Ackley, M.W.; Yang, R.T. Adsorption characteristics of high-exchange clinoptilolites. Ind. Eng. Chem. Res. 1991, 30, 2523–2530. [Google Scholar] [CrossRef]
- Tanaka, H.; Yamasaki, N.; Muratani, M.; Hino, R. Structure and formation process of (K, Na)-clinoptilolite. Mater. Res. Bull. 2003, 38, 713–722. [Google Scholar] [CrossRef]
- Osmanlioglu, A. Treatment of radioactive liquid waste by sorption on natural zeolite in Turkey. J. Hazard. Mater. 2006, 137, 332–335. [Google Scholar] [CrossRef]
- Petrovic, A.; Barrett, W.C.; Larsson-Kovach, I.-M.; Reid, C.M.; Lisk, D.J. Downward migration of metalaxyl fungicide in creeping bentgrass sand lysimeters as affected by organic waste, peat and zeolite amendments. Chemosphere 1998, 37, 249–256. [Google Scholar] [CrossRef]
- Reháková, M.; Čuvanová, S.; Dzivák, M.; Rimár, J.; Gaval’Ová, Z. Agricultural and agrochemical uses of natural zeolite of the clinoptilolite type. Curr. Opin. Solid State Mater. Sci. 2004, 8, 397–404. [Google Scholar] [CrossRef]
- Akgül, M. Enhancement of the anionic dye adsorption capacity of clinoptilolite by Fe3+-grafting. J. Hazard. Mater. 2014, 267, 1–8. [Google Scholar] [CrossRef]
- Stocker, K.; Ellersdorfer, M.; Lechleitner, A.; Lubensky, J.; Raith, J.G. Impact of concentrated acid, base and salt pretreatments on the characteristics of natural clinoptilolite and its ammonium uptake from model solution and real effluents. Microporous Mesoporous Mater. 2019, 288, 109553. [Google Scholar] [CrossRef]
- Lin, L.; Lei, Z.F.; Wang, L.; Liu, X.; Zhang, Y.; Wan, C.L.; Lee, D.J.; Tay, J.H. Adsorption mechanisms of high-levels of am-monium onto natural and NaCl-modified zeolites. Sep. Purif. Technol. 2013, 103, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Kalló, D.; Papp, J.; Valyon, J. Adsorption and catalytic properties of sedimentary clinoptilolite and mordenite from the Tokaj Hills, Hungary. Zeolites 1982, 2, 13–16. [Google Scholar] [CrossRef]
- Woo, H.C.; Lee, K.H.; Lee, J.S. Catalytic skeletal isomerization of n-butenes to isobutene over natural clinoptilolite zeolite. Appl. Catal. A Gen. 1996, 134, 147–158. [Google Scholar] [CrossRef]
- Khalilzadeh, M.A.; Sadeghifar, H.; Venditti, R. Natural Clinoptilolite/KOH: An Efficient Heterogeneous Catalyst for Car-boxymethylation of Hemicellulose. Ind. Eng. Chem. Res. 2019, 58, 11680–11688. [Google Scholar] [CrossRef]
- Jayaraman, A.; Hernandez-Maldonado, A.J.; Yang, R.T.; Chinn, D.; Munson, C.L.; Moh, D.H. Clinoptilolites for nitro-gen/methane separation. Chem. Eng. Sci. 2004, 59, 2407–2417. [Google Scholar] [CrossRef]
- Hao, X.F.; Hu, H.J.; Li, Z.; Wu, L.M.; Liu, X.Q.; Zhang, Y.N. Adsorption Properties of Modified Clinoptilolite for Methane and Nitrogen. Materials 2018, 11, 2024. [Google Scholar] [CrossRef] [Green Version]
- Kouvelos, E.; Kesore, K.; Steriotis, T.; Grigoropoulou, H.; Bouloubasi, D.; Theophilou, N.; Tzintzos, S.; Kanelopoulos, N. High pressure N2/CH4 adsorption measurements in clinoptilolites. Microporous Mesoporous Mater. 2007, 99, 106–111. [Google Scholar] [CrossRef]
- Kennedy, D.A.; Khanafer, M.; Tezel, F.H. The effect of Ag+ cations on the micropore properties of clinoptilolite and related adsorption separation of CH4 and N2 gases. Microporous Mesoporous Mater. 2019, 281, 123–133. [Google Scholar] [CrossRef]
- Li, J.; Ullah, R.; Jiao, J.; Sun, J.H.; Bai, S.Y. Ion exchange of cations from different groups with ammonium-modified clinoptilolite and selectivity for methane and nitrogen. Mater. Chem. Phys. 2020, 256, 123760. [Google Scholar] [CrossRef]
- Ouyang, T.; Zhai, C.W.; Sun, J.H.; Panezai, H.; Bai, S.Y. Nanosol precursor as structural promoter for clinoptilolite via hydrothermal synthesis and resulting effects on selective adsorption of CH4 and N2. Microporous Mesoporous Mater. 2020, 294, 109913–109920. [Google Scholar] [CrossRef]
- Abukhadra, M.R.; Basyouny, M.G.; El-Sherbeeny, A.M.; El-Meligy, M.A. The effect of different green alkali modification pro-cesses on the clinoptilolite surface as adsorbent for ammonium ions; characterization and application. Microporous Mesoporous Mater. 2020, 300, 110145. [Google Scholar] [CrossRef]
- Hedayati, M.S.; Li, L.Y. Removal of polycyclic aromatic hydrocarbons from aqueous media using modified clinoptilolite. J. Environ. Manag. 2020, 273, 111113. [Google Scholar] [CrossRef] [PubMed]
- Zamzow, M.J.; Eichbaum, B.R.; Sandgren, K.R.; Shanks, D.E. Removal of Heavy Metals and Other Cations from Wastewater Using Zeolites. Sep. Sci. Technol. 1990, 25, 1555–1569. [Google Scholar] [CrossRef]
- Colella, C. Ion exchange equilibria in zeolite minerals. Miner. Deposita 1996, 31, 554–562. [Google Scholar] [CrossRef]
- Kesraoui-Ouki, S.; Cheeseman, C.; Perry, R. Effects on conditioning treatment chabazite and clinoptilolite prior to lead, zinc and cadmium. Environ. Sci. Technol. 1993, 27, 1108–1116. [Google Scholar] [CrossRef]
- Li, Y.R.; Bai, P.; Yan, Y.; Yan, W.F.; Shi, W.; Xu, R.R. Removal of Zn2+, Pb2+, Cd2+, and Cu2+ from aqueous solution by synthetic clinoptilolite. Microporous Mesoporous Mater. 2019, 273, 203–211. [Google Scholar] [CrossRef]
- Argun, M.E. Use of clinoptilolite for the removal of nickel ions from water: Kinetics and thermodynamics. J. Hazard. Mater. 2008, 150, 587–595. [Google Scholar] [CrossRef]
- Pandey, S.; Fosso-Kankeu, E.; Spiro, M.J.; Waanders, F.; Kumar, N.; Ray, S.S.; Kim, J.; Kang, M. Equilibrium, kinetic, and thermodynamic studies of lead ion adsorption from mine wastewater onto MoS2-clinoptilolite composite. Mater. Today Chem. 2020, 18, 100376. [Google Scholar] [CrossRef]
- Wise, W.S.; Nokleberg, W.J.; Kokinos, M. Clinoptilolite and ferrierite from Agoura, California. Am. Mineral. 1969, 54, 887–895. [Google Scholar]
- Danuthai, T.; Sooknoi, T.; Jongpatiwut, S.; Rirksomboon, T.; Osuwan, S.; Resasco, D.E. Effect of extra-framework cesium on the deoxygenation of methylester over CsNaX zeolites. Appl. Catal. A Gen. 2011, 409–410, 74–81. [Google Scholar] [CrossRef]
- Rodríguez-Iznaga, I.; Petranovskii, V.; Castillón-Barraza, F.; Concepción-Rosabal, B. Copper-Silver Bimetallic System on Natural Clinoptilolite: Thermal Reduction of Cu2+ and Ag+ Exchanged. J. Nanosci. Nanotechnol. 2011, 11, 5580–5586. [Google Scholar] [CrossRef]
- Nightingale, E.R.J. Phenomenological theory of ion solvation. Effective radius of hydrated ions. J. Phys. Chem. 1959, 63, 1381–1387. [Google Scholar] [CrossRef]
- Maes, A.; Cremers, A. Ion exchange of synthetic zeolite X and Y with Co2+, Ni2+, Cu2+ and Zn2+ ions. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1975, 71, 265–277. [Google Scholar] [CrossRef]
- Corral-Capulin, N.; Vilchis-Nestor, A.; Gutiérrez-Segura, E.E.; Solache-Ríos, M. The influence of chemical and thermal treatments on the fluoride removal from water by three mineral structures and their characterization. J. Fluor. Chem. 2018, 213, 42–50. [Google Scholar] [CrossRef]
- Rodríguez-Iznaga, I.; Rodríguez-Fuentes, G.; Petranovskii, V. Ammonium modified natural clinoptilolite to remove manganese, cobalt and nickel ions from wastewater: Favorable conditions to the modification and selectivity to the cations. Microporous Mesoporous Mater. 2018, 255, 200–210. [Google Scholar] [CrossRef]
- Garcia-Basabe, Y.; Rodriguez-Iznaga, I.; de Menorval, L.-C.; Llewellyn, P.; Maurin, G.; Lewis, D.W.; Binions, R.; Autie, M.; Ruiz-Salvador, A.R. Step-wise dealumination of natural clinoptilolite: Structural and physicochemical characterization. Microporous Mesoporous Mater. 2010, 135, 187–196. [Google Scholar] [CrossRef]
- Tajeddine, L.; Gailhanou, H.; Blanc, P.; Lassin, A.; Gaboreau, S.; Vieillard, P. Hydration-dehydration behavior and thermo-dynamics of MX-80 montmorillonite studied using thermal analysis. Thermochim. Acta 2015, 604, 83–93. [Google Scholar] [CrossRef]
- Weidenthaler, C.; Schmidt, W. Thermal Stability and Thermal Transformations of Co2+- or Ni2+-Exchanged Zeolites A, X, and Y. Chem. Mater. 2000, 12, 3811–3820. [Google Scholar] [CrossRef]
- Alver, B.E.; Sakizci, M.; Yörükoğullari, E. Investigation of clinoptilolite rich natural zeolites from Turkey: A combined XRF, TG/DTG, DTA and DSC study. J. Therm. Anal. Calorim. 2010, 100, 19–26. [Google Scholar] [CrossRef]
- Alver, B.E.; Sakizci, M. Influence of acid treatment on structure of clinoptilolite tuff and its adsorption of methane. Adsorption 2015, 21, 391–399. [Google Scholar] [CrossRef]
- Güvenir, O.; Kalıpçılar, H.; Çulfaz, A. Crystallization field and rate study for the formation of single phase sodium-potassium and potassium clinoptilolite. Cryst. Res. Technol. 2011, 46, 345–350. [Google Scholar] [CrossRef]
- Lihareva, N.; Petrov, O.; Dimowa, L.; Tzvetanova, Y.; Piroeva, I.; Ublekov, F.; Nikolov, A. Ion exchange of Cs+ and Sr2+ by natural clinoptilolite from bi-cationic solutions and XRD control of their structural positioning. J. Radioanal. Nucl. Chem. 2020, 323, 1093–1102. [Google Scholar] [CrossRef]
- Rodríguez-Iznaga, I.; Petranovskii, V.; Rodríguez-Fuentes, G. Ion-exchange of amino- and aqua-complexes of nickel and cobalt in natural clinoptilolite. J. Environ. Chem. Eng. 2014, 2, 1221–1227. [Google Scholar] [CrossRef]
- Kennedy, D.A.; Tezel, F.H. Cation exchange modification of clinoptilolite—Screening analysis for potential equilibrium and kinetic adsorption separations involving methane, nitrogen, and carbon dioxide. Microporous Mesoporous Mater. 2018, 262, 235–250. [Google Scholar] [CrossRef]
- Alshameri, A.; Ibrahim, A.; Assabri, A.M.; Lei, X.R.; Wang, H.Q.; Yan, C.J. The investigation into the ammonium removal performance of Yemeni natural zeolite: Modification, ion exchange mechanism, and thermodynamics. Powder Technol. 2014, 258, 20–31. [Google Scholar] [CrossRef]
- Ho, Y.S.; Ng, J.C.; McKay, G. Kinetics of pollutant sorption by biosorbents: Review. Sep. Purif. Methods 2000, 29, 189–232. [Google Scholar] [CrossRef]
- Javanbakht, V.; Ghoreishi, S.M.; Habibi, N.; Javanbakht, M. A novel magnetic chitosan/clinoptilolite/magnetite nanocomposite for highly efficient removal of Pb(II) ions from aqueous solution. Powder Technol. 2016, 302, 372–383. [Google Scholar] [CrossRef]
- Barros, M.A.S.D.; Arroyo, P.A.; Sousa-Aguiar, E.F.; Tavares, C.R.G. Thermodynamics of the Exchange Processes between K+, Ca2+ and Cr3+ in Zeolite NaA. Adsorption 2004, 10, 227–235. [Google Scholar] [CrossRef]
- Tarasevich, Y.I.; Polyakov, V.E. Ion-exchange equilibria and exchange heats on clinoptilolite involving singly-charged cations. Theor. Exp. Chem. 1996, 32, 276–280. [Google Scholar] [CrossRef]
- Inglezakis, V.J.; Zorpas, A.A. Heat of adsorption, adsorption energy and activation energy in adsorption and ion exchange systems. Desalination Water Treat. 2011, 39, 149–157. [Google Scholar]
- Ijagbemi, C.O.; Baek, M.-H.; Kim, D.-S. Montmorillonite surface properties and sorption characteristics for heavy metal removal from aqueous solutions. J. Hazard. Mater. 2009, 166, 538–546. [Google Scholar] [CrossRef]
- Kennedy, D.A.; Mujčin, M.; Abou-Zeid, C.; Tezel, F.H. Cation exchange modification of clinoptilolite-thermodynamic effects on adsorption separations of carbon dioxide, methane, and nitrogen. Microporous Mesoporous Mater. 2019, 274, 327–341. [Google Scholar] [CrossRef]
- Sacerdoti, M.; Lucchetti, G. The crystal structure of heulandite-Sr from Campegli (Liguria, Italy). Microporous Mesoporous Mater. 2010, 131, 310–313. [Google Scholar] [CrossRef]
- Zhang, J.; Singh, R.; Webley, P.A. Alkali and alkaline-earth cation exchanged chabazite zeolites for adsorption based CO2 capture. Microporous Mesoporous Mater. 2008, 111, 478–487. [Google Scholar] [CrossRef]
- Mani, F.; Sawada, J.A.; Kuznicki, S.M. A comparison of the adsorptive behavior of ETS-10, 13X, and highly siliceous ZSM-5. Microporous Mesoporous Mater. 2015, 214, 32–40. [Google Scholar] [CrossRef]
- Vosoughi, M.; Maghsoudi, H.; Gharedaghi, S. Ion-exchanged ETS-10 adsorbents for CO2/CH4 separation: IAST assisted comparison of performance with other zeolites. J. Nat. Gas Sci. Eng. 2021, 88, 103862. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Chemical Formula | Relative Crystallinity |
|---|---|---|
| (Na, K)–CP | Na1.122K6.433Si28.387Al7.631O72 | 1.00 |
| NH4–CP | Na0.147K0.544(NH4)6.864Si28.387Al7.631O72 | 1.542 |
| Li–2–CP | Na0.147K0.544(NH4)3.869Li2.995Si28.387Al7.631O72 | 1.387 |
| Li–3–CP | Na0.147K0.544(NH4)2.683Li4.181Si28.387Al7.631O72 | 1.234 |
| Li–4–CP | Na0.147K0.544(NH4)1.671Li5.193Si28.387Al7.631O72 | 1.233 |
| Li–5–CP | Na0.147K0.544(NH4)1.145Li5.719Si28.387Al7.631O72 | 1.191 |
| Li–6–CP | Na0.147K0.544(NH4)0.679Li6.185Si28.387Al7.631O72 | 1.165 |
| Cs–2–CP | Na0.147K0.544(NH4)3.814Cs3.050Si28.387Al7.631O72 | 0.495 |
| Cs–3–CP | Na0.147K0.544(NH4)2.708Cs4.156Si28.387Al7.631O72 | 0.454 |
| Cs–4–CP | Na0.147K0.544(NH4)1.791Cs5.073Si28.387Al7.631O72 | 0.446 |
| Cs–5–CP | Na0.147K0.544(NH4)1.214Cs5.650Si28.387Al7.631O72 | 0.430 |
| Cs–6–CP | Na0.147K0.544(NH4)0.744Cs6.120Si28.387Al7.631O72 | 0.399 |
| Ca–2–CP | Na0.147K0.544(NH4)2.894Ca1.985Si28.387Al7.631O72 | 1.132 |
| Ca–3–CP | Na0.147K0.544(NH4)1.850Ca2.507Si28.387Al7.631O72 | 1.061 |
| Ca–4–CP | Na0.147K0.544(NH4)1.014Ca2.925Si28.387Al7.631O72 | 1.000 |
| Sr–2–CP | Na0.147K0.544(NH4)3.908Sr1.478Si28.387Al7.631O72 | 0.800 |
| Sr–4–CP | Na0.147K0.544(NH4)2.350Sr2.257Si28.387Al7.631O72 | 0.782 |
| Sr–6–CP | Na0.147K0.544(NH4)1.374Sr2.745Si28.387Al7.631O72 | 0.772 |
| Sr–8–CP | Na0.147K0.544(NH4)0.786Sr3.039Si28.387Al7.631O72 | 0.668 |
| Samples | Exchange Times | 313 K | 333 K | 353 K | Δr | Ea | |||
|---|---|---|---|---|---|---|---|---|---|
| Δr | Δr | Δr | Δr | Δr | Δr | ||||
| Li–x–CP | 2 | −4.603 | 30.59 | −4.897 | 29.64 | −5.796 | 30.50 | 4.976 | 6.220 |
| 3 | −3.226 | 22.69 | −3.602 | 22.45 | −4.138 | 22.70 | 3.878 | 7.349 | |
| 4 | −2.934 | 23.16 | −3.513 | 23.50 | −3.857 | 23.14 | 4.316 | 7.839 | |
| 5 | −2.390 | 14.39 | −2.631 | 14.25 | −2.968 | 14.40 | 2.117 | 8.380 | |
| 6 | −2.244 | 15.61 | −2.609 | 15.76 | −2.863 | 15.59 | 2.643 | 9.319 | |
| Cs–x–CP | 2 | −3.479 | 14.99 | −3.706 | 14.77 | −4.081 | 14.99 | 1.214 | 3.504 |
| 3 | −2.749 | 30.08 | −3.482 | 30.48 | −3.949 | 30.08 | 6.672 | 6.406 | |
| 4 | −2.694 | 23.12 | −3.105 | 22.97 | −3.623 | 23.13 | 4.547 | 8.230 | |
| 5 | −2.442 | 12.39 | −2.581 | 12.07 | −2.823 | 12.07 | 1.439 | 10.373 | |
| 6 | −2.273 | 13.22 | −2.497 | 13.10 | −2.804 | 13.23 | 1.867 | 11.370 | |
| Ca–x–CP | 2 | −1.593 | 8.641 | −1.749 | 8.591 | −1.868 | 8.441 | 1.113 | 5.082 |
| 3 | −1.217 | 14.16 | −1.406 | 13.87 | −1.578 | 13.57 | 3.216 | 8.866 | |
| 4 | −1.114 | 9.663 | −1.225 | 9.416 | −1.380 | 9.322 | 1.912 | 9.283 | |
| Sr–x–CP | 2 | −1.431 | 24.14 | −1.629 | 23.28 | −2.009 | 23.04 | 6.128 | 5.308 |
| 4 | −0.968 | 11.71 | −1.116 | 11.46 | −1.265 | 11.23 | 2.701 | 6.409 | |
| 6 | −0.803 | 9.976 | −0.912 | 9.703 | −1.055 | 9.560 | 2.321 | 9.836 | |
| 8 | −0.548 | 5.109 | −0.602 | 4.965 | −0.687 | 4.924 | 1.052 | 12.604 | |
| Samples | Exchange Times | Pseudo-First-Order | Pseudo-Second-Order | qe (exp) (mg·g−1) | ||||
|---|---|---|---|---|---|---|---|---|
| k1 (min−1) | R2 | qe (cal) (mg·g−1) | k2 (g·mg−1·min−1) | R2 | qe (cal) (mg·g−1) | |||
| Li–x–CP | 2 | 0.2003 | 0.8236 | 1.075 | 0.4291 | 0.9998 | 6.557 | 6.46 |
| 3 | 0.1567 | 0.9978 | 1.864 | 0.2556 | 0.9996 | 9.066 | 9.01 | |
| 4 | 0.1289 | 0.9470 | 1.610 | 0.3004 | 0.9999 | 11.186 | 11.19 | |
| 5 | 0.2853 | 0.9889 | 1.531 | 0.4800 | 0.9999 | 12.422 | 12.33 | |
| 6 | 0.1568 | 0.9003 | 0.744 | 0.7013 | 0.999 | 13.351 | 13.33 | |
| Cs–x–CP | 2 | 0.1801 | 0.9712 | 27.067 | 0.0179 | 0.9998 | 128.205 | 125.87 |
| 3 | 0.1131 | 0.8677 | 29.818 | 0.0158 | 0.9999 | 169.492 | 171.53 | |
| 4 | 0.2169 | 0.9788 | 24.039 | 0.0245 | 0.9999 | 212.766 | 209.38 | |
| 5 | 0.0954 | 0.8121 | 14.189 | 0.0462 | 0.9999 | 232.56 | 233.18 | |
| 6 | 0.1666 | 0.9286 | 14.326 | 0.0400 | 0.9999 | 250.00 | 252.57 | |
| Ca–x–CP | 2 | 0.1101 | 0.8656 | 4.754 | 0.0991 | 0.9999 | 24.510 | 24.70 |
| 3 | 0.0903 | 0.9237 | 5.325 | 0.0857 | 0.9994 | 30.675 | 31.20 | |
| 4 | 0.1315 | 0.9716 | 4.779 | 0.1022 | 0.9999 | 36.364 | 36.40 | |
| Sr–x–CP | 2 | 0.2217 | 0.9177 | 10.746 | 0.0434 | 0.9996 | 41.322 | 40.20 |
| 4 | 0.1716 | 0.9412 | 7.039 | 0.0729 | 0.9999 | 61.728 | 61.40 | |
| 6 | 0.197 | 0.9542 | 4.123 | 0.1474 | 0.9999 | 75.188 | 74.70 | |
| 8 | 0.1127 | 0.8494 | 2.188 | 0.2440 | 0.9999 | 82.644 | 82.70 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.; Jia, B.; Li, Y.; Sun, J.; Wu, X. Explorations on Thermodynamic and Kinetic Performances of Various Cationic Exchange Durations for Synthetic Clinoptilolite. Molecules 2022, 27, 2597. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082597
Wang K, Jia B, Li Y, Sun J, Wu X. Explorations on Thermodynamic and Kinetic Performances of Various Cationic Exchange Durations for Synthetic Clinoptilolite. Molecules. 2022; 27(8):2597. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082597
Chicago/Turabian StyleWang, Keling, Bingying Jia, Yehong Li, Jihong Sun, and Xia Wu. 2022. "Explorations on Thermodynamic and Kinetic Performances of Various Cationic Exchange Durations for Synthetic Clinoptilolite" Molecules 27, no. 8: 2597. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082597