
Computational Insights and In Vitro Validation of Antibacterial Potential of Shikimate Pathway-Derived Phenolic Acids as NorA Efflux Pump Inhibitors



Abstract
:1. Introduction
2. Results and Discussion
2.1. In Silico Studies
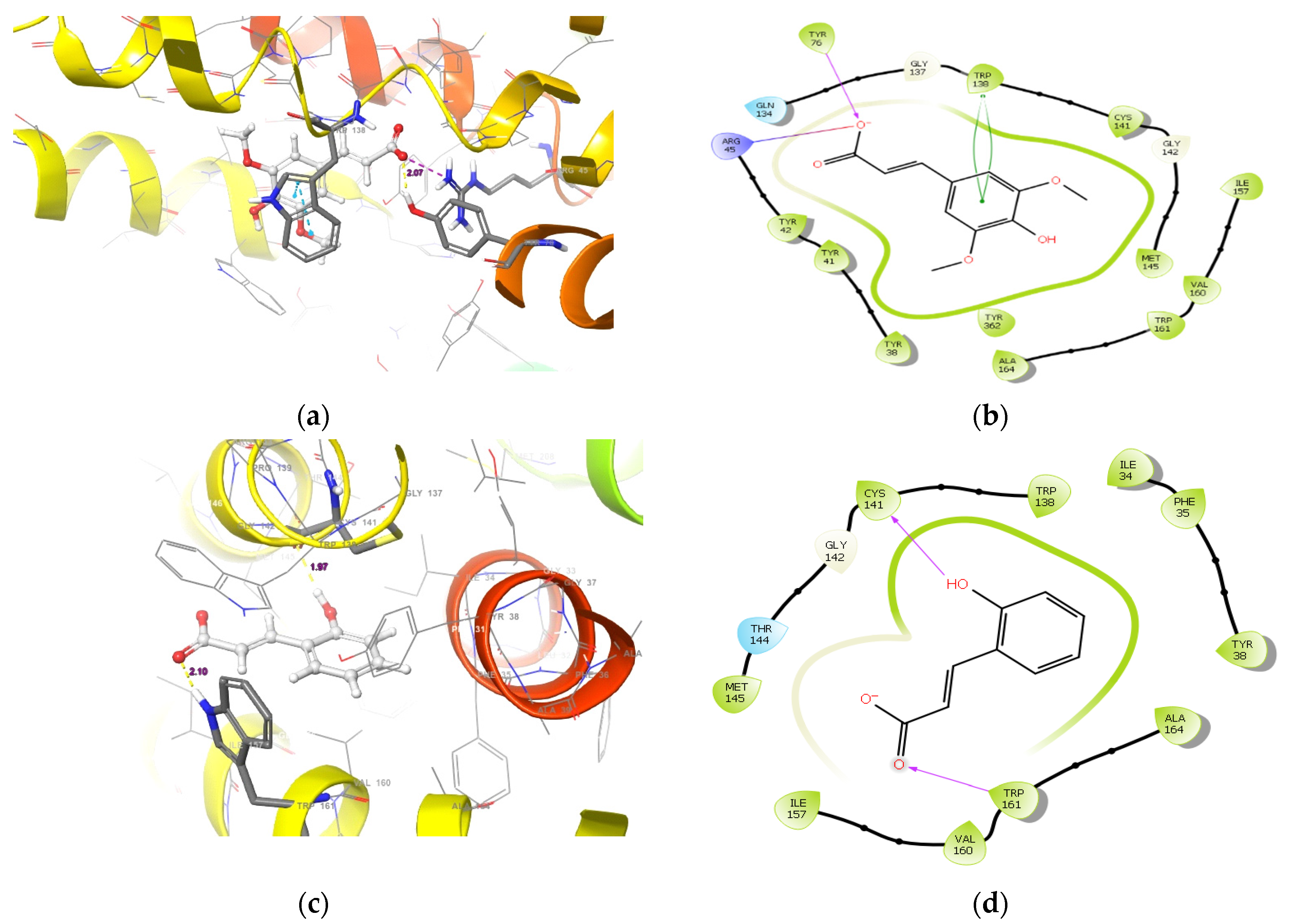
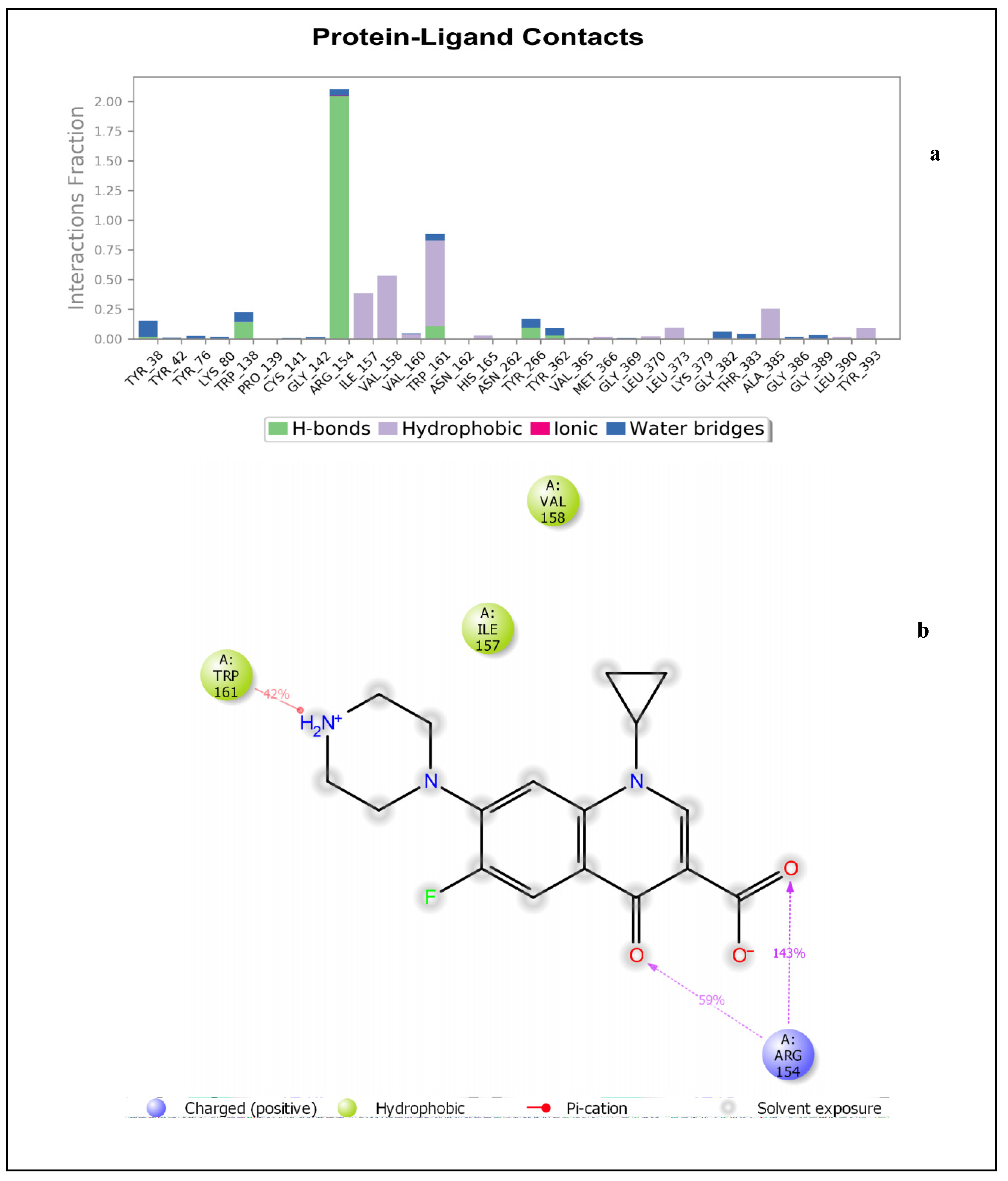
2.1.1. Molecular Docking
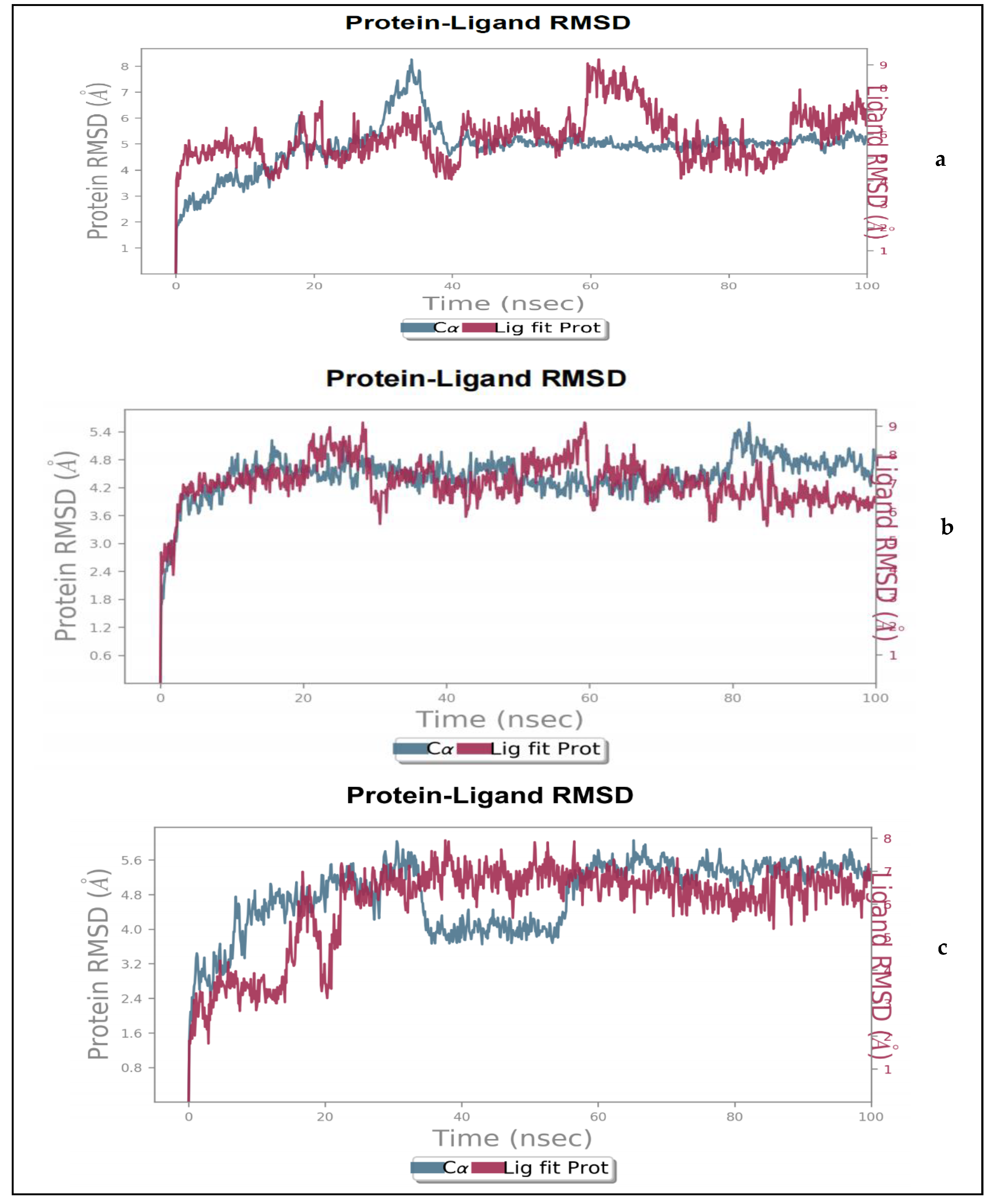
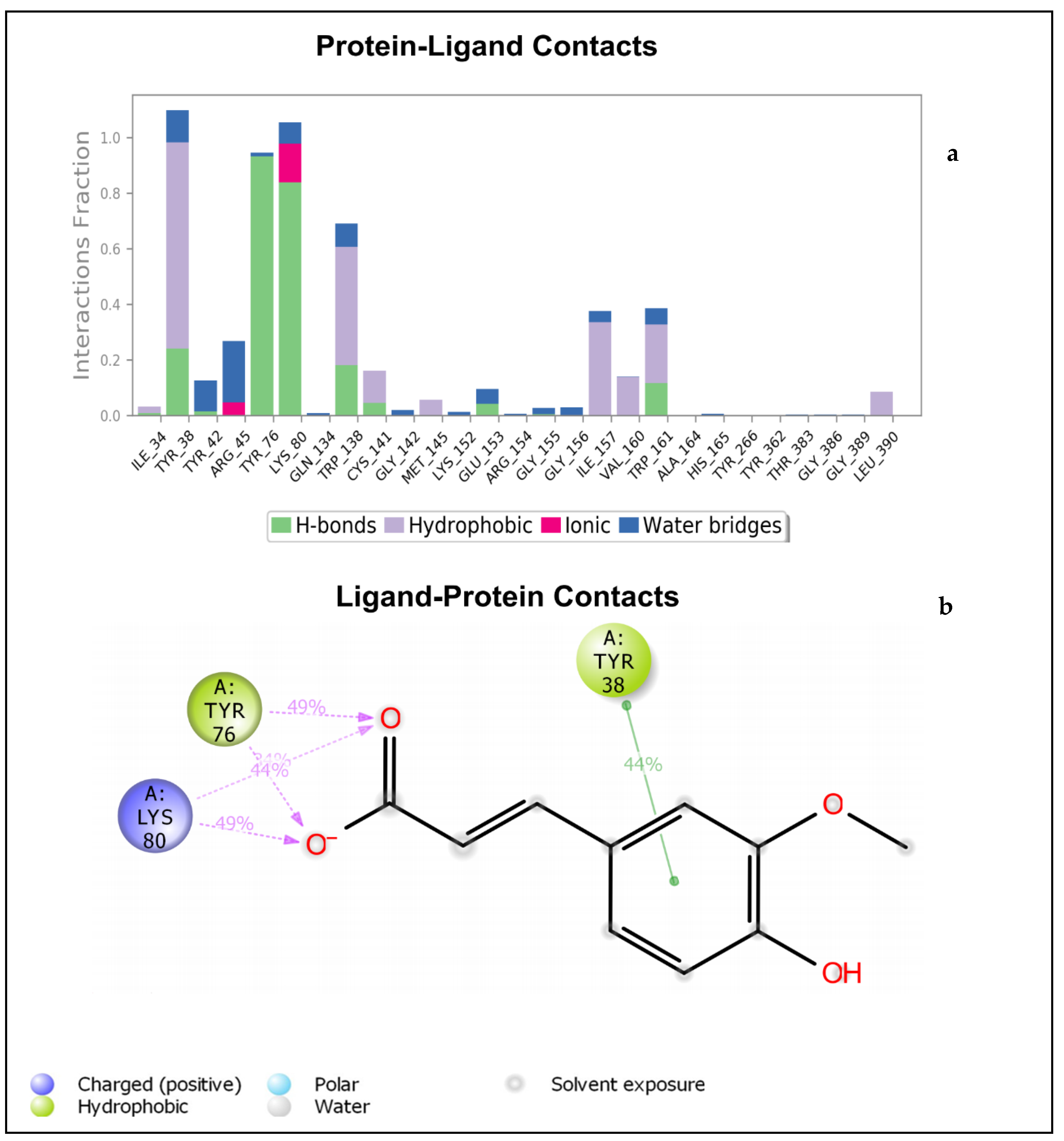
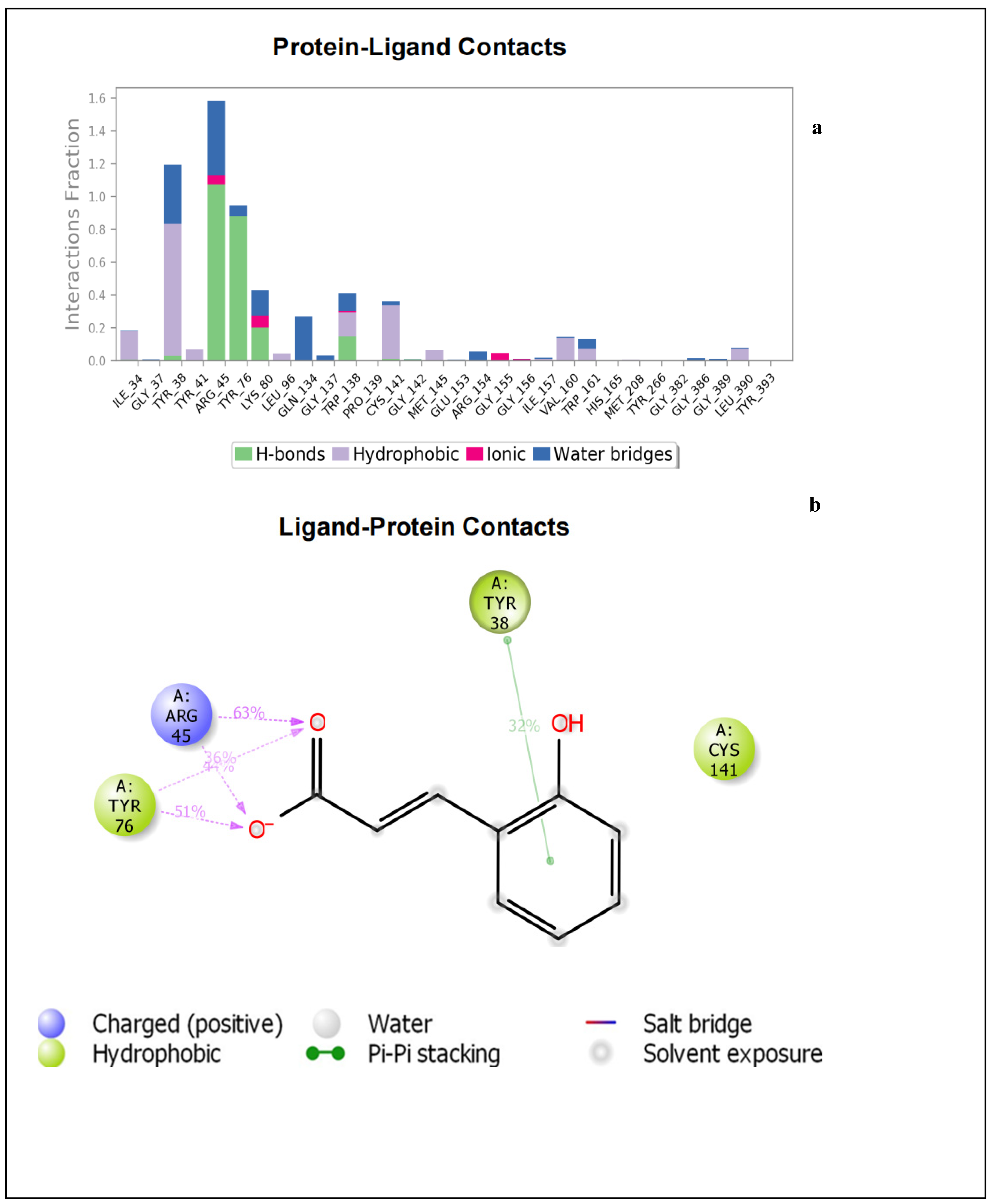
2.1.2. Molecular Dynamics
2.1.3. Pharmacokinetic Properties and Toxicity
2.2. In Vitro Antibacterial Evaluation
3. Materials and Methods
3.1. Molecular Modelling
3.2. Pharmacokinetic Properties and Toxicity Risk Assessment
3.3. In Vitro Antimicrobial Evaluations
3.3.1. Source and Preparation of Bacterial Cultures
3.3.2. Test Compounds
3.3.3. Determination of Minimum Inhibition Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
3.3.4. Time-Kill Assay
3.3.5. Combination Therapy (Checkerboard Assay)
3.4. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Subbaiah, S.G.P.; Dakappa, S.S.; Lakshmikantha, R.Y. Antibacterial and molecular docking studies of bioactive component from leaves of Stachytarpheta cayennensis (Rich.) Vahl. Res. J. Phytochem. 2016, 11, 28–34. [Google Scholar] [CrossRef]
- Seukep, A.J.; Kuere, V.; Nahar, L.; Sarkar, S.D.; Guo, M. Plant-derived secondary metabolites as the main source of efflux pump inhibitors and methods for identification. J. Pharm. Anal. 2020, 10, 277–290. [Google Scholar] [CrossRef]
- Papkou, A.; Hedge, J.; Kapel, N. Efflux pump activity potentiates the evolution of antibiotic resistance across S. aureus isolates. Nat. Commun. 2019, 11, 3970–3980. [Google Scholar] [CrossRef]
- Costa, S.S.; Sobkowial, B.; Parreira, R. Genetic diversity of NorA, coding for a main efflux pump of Staphylococcus aureus. Front. Genet. 2019, 9, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Palazzotti, D.; Bissaro, M.; Bolcato, G. Deciphering the molecular recognition mechanism of multidrug resistance Staphylococcus aureus NorA efflux pump using a supervised molecular dynamics approach. Int. J. Mol. Sci. 2019, 20, 4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.L.; Grinius, L.; Hoope, D.C. NorA Functions as a Multidrug Efflux Protein in both Cytoplasmic Membrane Vesicles and Reconstituted Proteoliposomes. J. Bacteriol. 2002, 184, 1370–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sri Sundaramoorthy, N.; Mitra, K.; Ganesh, J.S.; Makala, H. Ferulic acid derivative inhibits NorA efflux and in combination with ciprofloxacin curtails growth of MRSA in vitro and in vivo. Microb. Pathog. 2018, 124, 54–62. [Google Scholar] [CrossRef]
- dos Santos, J.F.S.; Tintino, S.R.; de Freitas, T.S. In vitro and in silico evaluation of the inhibition of Staphylococcus aureus efflux pumps by caffeic and gallic acid. Comp. Immunol. Microbiol. Infect. Dis. 2018, 57, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Sabiu, S.; Balogun, F.O.; Amoo, S.O. Phenolics Profiling of Carpobrotus edulis (L.) NE Br. and Insights into Molecular Dynamics of Their Significance in Type 2 Diabetes Therapy and Its Retinopathy Complication. Molecules 2021, 26, 4867. [Google Scholar] [CrossRef]
- Ghimire, B.K.; Seong, E.S.; Yu, C.Y.; Kim, S.H.; Chung, I.M. Evaluation of phenolic compounds and antimicrobial activities in transgenic Codonopsis lanceolata plants via overexpression of the γ-tocopherol methyltransferase (γ-tmt) gene. S. Afr. J. Bot. 2017, 109, 25–33. [Google Scholar] [CrossRef]
- Mandal, S.M.; Dias, R.O.; Franco, O.L. Phenolic compounds in antimicrobial therapy. J. Med. Food 2017, 20, 1031–1038. [Google Scholar] [CrossRef]
- Bouarab-Chibane, L.; Forquet, V.; Lanteri, P. Antibacterial properties of polyphenols: Characterization and QSAR (Quantitative structure-activity relationship) models. Front. Microbiol. 2019, 10, 829. [Google Scholar] [CrossRef]
- Ribeiro, J.; Silva, V.; Aires, A. Antimicrobial activity of phenolic compounds extracted from Platanus hybrid: Exploring alternative therapies for a post-antibiotic era. Proceedings 2020, 66, 18. [Google Scholar]
- Haq, A.; Siddiqi, M.; Batool, S.Z.; Islam, A.; Khan, A.; Khan, D.; Khan, S.; Khan, H.; Shah, A.A.; Hasan, F.; et al. Comprehensive investigation on the synergistic antibacterial activities of Jatropha curcas pressed cake and seed oil in combination with antibiotics. AMB Express 2019, 9, 67–88. [Google Scholar] [CrossRef] [Green Version]
- Zárate, S.G.; Morales, P.; Swiderek, A. A molecular modelling approach to identify novel inhibitors of the major facilitator superfamily of efflux pump transporters. Antibiotics 2019, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uniyal, A.; Mahapatra, M.K.; Tiwari, V. Targeting SARS-CoV-2 main protease: Structure based virtual screening, in silico, ADMET studies and molecular dynamics simulations for identification of potential inhibitors. J. Biomol. Struct. Dyn. 2020, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Husain, F.M.; Alhumaydhi, F.A.; Alwashmi, A.S.; Rehman, M.T.; Alruwetei, A.M.; Hassan, M.I.; Islam, A.; Shamsi, A. Exploring the molecular interactions of Galantamine with human Transferrin: In-silico and in vitro insight. J. Mol. Liq. 2021, 335, 116227. [Google Scholar] [CrossRef]
- Khan, A.; Ali, S.S.; Khan, M.T.; Saleem, S.; Ali, A.; Suleman, M.; Babar, Z.; Shafiq, A.; Khan, M.; Wei, D.Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro). J. Biomol. Struct. Dyn. 2020, 39, 4659–4670. [Google Scholar] [CrossRef]
- Kumar, N.; Goel, N. Phenolic acids: Natural versatile molecules with promising therapeutic applications. Biotechnol. Rep. 2019, 24, 370–380. [Google Scholar] [CrossRef]
- Lamazares, E.; MacLeod-Carey, D.; Miranda, F.P.; Mena-Ulecia, K. Theoretical evaluation of novel thermolysin inhibitors from Bacillus thermoproteolyticus. Possible antibacterial agents. Molecules 2021, 26, 386–406. [Google Scholar]
- Sabiu, S.; Idowu, K. An insight on the nature of biochemical interactions between glycyrrhizin, myricetin and CYP3A4 isoform. J. Food Biochem. 2021, 46, e13831. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Boshoff, H.I.M.; Arora, K. Synthesis, antitubular evaluation, molecular dynamics studies of 4,6-disubstituted-2-oxo-dihydropyridine-3-carbonitriles. J. Mol. Struct. 2019, 1197, 117–133. [Google Scholar] [CrossRef]
- Bhaskar, B.V.; Muni, T.; Babu, C. Homology modeling, molecular dynamics, and virtual screening of NorA efflux pump inhibitors of Staphylococcus aureus. Drug Des. Dev. Ther. 2016, 10, 3237–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khade, A.B.; Boshoff, H.I.M.; Arora, K. Design, synthesis, evaluation, and molecular dynamic simulation of triclosan mimic diphenyl ether derivatives as antitubercular and antibacterial agents. Struct. Chem. 2020, 31, 983–998. [Google Scholar] [CrossRef]
- Isa, M.A.; Mustapha, A.; Qazi, S. In silico molecular docking and molecular dynamic simulation of potential inhibitors of 3C-like main proteinase (3CLpro) from severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) using selected African medicinal plants. Adv. Tradit. Med. 2020, 22, 107–123. [Google Scholar] [CrossRef]
- Muniz, D.F.; dos Santos Barbosa, C.R.; de Menezes, I.R.A. In vitro and in silico inhibitory effects of synthetic and natural derivatives against the NorA efflux pump in Staphylococcus aureus. Food Chem. 2021, 337, 127776. [Google Scholar] [CrossRef]
- Sanhueza, L.; Melo, R.; Montero, R.; Maisey, K. Synergistic interactions between phenolic compounds identified in grape pomace extract with antibiotics of different classes against Staphylococcus aureus and Escherichia coli. PLoS ONE 2017, 12, e0172273. [Google Scholar] [CrossRef]
- Santos-Sánchez, N.F.; Salas-Coronado, R.; Hernández-Carlos, B.; Villanueva-Cañongo, C. Shikimic Acid Pathway in Biosynthesis of Phenolic Compounds. Plant Physiological Aspects of Phenolic Compounds. Plant Physiol. Asp. Phenolic Compd. 2019, 1, 1–15. [Google Scholar]
- Zimmermann, S.; Strobel, M.K.; Bohnert, J.A. Clinically approved drugs inhibit the Staphylococcus aureus multidrug NorA efflux pump and reduce biofilm formation. Front. Microbiol. 2019, 10, 2762–2772. [Google Scholar] [CrossRef] [Green Version]
- Mekinie, I.G.; Skroza, D.; Ljubenkov, I. Antioxidant and antimicrobial potential of phenolic metabolites from traditionally used mediterranean herbs and spices. Foods 2019, 8, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koivuniemi, A.; Fallarero, A.; Bunker, A. Insight into the antimicrobial mechanism of action of β2,2-amino acid derivatives from molecular dynamics simulation: Dancing the can-can at the membrane surface. BBA-Biomembr. 2019, 1861, 183028–183036. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. Schrödinger Release 2021-2; Prime: New York, NY, USA, 2021. [Google Scholar]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W.J. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Clark, A.J.; Tiwary, P.; Borrelli, K.; Feng, S.; Miller, E.B.; Abel, R.; Friesner, R.A.; Berne, B.J. Prediction of Protein−Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations. J. Chem. Theory Comput. 2016, 12, 2990–2998. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2014, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B.J. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem 2006, 14, 3160–3173. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Smiesko, M.; Sellner, M.; Lill, M.A. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T.; Frye, L.L. Greenwood JR Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Cappel, D.; Jerome, S.; Hessler, G.; Matter, H. Impact of Different Automated Binding Pose Generation Approaches on Relative Binding Free Energy Simulations. J. Chem. Inf. Model. 2020, 60, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kokubo, H. Exploring the Stability of Ligand Binding Modes to Proteins by Molecular Dynamics Simulations: A Cross-docking Study. J. Chem. Inf. Model. 2017, 57, 2514–2522. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.D.; Unal, H.; Desnoyer, R.; Karnik, S.S. Divergent Spatiotemporal Interaction of Angiotensin Receptor Blocking Drugs with Angiotensin Type 1 Receptor. J. Chem. Inf. Model. 2018, 58, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.B.; Murphy, R.B.; Sindhikara, D.; Borrelli, K.W.; Grisewood, M.J.; Ranalli, F.; Dixon, S.L.; Jerome, S.; Boyles, N.A.; Day, T.; et al. Reliable and Accurate Solution to the Induced Fit Docking Problem for Protein–Ligand Binding. J. Chem. Theory Comput. 2021, 17, 2630–2639. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregerson, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06). Tampa, FL, USA; 2006. Available online: http://sc06.supercomputing.org/schedule/event_detail.php?evid=9088 (accessed on 28 August 2021).
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basri, D.F.; Xian, L.W.; Abdul Shukor, N.I.; Latip, J. Bacteriostatic antimicrobial combination: Antagonistic interaction between epsilon-viniferin and vancomycin against methicillin-resistant. Staphylococcus Aureus. BioMed Res. Int. 2014, 3, 461756. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Title | Docking Score | Glide Emodel | XP G Score | IFD Score |
|---|---|---|---|---|---|
| (kcal/mol) | (kcal/mol) | (kcal/mol) | (kcal/mol) | ||
| 1 | Sinapic Acid | −9.04 | −42.02 | −9.04 | −771.92 |
| 2 | Sinapic Acid | −7.27 | −38.66 | −7.27 | −770.04 |
| 3 | Sinapic Acid | −6.45 | −31.35 | −6.45 | −768.67 |
| 4 | Sinapic Acid | −2.04 | −36.05 | −2.04 | −765.21 |
| 5 | Sinapic Acid | −1.21 | −35.90 | −1.21 | −763.33 |
| 6 | p-Coumaric Acid | −6.91 | −39.45 | −6.91 | −767.95 |
| 7 | p-Coumaric Acid | −6.32 | −40.26 | −6.32 | −767.68 |
| 8 | p-Coumaric Acid | −6.31 | −37.59 | −6.31 | −767.32 |
| 9 | p-Coumaric Acid | −6.45 | −34.56 | −6.45 | −767.30 |
| 10 | p-Coumaric Acid | −6.24 | −37.08 | −6.24 | −767.24 |
| 11 | Ciprofloxacin | −4.31 | −59.19 | −4.36 | −760.89 |
| 12 | Ciprofloxacin | −3.25 | −42.74 | −3.30 | −759.56 |
| Properties | Sinapic Acid | p-Coumaric Acid | Ciprofloxacin |
|---|---|---|---|
| Molecular formula | C11H12O5 | C9H8O3 | C17H18FN3O3 |
| Molecular weight (g/mol) | 224.21 | 164.16 | 331.34 |
| Bioavailability score | 0.56 | 0.85 | 0.55 |
| Water solubility | Soluble | Soluble | Moderate |
| Lipophilicity (ilogP) | 1.63 | 0.95 | 2.24 |
| GIT absorption | High | High | High |
| Hydrogen bond acceptors | 5 | 3 | 5 |
| Hydrogen bond donors | 2 | 2 | 2 |
| Lipinski’s rule | Yes | Yes | Yes |
| CYP1A2 | No | No | No |
| CYP2C19 | |||
| CYP2C9 | No | No | No |
| CYP2D6 | No | No | No |
| CYP3A4 | No | No | No |
| Compounds | LD50 (mg/Kg) | Hepatotoxicity | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity |
|---|---|---|---|---|---|---|
| Sinapic acid | 1190 (class 4) | Active | Inactive | Active | Inactive | Inactive |
| p-coumaric acid | 1190 (class 4) | Active | Inactive | Active | Inactive | Inactive |
| Ciprofloxacin | 2000 (class 4) | Inactive | Inactive | Inactive | Inactive | inactive |
| Compounds | ||||||
|---|---|---|---|---|---|---|
| Bacterial Strains | Sinapic Acid | p-Coumaric Acid | Ciprofloxacin | |||
| MIC (μg/mL) | MBC (μg/mL) | MIC (μg/mL) | MBC (μg/mL) | MIC (μg/mL) | MBC (μg/mL) | |
| Staphylococcus aureus (Gram-positive) | 31.25 | 62.50 | 31.25 | 62.50 | 7.81 | 15.63 |
| Escherichia coli (Gram-negative) | 125.00 | 250.00 | 62.50 | 125.00 | 15.63 | 31.25 |
| Bacterial Strains | MIC (μg/mL) | FICI | ||
|---|---|---|---|---|
| Treatments | Sinapic Acid | Ciprofloxacin | ||
| Sinapic acid + ciprofloxacin | S. aureus | 1.9 | 0.4 | 0.3 |
| E. coli | 2.6 | 7.8 | 0.6 | |
| p-coumaric acid + ciprofloxacin | S. aureus | 7.8 | 1.9 | 0.5 |
| E. coli | 3.9 | 0.9 | 0.3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, K.; Coopoosamy, R.M.; Gumede, N.J.; Sabiu, S. Computational Insights and In Vitro Validation of Antibacterial Potential of Shikimate Pathway-Derived Phenolic Acids as NorA Efflux Pump Inhibitors. Molecules 2022, 27, 2601. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082601
Singh K, Coopoosamy RM, Gumede NJ, Sabiu S. Computational Insights and In Vitro Validation of Antibacterial Potential of Shikimate Pathway-Derived Phenolic Acids as NorA Efflux Pump Inhibitors. Molecules. 2022; 27(8):2601. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082601
Chicago/Turabian StyleSingh, Karishma, Roger M. Coopoosamy, Njabulo J. Gumede, and Saheed Sabiu. 2022. "Computational Insights and In Vitro Validation of Antibacterial Potential of Shikimate Pathway-Derived Phenolic Acids as NorA Efflux Pump Inhibitors" Molecules 27, no. 8: 2601. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082601