
Mechanochemical Dimerization of Aldoximes to Furoxans


1
Hefei National Laboratory for Physical Sciences, Microscale and Department of Chemistry, University of Science and Technology of China, Hefei 230026, China
2
State Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou 730000, China
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(8), 2604; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082604
Submission received: 13 March 2022
/
Revised: 12 April 2022
/
Accepted: 15 April 2022
/
Published: 18 April 2022
(This article belongs to the Special Issue Mechanochemical Synthesis of Organic Compounds)
Abstract
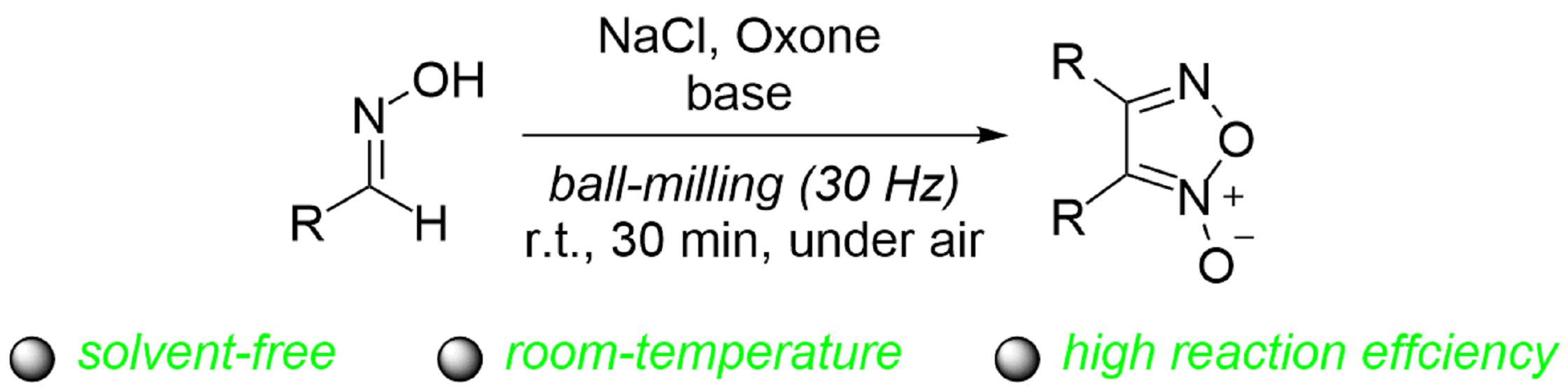
:Solvent-free mechanical milling is a new, environmentally friendly and cost-effective technology that is now widely used in the field of organic synthesis. The mechanochemical solvent-free synthesis of furoxans from aldoximes was achieved through dimerization of the in situ generated nitrile oxides in the presence of sodium chloride, Oxone and a base. A variety of furoxans was obtained with up to a 92% yield. The present protocol has the advantages of high reaction efficiency and mild reaction conditions.

1. Introduction
The furoxans (1,2,5-oxadiazole 2-oxides) are an important class of heterocyclic compounds with a long history [1]. Because of their ability to release NO [2,3], they play an important role in biochemistry, pharmaceuticals and other fields [4,5,6,7,8]. Over the past few decades, extensive work has been devoted to their synthesis. Among them, the commonly used approaches are oxidation of aldoximes, dehydration of nitrobenzenes and pyrolysis of o-nitroazidobenzenes [9,10,11,12,13,14,15,16,17,18]. In general, most of these preparation methods are very useful, but they often suffer from some drawbacks, such as the use of complicated starting materials, special oxidants, toxic organic solvents and so on. Therefore, it is necessary to develop an efficient and environmentally friendly method to synthesize furoxans from readily available starting materials.
Solvent-free reactions have drawn much attention in recent years. As one attractive type of solvent-free reactions mechanochemistry does not use organic solvents in the reaction process, or only a small amount of liquid-assisted grinding (LAG) is used, which can greatly reduce waste discharge [19,20,21,22,23,24,25,26]. In addition, mechanochemical protocols have the advantages of shortening the reaction time, reducing the reaction temperature, improving reaction selectivity, and can even provide products that are difficult or impossible to access in liquid-phase reactions [27,28,29,30,31]. Therefore, solvent-free mechanochemical reactions have been effectively used in organic synthesis [32,33,34,35,36,37,38].
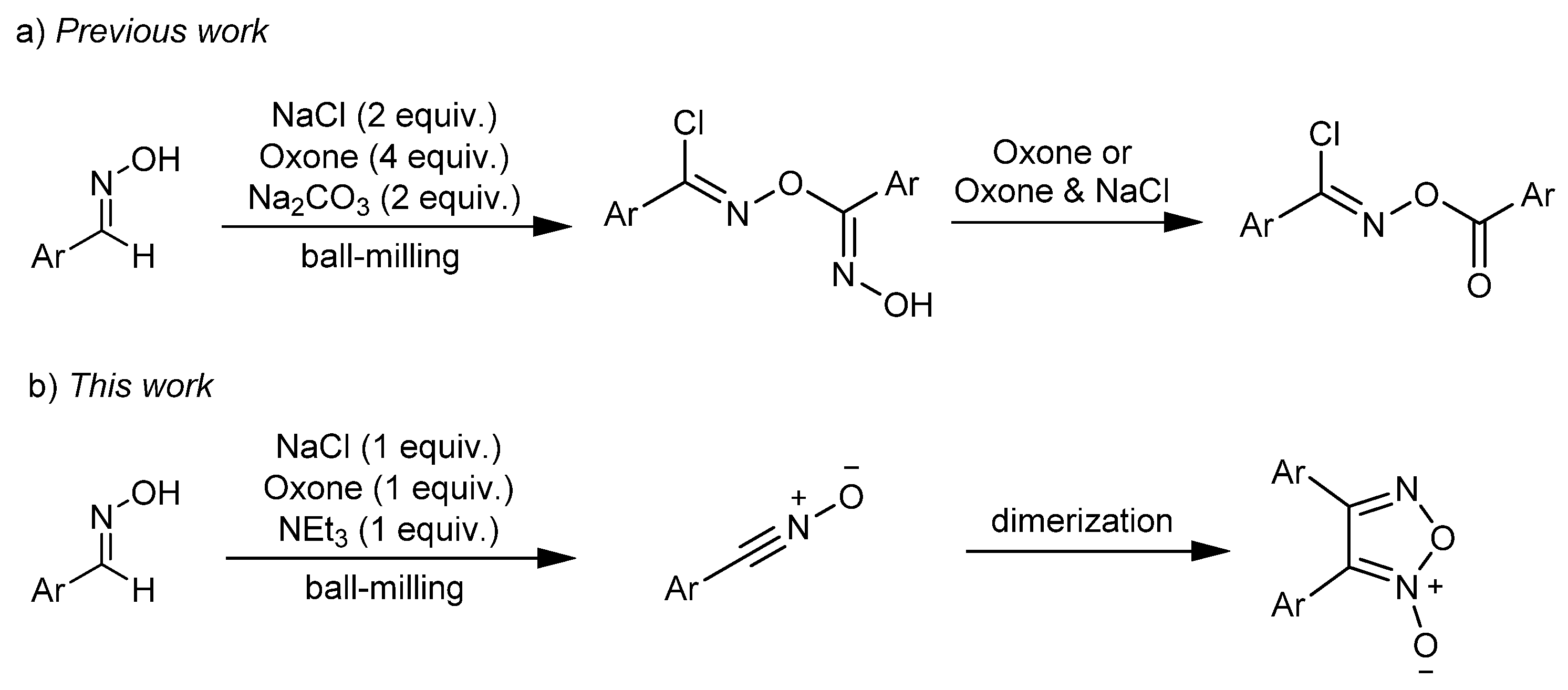
Aldoximes have drawn increasing attention in organic synthesis due to their easy availability, better selectivity and tolerance to various functional groups. Oxone (2KHSO5·KHSO4·K2SO4) is a stable and non-toxic inorganic oxidant, as demonstrated in reactions involving aldoximes [39,40,41,42]. Oxone has also been employed in mechanochemical reactions [42,43,44,45]. In 2019, the Tong group reported a protocol to generate nitrile oxides through NaCl/Oxone oxidation of aldoximes [41]. We previously reported the formation of N-acyloxyimidoyl chlorides from the mechanochemical solvent-free reaction of aldoximes with NaCl and Oxone in the presence of Na2CO3 (Scheme 1a) [42]. It was found that product distribution and product yield were sensitive to the molar ratio of the reagents as well as the employed base. Interestingly, certain amounts of furoxans could be generated from aldoximes under the modified conditions. To further study this new reaction, we decided to optimize the reaction conditions. To our satisfaction, a mild and efficient method was established for the formation of furoxans after detailed explorations (Scheme 1b).
2. Results and Discussion
Our initial investigation was started by using (E)-4-methylbenzaldehyde oxime (1a) as a representative substrate. A mixture of 1a (0.2 mmol), NaCl (1.0 equiv.), Oxone (1.0 equiv.) and Na2CO3 (1.0 equiv.) together with four stainless-steel balls (5 mm in diameter) was introduced into a stainless-steel jar (5 mL) and milled (30 Hz) in a Retsch MM400 mixer mill (Retsch GmbH, Haan, Germany) at room temperature for 30 min. After separation, an 8% yield of 2a was obtained (Table 1, entry 1). The product distribution was significantly affected by the choice of the used base. When Na2CO3 was replaced by other inorganic bases, including NaOtBu, NaOAc and NaHCO3, only a trace amount of 2a was obtained (Table 1, entries 2–4). Satisfyingly, the desired product 2a was isolated in 36% yield when K2CO3 was employed as the base (Table 1, entry 5). However, Cs2CO3 afforded 2a in only a 7% yield (Table 1, entry 6). Then, we tried to use organic bases, such as 4-dimethylaminopyridine (DMAP), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 1,4-diazabicyclo[2.2.2]octane (DABCO), which did not afford the product at all (Table 1, entries 7–9). To our delight, when triethylamine (NEt3) was employed, 2a could be obtained in a 79% yield (Table 1, entry 10). Based on this result, the influence of the amount of NEt3 on the product yield was investigated. The product yield was decreased to 69% and 53% when the amount of NEt3 was increased to 1.25 equiv. and 1.5 equiv., respectively (Table 1, entries 11 and 12), showing a detrimental effect of excess NEt3. The exact reason is unknown so far. On the other hand, the yield was reduced to 62% and 38% when the equivalent of NEt3 was less than the required stoichiometric amount (0.75 equiv. and 0.5 equiv., respectively) (Table 1, entries 13 and 14). When a mixture of 1a (0.2 mmol), NaCl (1.0 equiv.), Oxone (1.0 equiv.) and NEt3 (1.0 equiv.) was magnetically stirred at room temperature for 2 h, only a trace amount of 2a could be obtained (Table 1, entry 15). This result demonstrated the great advantage of the current reaction by ball milling over magnetic stirring. Then, the reaction time was investigated. When the reaction time was shortened from 30 min to 15 min, the desired product 2a was obtained in only a 47% yield (Table 1, entry 16). When prolonging the reaction time to 40 min, the yield had no further improvement (Table 1, entry 17). When the amounts of NaCl and Oxone were increased, the yield essentially remained the same (Table 1, entries 18 and 19). The LAG protocol has been shown to improve reaction efficiency in mechanochemical reactions [31,32,46,47,48,49,50]. Accordingly, several liquids were added to the reaction mixture as LAG agents. However, ethyl alcohol (EtOH), dichloromethane (DCM), ethyl acetate (EtOAc) and acetonitrile (CH3CN) were detrimental to the reaction, and 2a were obtained in 50–71% yields (Table 1, entries 20–23). Therefore, optimized reaction conditions were established as follows: 0.2 mmol of 1a, 1.0 equiv. of NaCl, 1.0 equiv. of Oxone and 1.0 equiv. of NEt3 at 30 Hz for 30 min (Table 1, entry 10).
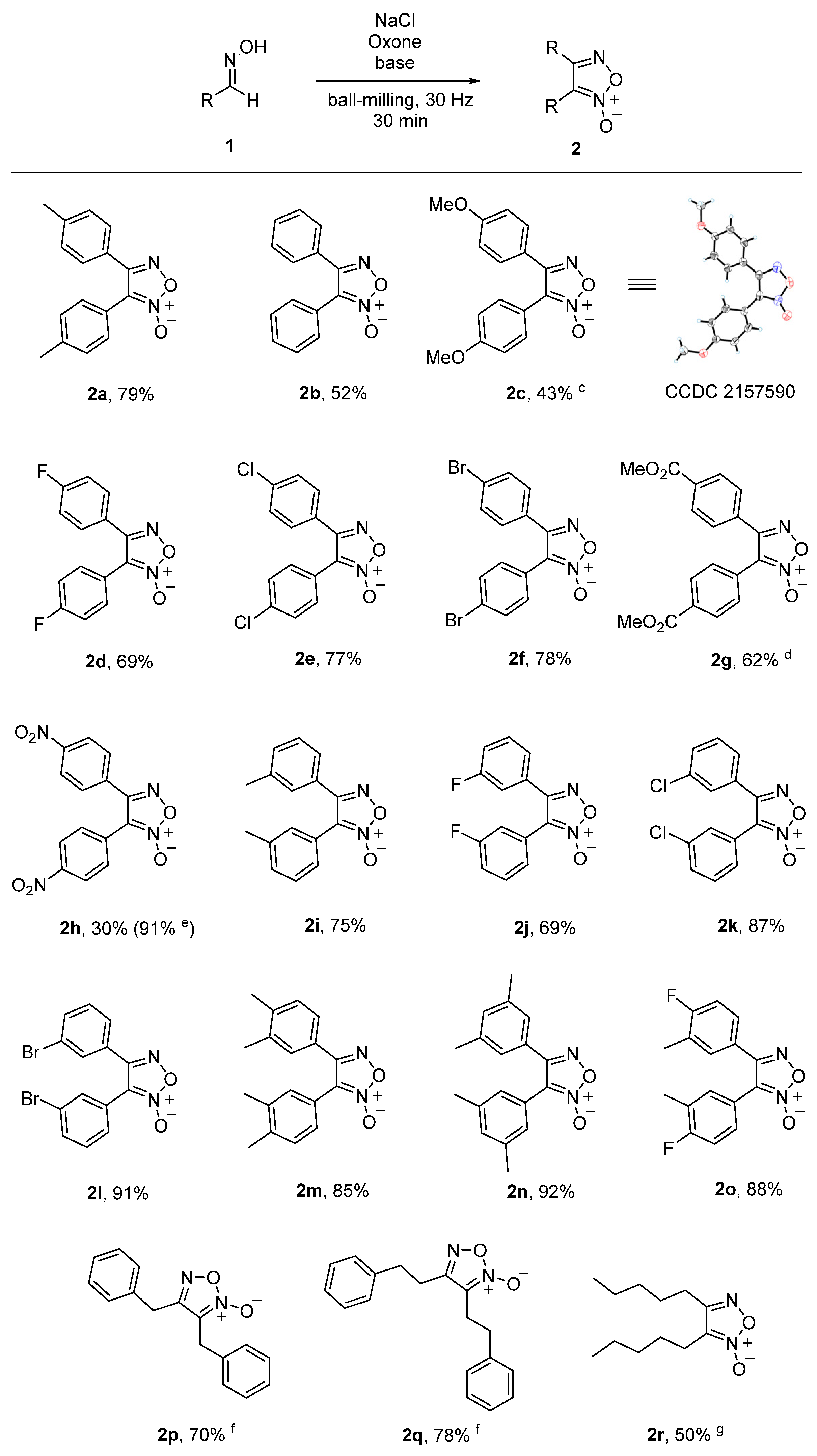
With the optimized reaction conditions in hand, the scope and generality of this reaction were then examined, and the results are shown in Scheme 2. At first, a variety of aromatic aldoximes (1a–o) were explored and found to be compatible under the optimized reaction conditions. The substrate 1b with no substituent on the phenyl ring gave the corresponding product 2b in a 52% yield. For the aldoxime 1c containing the strong electron-rich para-OMe group, very low efficiency was observed with NEt3 as a base. To our delight, product 2c could be obtained in a 43% yield when NEt3 was replaced by Na2CO3. As for the para-halogen-substituted (E)-benzaldehyde oximes 1d–f, the desired products 2d–f were synthesized in 69–78% yields. For the substrate 1g with the para-substituted CO2Me group, the corresponding product 2g was isolated in a 62% yield after prolonging the reaction time to 60 min. When the aldoxime 1h bearing the strong electron-deficient para-NO2 was investigated, the desired product 2h was obtained in only a 30% yield. However, NaOtBu could replace NEt3 to achieve a high yield of 91% for product 2h. As for the meta-substituted substrates 1i–l bearing Me, F, Cl and Br, the desired products 2i–l were isolated in 69–91% yields. The disubstituted substrates 1m–o were also compatible under the standard reaction conditions, affording products 2m–o in good yields of 85–92%. Unfortunately, it was found that heteroaromatic aldoximes, including (E)-nicotinaldehyde oxime, (E)-thiophene-2-carbaldehyde oxime and (E)-benzofuran-2-carbaldehyde oxime, were not suitable substrates for the current reaction. To further illustrate the substrate scope of this reaction, the substrates were extended from the aromatic aldoximes to aliphatic aldoximes with Na2CO3 as the base. Gratifyingly, the (E)-2-phenylacetaldehyde oxime 1p gave the corresponding product 2p in a 70% yield. When (E)-3-phenylpropanal oxime 1q was employed under the newly modified reaction conditions, the desired 2q was obtained in a 78% yield. Another aliphatic aldoxime 1r formed from hexanal was also applicable to our reaction and provided 2r in a 50% yield. The structures of products were unambiguously confirmed by single-crystal X-ray diffraction analysis with 2c as an example (see the Supplementary Materials for details).
It is noteworthy that NEt3 was used as the base for the efficient formation of furoxans in most cases. For the strong electron-deficient aldoxime 1h bearing 4-NO2Ph group, a stronger base NaOtBu could dramatically increase the product yield. In contrast, for the electron-rich substrates including aldoxime 1c containing the 4-OMePh group and aliphatic aldoximes 1p–r, a weaker base Na2CO3 was required. The exact reasons for these phenomena are not yet clear but it is likely that the different basicity of the employed three bases matches the formation of the corresponding 1,3-dipolar nitrile oxides and the subsequent dimerization.
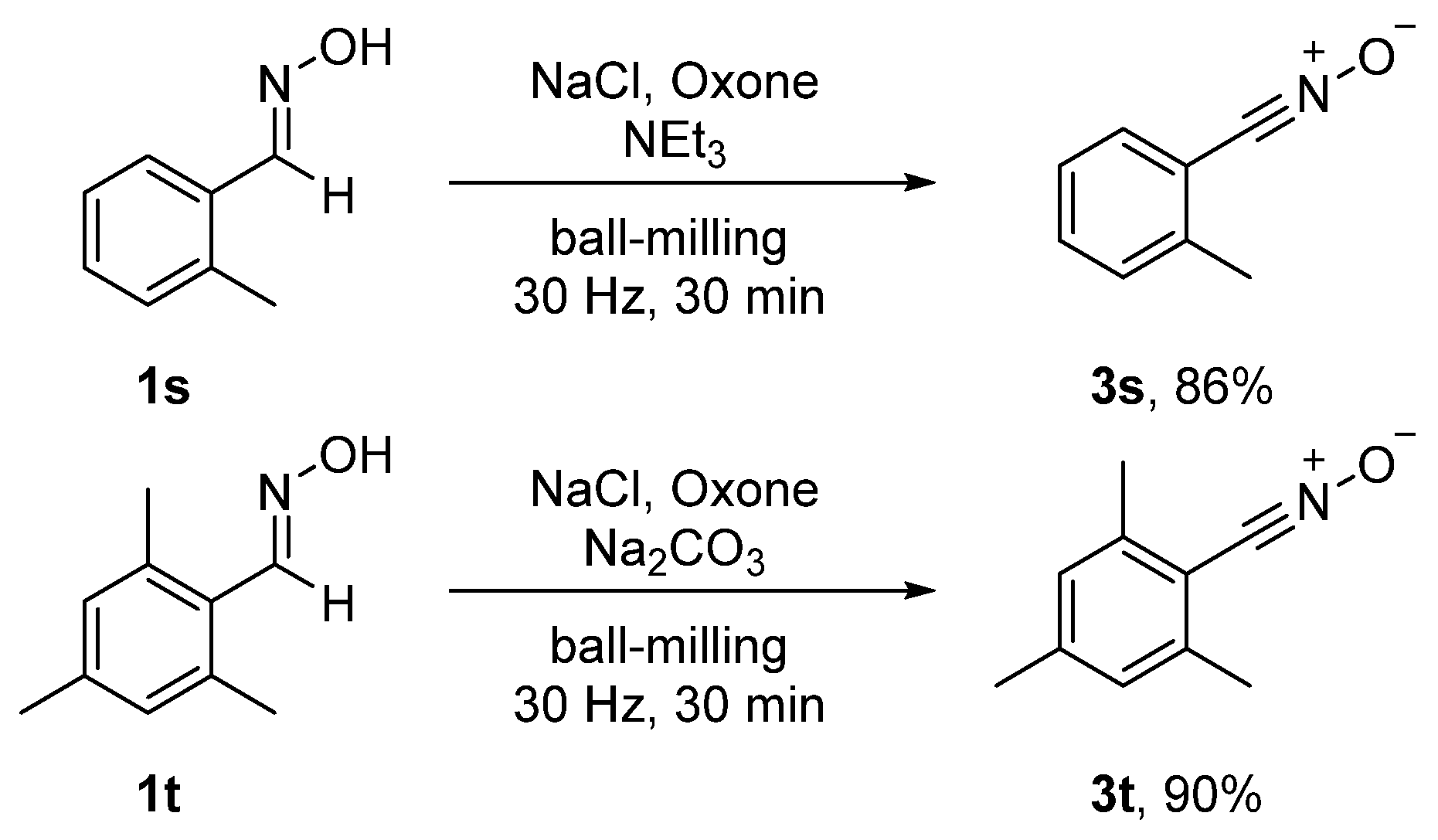
During the course of our studies, we tried to use the ortho-substituted (E)-2-methylbenzaldehyde oxime (1s) as the substrate. Intriguingly, the nitrile oxide 3s rather than its dimer was obtained in an 86% yield, probably due to the steric hindrance caused by the ortho-substituent. Similarly, the 1,3-dipole 3t was isolated in a 90% yield when (E)-2,4,6-trimethylbenzaldehyde oxime (1t) was employed (Scheme 3).
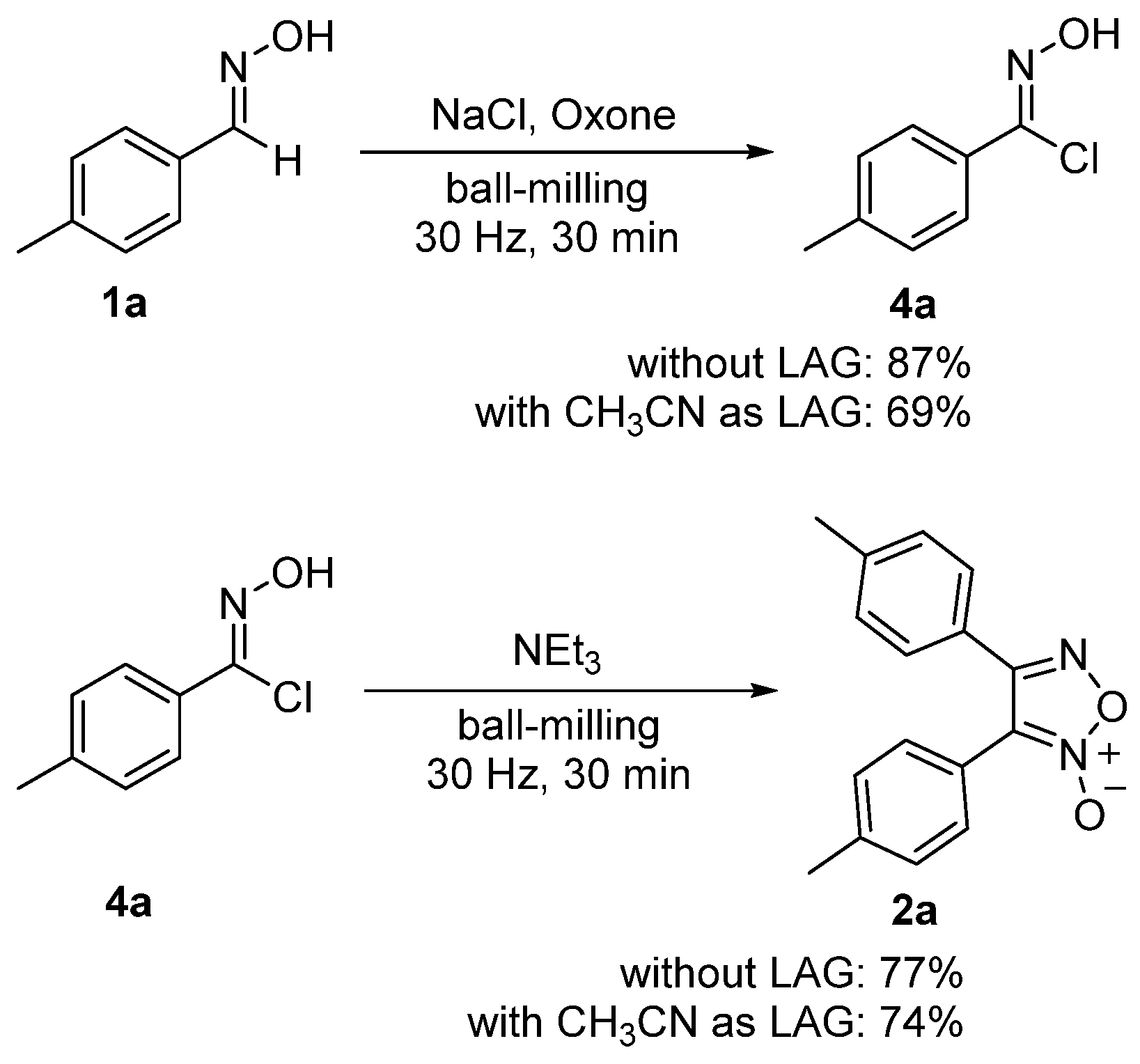
To gain insight into the reaction mechanism of this transformation, control experiments were performed (Scheme 4). The reaction of 1a (0.2 mmol), NaCl (1 equiv.) and Oxone (1 equiv.) afforded hydroximoyl chloride 4a in an 87% yield under our solvent-free ball-milling conditions. Then, 4a was allowed to react with NEt3 (1 equiv.) under the ball-milling conditions and produced 2a in a 77% yield. The effect of LAG on these two reactions was also examined. It was found that CH3CN as LAG seemed to retard the formation of 4a to a certain degree and showed nearly no effect on the subsequent dimerization process. Thus, these control experiments demonstrated that 4a should be the key intermediate for the transformation to 2a and explained why a slightly overall lower yield for the formation of 2a was observed with CH3CN as the LAG agent (71% vs. 79%, Table 1, entry 23 vs. entry 10).
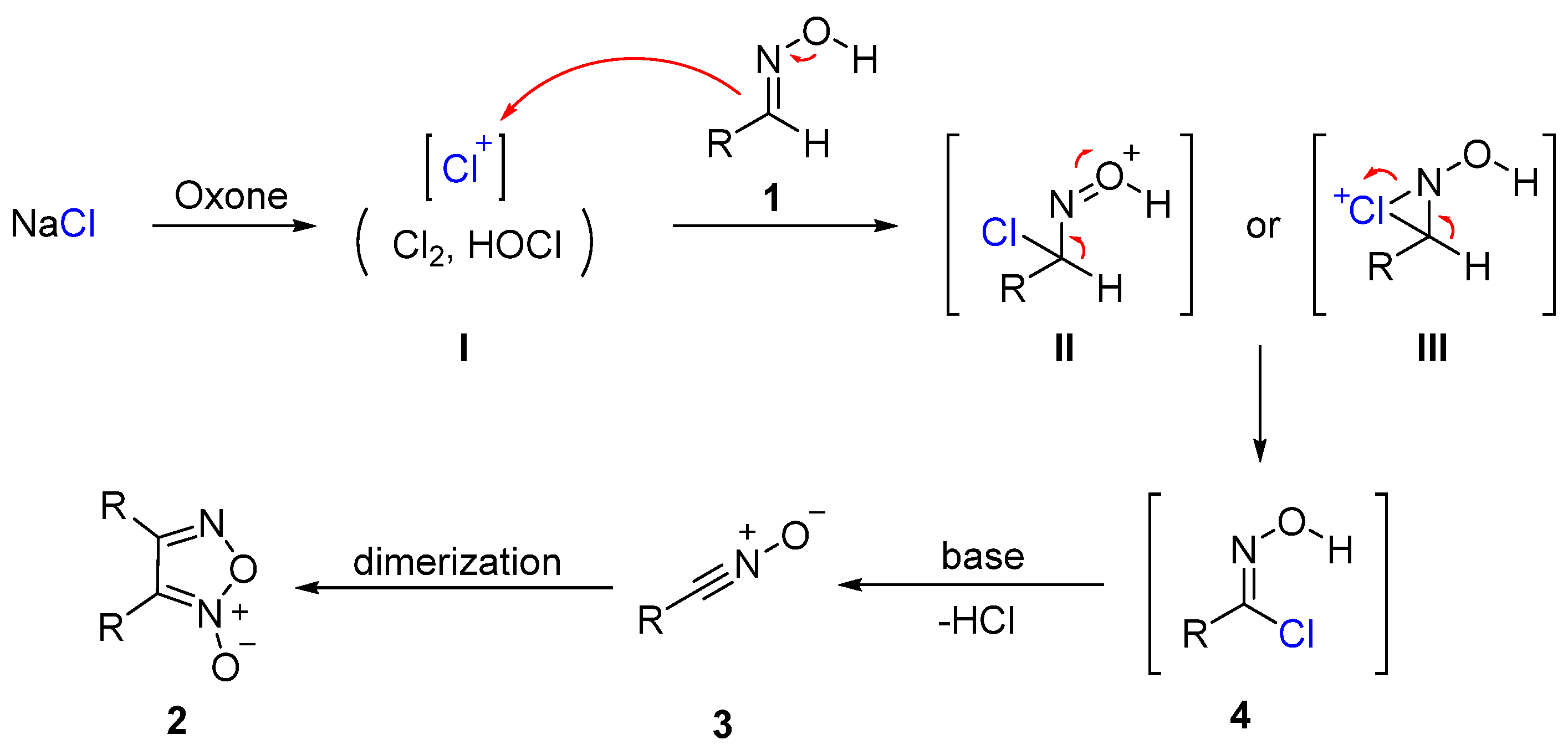
On the basis of the above experimental results and previous literature [41,42], a plausible mechanism is proposed (Scheme 5). First, NaCl is oxidized by Oxone to generate the chlorinating species I. Then, aldoxime 1 undergoes a chlorination reaction with I to provide the hydroximoyl chloride 4 via the possible intermediate II or III. Subsequently, 4 eliminates HCl with the aid of base, and the resulting nitrile oxide 3 undergoes 1,3-dipolar addition to the C≡N bond of another 3 to give the dimerization product 2.
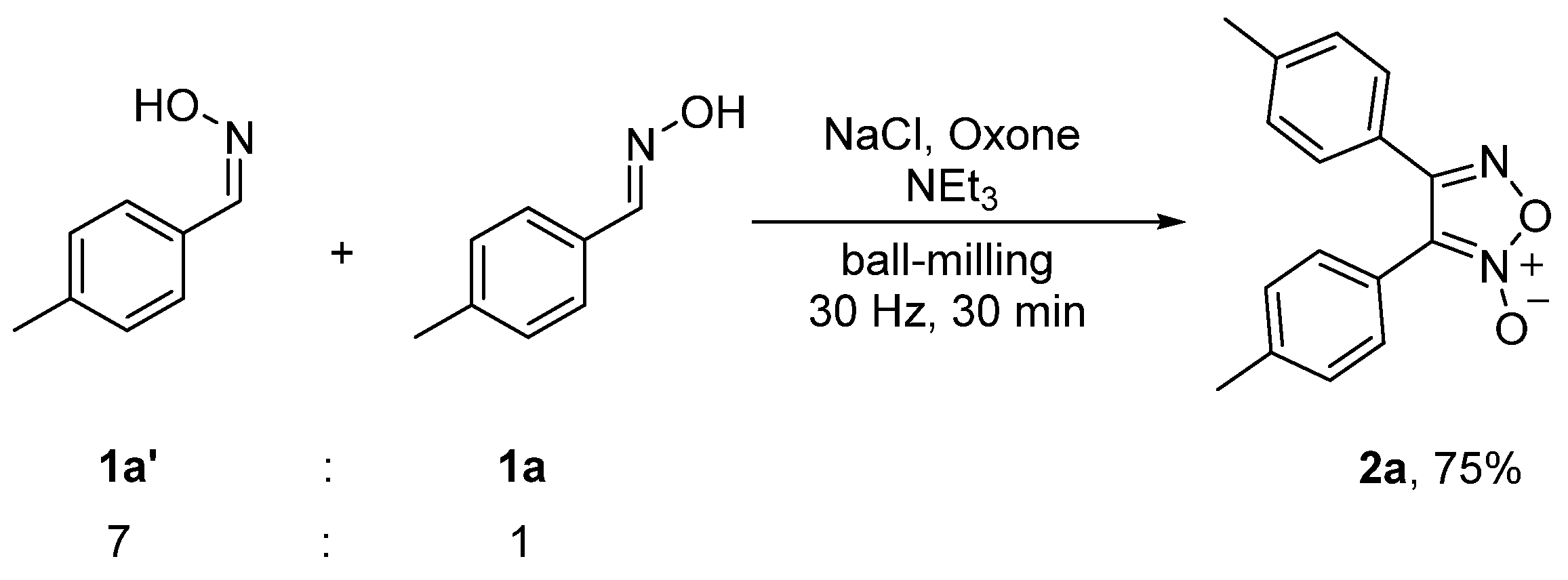
The aldoximes used in the above experiments were prepared according to the reported procedure [41] and were determined as the E-isomers [51]. The E-isomers of aldoximes could be converted into the corresponding Z-isomers under acidic conditions [51]. When a mixture of (Z)-4-methylbenzaldehyde oxime (1a’) and the E-isomer 1a in a molar ratio of 7:1 was employed to replace the single isomer 1a, 2a was isolated in a 75% yield (Scheme 6), indicating that both E- and Z-isomers of aldoximes could provide furoxans in essentially the same yields.
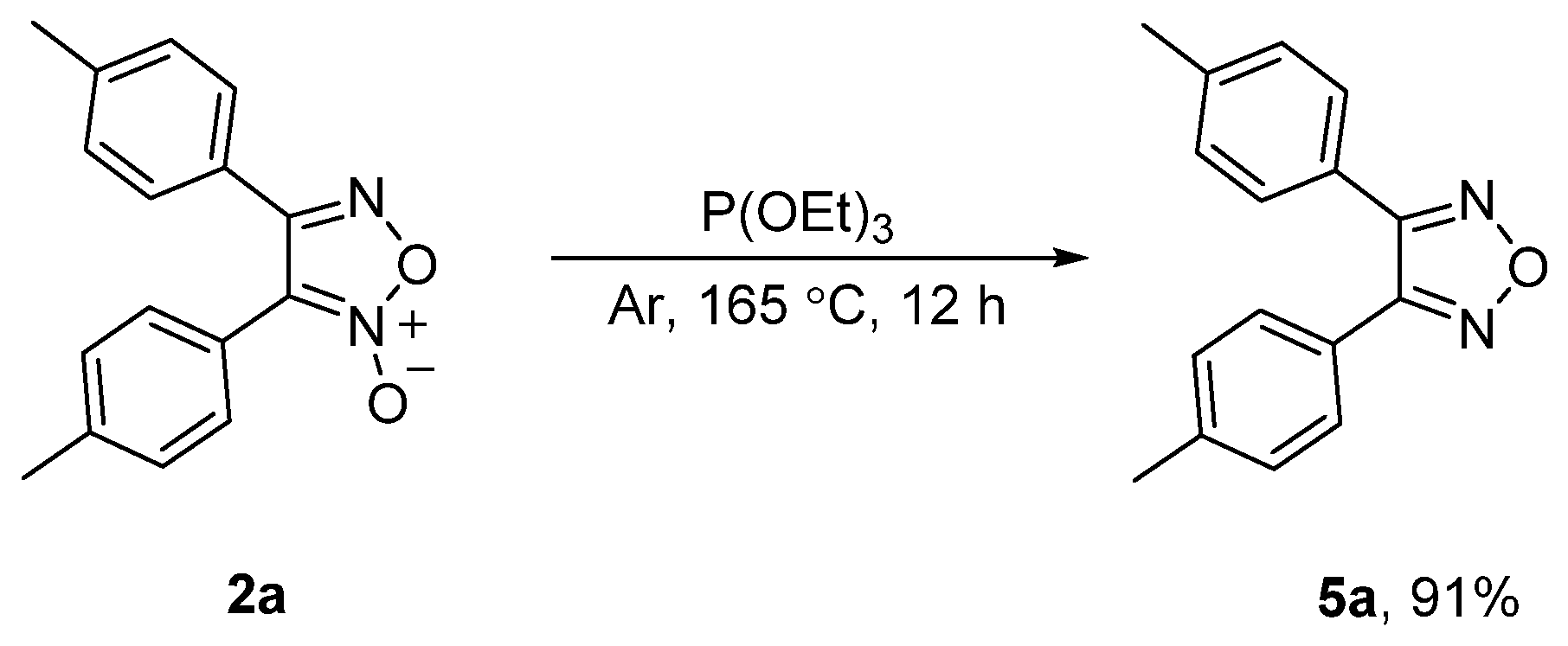
To demonstrate the utility of the obtained furoxans, 2a could be deoxygenated by triethyl phosphite (P(OEt)3) to provide 1,2,5-oxadiazole 5a in 91% yield at 165 °C for 12 h under an argon atmosphere (Scheme 7) [52].
For the purpose of comparing the present solvent-free reaction with its liquid-phase counterpart, we performed the reaction of 1a (0.2 mmol) with NaCl (1.0 equiv.), Oxone (1.0 equiv.) and NEt3 (1.0 equiv.) in several organic solvents including dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF), acetonitrile (MeCN) and 1,2-dichloroethane (DCE) at room temperature for 2 h. The results showed that MeCN was the best solvent, and the yield of product 2a was 47%. It is obvious that the present mechanochemical protocol has the higher yield (79% vs. 47%) and shorter reaction time (30 min vs. 120 min) under solvent-free conditions compared to the liquid-phase counterpart reaction. The possible reason is that the possibility of close contact of 1,3-dipoles for dimerization under the solvent-free mechanical milling conditions is much higher than that in the liquid phase.
Green chemistry metrics, such as complete and simple environmental factors (cEF and sEF), atom economy (AE) and reaction mass efficiency (RME), for the mechanosynthesis of 2a were quantified, showing advantages in greenness compared with those of its liquid-phase counterpart (see the Supplementary Materials for details).
3. Materials and Methods
3.1. General Information
All reagents were obtained from commercial sources and used without further purification. NMR spectra were recorded on a Bruker Advance III HD 400 NMR spectrometer (Bruker BioSpin AG, Fällanden, Switzerland; 400 MHz for 1H NMR; 101 MHz for 13C NMR; 376 MHz for 19F NMR) and a Bruker Advance III HD 500 NMR spectrometer (Bruker BioSpin AG, Fällanden, Switzerland; 500 MHz for 1H NMR; 126 MHz for 13C NMR; 471 MHz for 19F NMR). 1H NMR chemical shifts were determined relative to TMS at 0.00 ppm, CDCl3 at δ 7.26 ppm or DMSO-d6 at δ 2.50 ppm. 13C NMR chemical shifts were determined relative to TMS at 0.00 ppm, CDCl3 at δ 77.16 ppm or DMSO-d6 at δ 39.52 ppm. Data for 1H NMR and 13C NMR are reported as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet). High-resolution mass spectra (HRMS) were taken on a Waters Acquity UPLC-Xevo G2 QTof mass spectrometer (Waters, Milford, MA, USA) with FTMS-ESI in positive mode. Ball-milling reactions were performed in a MM400 mixer mill (Retsch GmbH, Haan, Germany), using a 5 mL stainless-steel jar with four 5 mm diameter stainless-steel balls and were milled at a frequency of 1800 rounds per minute (30 Hz) at room temperature. E-Aldoximes 1 were prepared according to the reported protocol [41]. A mixture of Z-isomer 1a’ and E-isomer 1a was obtained in molar ratio of 7:1 by stirring E-1a in trifluoroacetic acid (TFA)-CHCl3 at 0 °C for 20 min followed by removal of the volatiles in vacuo [51]. Single crystals of 2c were grown from dichloromethane/n-hexane at 4 °C.
3.2. Mechanochemical Synthesis and Characterization of Products 2a–r, 3s and 3t
A mixture of aldoximes 1a–t (0.2 mmol), NaCl (0.2 mmol), Oxone (0.2 mmol) and base (0.2 mmol) together with four stainless-steel balls (5 mm in diameter) was introduced into a stainless-steel jar (5 mL). The reaction vessel along with another identical vessel was closed and fixed on the vibration arms of a Retsch MM400 mixer mill, and was vibrated at a rate of 1800 rounds per minute (30 Hz) at room temperature for 30 min. After completion of the reaction, the resulting mixtures from the two runs were combined and extracted with dichloromethane and water. The organic layer was decanted, and the aqueous layer was extracted by dichloromethane (2 × 20 mL). The combined organic extracts were evaporated to remove the solvent in vacuo. The residue was separated by flash column chromatography on silica gel with ethyl acetate/petroleum ether as the eluent to afford products 2a–r, 3s and 3t.
3,4-Di-p-tolyl-1,2,5-oxadiazole 2-oxide (2a). By following the general procedure, the reaction of 1a (54.8 mg, 0.4 mmol) with NaCl (24.6 mg, 0.4 mmol), Oxone (246.7 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2a (42.6 mg, 79% yield). White solid; 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 8.1 Hz, 2H), 7.40 (d, J = 7.9 Hz, 2H), 7.24 (d, J = 7.9 Hz, 4H), 2.41 (s, 3H), 2.39 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 156.4, 141.4, 141.0, 129.82 (2C), 129.78 (2C), 128.7 (2C), 128.3 (2C), 124.1, 120.2, 114.5, 20.66, 20.63; HRMS (FTMS-ESI) Calcd for C16H15N2O2 [M + H]+ 267.1128; found 267.1133.
3,4-Diphenyl-1,2,5-oxadiazole 2-oxide (2b). By following the general procedure, the reaction of 1b (44 µL, 0.4 mmol) with NaCl (24.4 mg, 0.4 mmol), Oxone (247.4 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2b (24.8 mg, 52% yield). White solid; 1H NMR (500 MHz, CDCl3) δ 7.56–7.49 (m, 5H), 7.48–7.40 (m, 5H); 13C NMR (126 MHz, CDCl3) δ 156.4, 131.1, 130.7, 129.2 (2C), 129.1 (2C), 128.9 (2C), 128.5 (2C), 126.8, 123.1, 114.4. The NMR data agreed with those in a literature report [11].
3,4-Bis(4-methoxyphenyl)-1,2,5-oxadiazole 2-oxide (2c). By following the general procedure, the reaction of 1c (61.4 mg, 0.4 mmol) with NaCl (94.0 mg, 1.6 mmol), Oxone (249.6 mg, 0.4 mmol) and Na2CO3 (85.4 mg, 0.8 mmol) afforded 2c (25.8 mg, 43% yield). White solid; 1H NMR (500 MHz, CDCl3) δ 7.47 (d, J = 8.9 Hz, 2H), 7.46 (d, J = 8.9 Hz, 2H), 6.95 (d, J = 8.9 Hz, 2H), 6.94 (d, J = 8.9 Hz, 2H), 3.85 (s, 3H), 3.84 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 161.7, 161.1, 156.1, 130.4 (2C), 129.9 (2C), 119.1, 115.0, 114.6 (4C), 114.3, 55.52, 55.51. The NMR data agreed with those in a literature report [53].
3,4-Bis(4-fluorophenyl)-1,2,5-oxadiazole 2-oxide (2d). By following the general procedure, the reaction of 1d (56.6 mg, 0.4 mmol) with NaCl (25.8 mg, 0.4 mmol), Oxone (248.3 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2d (38.6 mg, 69% yield). White solid; 1H NMR (400 MHz, DMSO-d6) δ 7.62–7.52 (m, 4H), 7.42–7.35 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 163.6 (d, JC–F = 249.1 Hz), 163.0 (d, JC–F = 249.2 Hz), 155.9, 131.6 (d, JC–F = 8.9 Hz, 2C), 130.9 (d, JC–F = 9.0 Hz, 2C), 122.7 (d, JC–F = 3.3 Hz), 119.0 (d, JC–F = 3.3 Hz), 116.4 (d, JC–F = 22.2 Hz, 2C), 116.3 (d, JC–F = 22.3 Hz, 2C), 114.2; 19F NMR (376 MHz, DMSO-d6) δ –108.83 to –108.98 (m, 2F); HRMS (FTMS-ESI) Calcd for C14H8F2N2O2Na [M + Na]+ 297.0446; found 297.0454.
3,4-Bis(4-chlorophenyl)-1,2,5-oxadiazole 2-oxide (2e). By following the general procedure, the reaction of 1e (63.0 mg, 0.4 mmol) with NaCl (23.6 mg, 0.4 mmol), Oxone (248.1 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2e (47.8 mg, 77% yield). White solid; 1H NMR (400 MHz, CDCl3) δ 7.49–7.38 (m, 8H); 13C NMR (126 MHz, CDCl3) δ 155.2, 137.7, 137.1, 130.0 (2C), 129.72 (2C), 129.70 (2C), 129.6 (2C), 125.0, 121.2, 113.5. The NMR data agreed with those in a literature report [54].
3,4-Bis(4-bromophenyl)-1,2,5-oxadiazole 2-oxide (2f). By following the general procedure, the reaction of 1f (81.0 mg, 0.4 mmol) with NaCl (24.6 mg, 0.4 mmol), Oxone (247.6 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2f (62.5 mg, 78% yield). White solid; 1H NMR (500 MHz, CDCl3) δ 7.58 (d, J = 8.6 Hz, 4H), 7.38 (d, J = 8.6 Hz, 4H); 13C NMR (126 MHz, CDCl3) δ 155.2, 137.8, 137.1, 130.0 (2C), 129.73 (2C), 129.71 (2C), 129.66 (2C), 125.0, 121.2, 113.5. The NMR data agreed with those in a literature report [55].
3,4-Bis(4-(methoxycarbonyl)phenyl)-1,2,5-oxadiazole 2-oxide (2g). By following the general procedure and prolonging the reaction time to 60 min, the reaction of 1g (72.8 mg, 0.4 mmol) with NaCl (25.2 mg, 0.4 mmol), Oxone (247.5 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2g (44.6 mg, 62% yield). White solid; 1H NMR (500 MHz, CDCl3) δ 8.13 (d, J = 8.4 Hz, 2H), 8.11 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 8.3 Hz, 4H), 3.96 (s, 3H), 3.95 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.1, 166.0, 155.4, 132.8, 132.2, 130.6, 130.5 (2C), 130.3 (2C), 128.7 (2C), 128.5 (2C), 127.0, 113.6, 52.69, 52.67; HRMS (FTMS-ESI) Calcd for C18H15N2O6 [M + H]+ 355.0925; found 355.0930.
3,4-Bis(4-nitrophenyl)-1,2,5-oxadiazole 2-oxide (2h). By following the general procedure, the reaction of 1h (67.8 mg, 0.4 mmol) with NaCl (26.2 mg, 0.4 mmol), Oxone (248.0 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2h (20.1 mg, 30% yield). When NEt3 is replaced by NaOtBu, a much better result was obtained. Thus, the reaction of 1h (68.0 mg, 0.4 mmol) with NaCl (47.6 mg, 0.8 mmol), Oxone (739.6 mg, 1.2 mmol) and NaOtBu (39.0 mg, 0.4 mmol) afforded 2h (61.2 mg, 91% yield). White solid; 1H NMR (400 MHz, CD2Cl2) δ 8.34 (d, J = 8.7 Hz, 2H), 8.32 (d, J = 8.7 Hz, 2H), 7.73 (d, J = 8.7 Hz, 2H), 7.72 (d, J = 8.7 Hz, 2H); 13C NMR (101 MHz, CD2Cl2) δ 153.7, 148.8, 148.1, 131.2, 128.9 (2C), 128.8 (2C), 127.8, 123.8 (2C), 123.6 (2C), 112.0. The NMR data agreed with those in a literature report [56].
3,4-Di-m-tolyl-1,2,5-oxadiazole 2-oxide (2i). By following the general procedure, the reaction of 1i (56.4 mg, 0.4 mmol) with NaCl (25.7 mg, 0.4 mmol), Oxone (247.2 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2i (41.5 mg, 75% yield). White solid; 1H NMR (500 MHz, DMSO-d6) δ 7.42–7.32 (m, 6H), 7.26–7.19 (m, 2H), 2.31 (s, 3H), 2.30 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 156.7, 138.5, 138.3, 131.7, 131.4, 129.1, 128.9, 128.8, 128.5, 126.2, 126.1, 125.4, 122.6, 114.6, 20.90, 20.86; HRMS (FTMS-ESI) Calcd for C16H15N2O2 [M + H]+ 267.1128; found 267.1138.
3,4-Bis(3-fluorophenyl)-1,2,5-oxadiazole 2-oxide (2j). By following the general procedure, the reaction of 1j (56.5 mg, 0.4 mmol) with NaCl (24.5 mg, 0.4 mmol), Oxone (248.0 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2j (38.5 mg, 69% yield). White solid; 1H NMR (500 MHz, DMSO-d6) δ 7.62–7.55 (m, 2H), 7.50–7.36 (m, 4H), 7.36–7.29 (m, 2H); 13C NMR (126 MHz, DMSO-d6) δ 161.9 (d, JC–F = 245.5 Hz), 161.8 (d, JC–F = 244.9 Hz), 155.6 (d, JC–F = 2.8 Hz), 131.5 (d, JC–F = 8.6 Hz), 131.3 (d, JC–F = 8.6 Hz), 128.2 (d, JC–F = 8.7 Hz), 125.4 (d, JC–F = 3.1 Hz), 124.7 (d, JC–F = 3.2 Hz), 124.6 (d, JC–F = 9.1 Hz), 118.2 (d, JC–F = 21.0 Hz), 117.9 (d, JC–F = 21.1 Hz), 115.8 (d, JC–F = 24.2 Hz), 115.3 (d, JC–F = 23.8 Hz), 114.0 (d, JC–F = 2.7 Hz); 19F NMR (471 MHz, DMSO-d6) δ –111.42 to –111.50 (m, 2F); HRMS (FTMS-ESI) Calcd for C14H8F2N2O2Na [M + Na]+ 297.0446; found 297.0456.
3,4-Bis(3-chlorophenyl)-1,2,5-oxadiazole 2-oxide (2k). By following the general procedure, the reaction of 1k (63.4 mg, 0.4 mmol) with NaCl (24.0 mg, 0.4 mmol), Oxone (249.4 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2k (54.6 mg, 87% yield). White solid; 1H NMR (500 MHz, DMSO-d6) δ 7.68 (ddd, J = 8.1, 2.3, 1.1 Hz, 1H), 7.66–7.61 (m, 2H), 7.59 (t, J = 1.8 Hz, 1H), 7.59–7.53 (m, 2H), 7.45 (dd, J = 7.7, 1.4 Hz, 1H), 7.43 (dd, J = 7.9, 1.4 Hz, 1H); 13C NMR (126 MHz, DMSO-d6) δ 155.5, 133.7, 133.5, 131.16, 131.15, 130.9, 130.8, 128.6, 128.1, 128.0, 127.8, 127.2, 124.6, 113.9; HRMS (FTMS-ESI) Calcd for C14H935Cl2N2O2 [M + H]+ 307.0036; found 307.0031.
3,4-Bis(3-bromophenyl)-1,2,5-oxadiazole 2-oxide (2l). By following the general procedure, the reaction of 1l (81.1 mg, 0.4 mmol) with NaCl (25.5 mg, 0.4 mmol), Oxone (248.1 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2l (72.8 mg, 91% yield). White solid; 1H NMR (400 MHz, DMSO-d6) δ 7.84–7.79 (m, 1H), 7.79–7.74 (m, 2H), 7.74–7.70 (m, 1H), 7.52–7.46 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 155.4, 134.0, 133.6, 131.4, 131.3, 131.1, 130.9, 128.3, 128.1, 127.5, 124.8, 122.0, 121.8, 113.8; HRMS (FTMS-ESI) Calcd for C14H979Br2N2O2 [M + H]+ 394.9025; found 394.9025.
3,4-Bis(3,4-dimethylphenyl)-1,2,5-oxadiazole 2-oxide (2m). By following the general procedure, the reaction of 1m (61.2 mg, 0.4 mmol) with NaCl (24.6 mg, 0.4 mmol), Oxone (247.9 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2m (51.6 mg, 85% yield). White solid; 1H NMR (400 MHz, DMSO-d6) δ 7.38 (d, J = 1.9 Hz, 1H), 7.37 (d, J = 1.9 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 7.24 (d, J = 7.9 Hz, 1H), 7.14 (dd, J = 7.9, 1.9 Hz, 1H), 7.12 (dd, J = 8.0, 1.9 Hz, 1H), 2.272 (s, 3H), 2.267 (s, 3H), 2.23 (s, 3H), 2.22 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 156.6, 139.8, 139.4, 137.3, 137.1, 129.94, 129.87, 129.4, 128.8, 126.4, 125.7, 123.8, 120.0, 114.5, 19.40, 19.36 (2C), 19.3; HRMS (FTMS-ESI) Calcd for C18H18N2O2Na [M + Na]+ 317.1260; found 317.1257.
3,4-Bis(3,5-dimethylphenyl)-1,2,5-oxadiazole 2-oxide (2n). By following the general procedure, the reaction of 1n (60.6 mg, 0.4 mmol) with NaCl (24.4 mg, 0.4 mmol), Oxone (247.8 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2n (55.1 mg, 92% yield). White solid; 1H NMR (400 MHz, DMSO-d6) δ 7.21 (s, 1H), 7.17 (s, 1H), 7.13–7.09 (m, 4H), 2.25 (s, 6H), 2.24 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 156.6, 138.3 (2C), 138.1 (2C), 132.5, 132.1, 126.4 (2C), 126.2, 125.8 (2C), 122.5, 114.4, 20.8 (2C), 20.7 (2C); HRMS (FTMS-ESI) Calcd for C18H19N2O2 [M + H]+ 295.1441; found 295.1427.
3,4-Bis(4-fluoro-3-methylphenyl)-1,2,5-oxadiazole 2-oxide (2o). By following the general procedure, the reaction of 1o (62.8 mg, 0.4 mmol) with NaCl (24.8 mg, 0.4 mmol), Oxone (247.4 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2o (54.8 mg, 88% yield). White solid; 1H NMR (500 MHz, DMSO-d6) δ 7.57–7.49 (m, 2H), 7.33–7.26 (m, 4H), 2.25 (d, J = 2.0 Hz, 3H), 2.24 (d, J = 2.0 Hz, 3H); 13C NMR (126 MHz, DMSO-d6) δ 162.1 (d, JC–F = 248.5 Hz), 161.6 (d, JC–F = 248.6 Hz), 155.9, 132.3 (d, JC–F = 5.7 Hz), 131.6 (d, JC–F = 5.9 Hz), 129.1 (d, JC–F = 9.0 Hz), 128.2 (d, JC–F = 8.9 Hz), 125.6 (d, JC–F = 20.5 Hz), 125.5 (d, JC–F = 20.3 Hz), 122.4 (d, JC–F = 3.6 Hz), 118.7 (d, JC–F = 3.4 Hz) 115.9 (d, JC–F = 23.0 Hz, 2C), 114.1, 14.16 (d, JC–F = 3.2 Hz), 14.08 (d, JC–F = 3.1 Hz); 19F NMR (471 MHz, DMSO-d6) δ –113.08 to –113.26 (m, 2F); HRMS (FTMS-ESI) Calcd for C16H12F2N2O2Na [M + Na]+ 325.0759; found 325.0752.
3,4-Dibenzyl-1,2,5-oxadiazole 2-oxide (2p). By following the general procedure, the reaction of 1p (55.6 mg, 0.4 mmol) with NaCl (48.0 mg, 0.8 mmol), Oxone (251.8 mg, 0.4 mmol) and Na2CO3 (85.0 mg, 0.8 mmol) afforded 2p (38.4 mg, 70% yield). White solid; 1H NMR (500 MHz, CDCl3) δ 7.33–7.26 (m, 3H), 7.25–7.21 (m, 3H), 7.13–7.08 (m, 2H), 7.00–6.95 (m, 2H), 3.86 (s, 2H), 3.61 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 156.8, 134.0, 133.8, 129.1 (2C), 129.0 (2C), 128.8 (2C), 128.4 (2C), 127.72, 127.70, 115.4, 32.1, 28.4; HRMS (FTMS-ESI) Calcd for C16H15N2O2 [M + Na]+ 267.1128; found 267.1125.
3,4-Diphenethyl-1,2,5-oxadiazole 2-oxide (2q). By following the general procedure, the reaction of 1q (61.0 mg, 0.4 mmol) with NaCl (47.6 mg, 0.8 mmol), Oxone (246.2 mg, 0.4 mmol) and Na2CO3 (85.8 mg, 0.8 mmol) afforded 2q (47.2 mg, 78% yield). White solid; 1H NMR (500 MHz, CD2Cl2) δ 7.28 (t, J = 7.2 Hz, 4H), 7.22 (t, J = 7.0 Hz, 2H), 7.10 (d, J = 7.1 Hz, 4H), 2.85 (t, J = 7.3 Hz, 2H), 2.82 (t, J = 7.9 Hz, 2H), 2.64 (t, J = 7.2 Hz, 2H), 2.49 (t, J = 7.9 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 157.7, 139.7, 139.4, 129.0 (2C), 128.8 (2C), 128.5 (2C), 128.4 (2C), 127.0, 126.8, 115.5, 32.7, 30.8, 27.2, 24.7; HRMS (FTMS-ESI) Calcd for C18H19N2O2 [M + H]+ 295.1441; found 295.1444.
3,4-Dipentyl-1,2,5-oxadiazole 2-oxide (2r). By following the general procedure, the reaction of 1r (48.4 mg, 0.4 mmol) with NaCl (47.6 mg, 0.8 mmol), Oxone (252.0 mg, 0.4 mmol) and Na2CO3 (127.2 mg, 1.2 mmol) afforded 2r (23.6 mg, 50% yield). Colourless liquid; 1H NMR (500 MHz, CDCl3) δ 2.62 (t, J = 7.7 Hz, 1H), 2.50 (t, J = 7.7 Hz, 1H), 1.79–1.69 (m, 2H), 1.67–1.57 (m, 3H), 1.41–1.27 (m, 9H), 0.95–0.86 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 158.1, 116.2, 31.4, 31.3, 26.5, 25.8, 25.2, 22.5, 22.38, 22.35, 14.00, 13.98; HRMS (FTMS-ESI) Calcd for C12H22N2O2Na [M + Na]+ 249.1573; found 249.1570.
2-Methylbenzonitrile nitrile oxide (3s). By following the general procedure, the reaction of 1s (55.4 mg, 0.4 mmol) with NaCl (24.4 mg, 0.4 mmol), Oxone (249.6 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 3s (46.9 mg, 86% yield). White solid; 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 7.8 Hz, 1H), 7.38 (t, J = 7.6 Hz, 1H), 7.28 (d, J = 7.2 Hz, 1H), 7.23 (t, J = 7.6 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 141.7, 132.6, 131.0, 130.4, 126.5, 113.9, 20.8; HRMS (FTMS-ESI) Calcd for C8H7NONa [M + Na]+ 156.0420; found 156.0425.
2,4,6-Trimethylbenzonitrile nitrile oxide (3t). By following the general procedure, the reaction of 1t (66.4 mg, 0.4 mmol) with NaCl (24.4 mg, 0.4 mmol), Oxone (369.2 mg, 0.6 mmol) and Na2CO3 (86.6 mg, 0.8 mmol) afforded 3t (59.0 mg, 90% yield). White solid; 1H NMR (400 MHz, DMSO-d6) δ 7.03 (s, 2H), 2.36 (s, 6H), 2.27 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 141.4, 140.9, 128.2, 110.3, 21.0, 20.2. The NMR data agreed with those in a literature report [41].
3.3. Mechanochemial Synthesis of 2a from 1a and 1a’
A mixture of Z-isomer 1a’ and E-isomer 1a (7:1) was used to replace 1a. By following the same procedure for the mechanochemical synthesis of 2a from 1a, the reaction of isomeric 1a’ and 1a (7:1) (54.6 mg, 0.4 mmol) with NaCl (24.2 mg, 0.4 mmol), Oxone (247.2 mg, 0.4 mmol) and NEt3 (56 μL, 0.4 mmol) afforded 2a (40.4 mg, 75% yield).
3.4. Mechanochemial Synthesis of 4a from 1a and 2a from 4a
A mixture of 1a (27.3 mg, 0.2 mmol), Oxone (124.4 mg, 0.2 mmol) and NaCl (12.3 mg, 0.2 mmol) together with four stainless-steel balls (5 mm in diameter) was introduced into a stainless-steel jar (5 mL). The reaction vessel and another same vessel were closed and fixed on the vibration arms of a Retsch MM400 mixer mill and were vibrated at a rate of 1800 rounds per minute (30 Hz) at room temperature for 30 min. After completion of the reaction, the resulting mixtures from the two runs were combined and extracted with dichloromethane and water. The organic layer was decanted, and the aqueous layer was extracted by dichloromethane (2 × 20 mL). The combined organic extracts were evaporated to remove the solvent in vacuo. The residue was separated by flash column chromatography on silica gel with ethyl acetate/petroleum ether as the eluent to afford 4a (59.8 mg, 87% yield). CH3CN, the best LAG in the overall dimerization reaction, was also examined for the synthesis of 4a. By following the above procedure, the reaction of 1a (56.8 mg, 0.4 mmol) with NaCl (23.8 mg, 0.4 mmol) and Oxone (245.8 mg, 0.4 mmol) in the presence of CH3CN (44 µL) afforded 4a (49.0 mg, 69% yield).
A mixture of 4a (34.2 mg, 0.2 mmol) and NEt3 (28 μL, 0.2 mmol) together with four stainless-steel balls (5 mm in diameter) was introduced into a stainless-steel jar (5 mL). The reaction vessel and another same vessel were closed and fixed on the vibration arms of a Retsch MM400 mixer mill and were vibrated at a rate of 1800 rounds per minute (30 Hz) at room temperature for 30 min. After completion of the reaction, the reaction vessels were washed with ethyl acetate three times (3 × 5 mL) and the combined solution was evaporated to remove the solvent in vacuo. The residue was separated by flash column chromatography on silica gel with ethyl acetate/petroleum ether as the eluent to afford 2a (41.2 mg, 77% yield). CH3CN, the best LAG in the overall dimerization reaction, was also examined for the synthesis of 2a from 4a. By following the above procedure, the reaction of 4a (68.2 mg, 0.4 mmol) with NEt3 (56 μL, 0.4 mmol) in the presence of CH3CN (12 µL) afforded 2a (39.6 mg, 74% yield).
3.5. Deoxygenation Reaction of 2a
A mixture of 2a (27.0 mg, 0.1 mmol) and triethyl phosphite (0.5 mL) was heated at 165 °C under an argon atmosphere for 12 h. After completion of the reaction, the reaction vessels were washed with ethyl acetate three times (3 × 5 mL), and the combined solution was evaporated to remove the solvent in vacuo. The residue was separated by flash column chromatography on silica gel with ethyl acetate/petroleum ether as the eluent to afford 5a (23.1 mg, 91% yield). 1H NMR (500 MHz, CDCl3) δ 7.43 (d, J = 8.0 Hz, 4H), 7.23 (d, J = 8.0 Hz, 4H), 2.41 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 153.2 (2C), 140.8 (2C), 129.7 (4C), 128.9 (4C), 123.1 (2C), 21.6 (2C); HRMS (FTMS-ESI) Calcd for C16H15N2O [M + H]+ 251.1179; found 251.1175.
3.6. Synthesis of 2a in Liquid Phase
To a stirred solution of 1a (27.5 mg, 0.2 mmol) in CH3CN (2 mL) were added NaCl (12.1 mg, 0.2 mmol), Oxone (123.4 mg, 0.2 mmol) and NEt3 (28 μL, 0.2 mmol). The reaction mixture was allowed to stir at room temperature for 2 h. Then, the reaction mixture was filtered through a silica gel plug with ethyl acetate as the eluent and, subsequently, the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel with petroleum ether/ethyl acetate as eluent to give product 2a (12.7 mg, 47% yield).
4. Conclusions
In conclusion, we have successfully developed a solvent-free dimerization reaction of aldoximes to obtain furoxans in the presence of sodium chloride, Oxone and a base under solvent-free ball-milling conditions. The starting materials are easily available, and various aromatic and aliphatic aldoximes can be employed as the substrates. This protocol has advantages of ambient reaction conditions, high yields, solvent-free and catalyst-free conditions. Finally, a plausible reaction mechanism is proposed to explain the formation of furoxans.
Supplementary Materials
Author Contributions
G.-W.W. supervised the project, analyzed data, discussed with R.-K.F. and wrote the manuscript; R.-K.F. and K.C. did experiments and provided a draft; C.N. characterized the X-ray structure of 2c. All authors contributed to the revision. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China, grant number 21372211.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Kaufman, J.V.R.; Picard, J.P. The furoxans. Chem. Rev. 1959, 59, 429–461. [Google Scholar] [CrossRef]
- Feelisch, M.; Schönafinger, K.; Noack, E. Thiol-mediated generation of nitric oxide accounts for the vasodilator action of furoxans. Biochem. Pharmacol. 1992, 44, 1149–1157. [Google Scholar] [CrossRef] [Green Version]
- Medana, C.; Ermondi, G.; Fruttero, R.; Stilo, A.D.; Ferretti, C.; Gasco, A. Furoxans as nitric oxide donors. 4-Phenyl-3-furoxancarbonitrile: Thiol-mediated nitric oxide release and biological evaluation. J. Med. Chem. 1994, 37, 4412–4416. [Google Scholar] [CrossRef] [PubMed]
- Takayama, H.; Shirakawa, S.; Kitajima, M.; Aimi, N.; Yamaguchi, K.; Hanasaki, Y.; Ide, T.; Katsuura, K.; Fujiwara, M.; Ijichi, K.; et al. Utilization of wieland furoxan synthesis for preparation of 4-aryl-1,2,5-oxadiazole-3-yl carbamate derivatives having potent anti-HIV activity. Bioorg. Med. Chem. Lett. 1996, 6, 1993–1996. [Google Scholar] [CrossRef]
- Lolli, M.L.; Cena, C.; Medana, C.; Lazzarato, L.; Morini, G.; Coruzzi, G.; Manarini, S.; Fruttero, R.; Gasco, A. A new class of ibuprofen derivatives with reduced gastrotoxicity. J. Med. Chem. 2001, 44, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- Boiani, M.; Cerecetto, H.; González, M.; Risso, M.; Olea-Azar, C.; Piro, O.E.; Castellano, E.E.; De Ceráin, A.L.; Ezpeleta, O.; Monge-Vega, A. 1,2,5-Oxadiazole N-oxide derivatives as potential anti-cancer agents: Synthesis and biological evaluation. Part IV. Eur. J. Med. Chem. 2001, 36, 771–782. [Google Scholar] [CrossRef]
- Santos, J.L.D.; Lanaro, C.; Chelucci, R.C.; Gambero, S.; Bosquesi, P.L.; Reis, J.S.; Lima, L.M.; Cerecetto, H.; González, M.; Costa, F.F.; et al. Design, synthesis, and pharmacological evaluation of novel hybrid compounds to treat sickle cell disease symptoms. Part II: Furoxan derivatives. J. Med. Chem. 2012, 55, 7583–7592. [Google Scholar] [CrossRef]
- Jovené, C.; Chugunova, E.A.; Goumont, R. The properties and the use of substituted benzofuroxans in pharmaceutical and medicinal chemistry: A comprehensive review. Mini Rev. Med. Chem. 2013, 13, 1089–1136. [Google Scholar] [CrossRef]
- Godovikova, T.I.; Golova, S.P.; Strelenko, Y.A.; Antipin, M.Y.; Struchkov, Y.T.; Khmel’nitskii, L.I. Synthesis and properties of unsubstituted furoxan. Mendeleev Commun. 1994, 4, 7–9. [Google Scholar] [CrossRef]
- Curini, M.; Epifano, F.; Marcotullio, M.C.; Rosati, O.; Ballini, R.; Bosica, G. Alumina promoted cyclization of α-nitro-oximes: A new entry to the synthesis of 1,2,5-oxadiazoles N-oxides(furoxans). Tetrahedron Lett. 2000, 41, 8817–8820. [Google Scholar] [CrossRef]
- Das, O.; Paria, S.; Paine, T.K. Copper(II)-mediated oxidation of 1,2-dioxime to furoxan. Tetrahedron Lett. 2008, 49, 5924–5927. [Google Scholar] [CrossRef]
- Matsubara, R.; Ando, A.; Hasebe, H.; Kim, H.; Tsuneda, T.; Hayashi, M. Synthesis and synthetic application of chloro- and bromofuroxans. J. Org. Chem. 2020, 85, 5959–5972. [Google Scholar] [CrossRef] [PubMed]
- Dyall, L.K. Oxidative cyclizations. VII. Cyclization of 2-substituted anilines with alkaline hypohalite. Aust. J. Chem. 1984, 37, 2013–2026. [Google Scholar] [CrossRef]
- Dyall, L.K.; Harvey, J.J.; Jarman, T.B. Oxidative cyclizations. VIII. Mechanisms of oxidation of ortho-substituted benzenamines and improved cyclizations by bis(acetato-O)phenyliodine. Aust. J. Chem. 1992, 45, 371–384. [Google Scholar] [CrossRef]
- Sun, T.; Hao, A.; Shen, J.; Song, L. Simple and practical procedure for the preparation of benzofurazan-N-oxides in the presence of cyclodextrin in neutral condition. Synth. Commun. 2009, 39, 4309–4314. [Google Scholar] [CrossRef]
- Gaughran, R.J.; Picard, J.P.; Kaufman, J.V.R. Contribution to the chemistry of benzfuroxan and benzfurazan derivatives. J. Am. Chem. Soc. 1954, 76, 2233–2236. [Google Scholar] [CrossRef]
- Sheremetev, A.B.; Aleksandrova, N.S.; Ignat’ev, N.V.; Schulte, M. Straightforward one-pot synthesis of benzofuroxans from o-halonitrobenzenes in ionic liquids. Mendeleev Commun. 2012, 22, 95–97. [Google Scholar] [CrossRef]
- Leyva, E.; Leyva-Ramos, S.; Jiménez-Cataño, R.; De Luna-Méndez, T.A.; Cárdenas-Chaparro, A. One-pot methodology for conversion of o-halogen nitrobenzenes to benzofuroxans. Synth. Commun. 2017, 47, 604–608. [Google Scholar] [CrossRef]
- Stolle, A.; Szuppa, T.; Leonhardt, S.E.S.; Ondruschka, B. Ball milling in organic synthesis: Solutions and challenges. Chem. Soc. Rev. 2011, 40, 2317–2329. [Google Scholar] [CrossRef]
- James, S.L.; Adams, C.J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K.D.M.; Hyett, G.; Jones, W.; et al. Mechanochemistry: Opportunities for new and cleaner synthesis. Chem. Soc. Rev. 2012, 41, 413–447. [Google Scholar] [CrossRef] [Green Version]
- Friščić, T. Supramolecular concepts and new techniques in mechanochemistry: Cocrystals, cages, rotaxanes, open metal–organic frameworks. Chem. Soc. Rev. 2012, 41, 3493–3510. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.-E.; Li, F.; Wang, G.-W. Mechanochemistry of fullerenes and related materials. Chem. Soc. Rev. 2013, 42, 7535–7570. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-W. Mechanochemical organic synthesis. Chem. Soc. Rev. 2013, 42, 7668–7700. [Google Scholar] [CrossRef] [PubMed]
- Hernández, J.G.; Bolm, C. Altering product selectivity by mechanochemistry. J. Org. Chem. 2017, 82, 4007–4019. [Google Scholar] [CrossRef]
- Andersen, J.; Mack, J. Mechanochemistry and organic synthesis: From mystical to practical. Green Chem. 2018, 20, 1435–1443. [Google Scholar] [CrossRef]
- Bolm, C.; Hernándezm, J.G. Mechanochemistry of gaseous reactants. Angew. Chem. Int. Ed. 2019, 58, 3285–3299. [Google Scholar] [CrossRef]
- Wang, G.-W.; Komatsu, K.; Murata, Y.; Shiro, M. Synthesis and X-ray structure of dumb-bell-shaped C120. Nature 1997, 387, 583–586. [Google Scholar] [CrossRef]
- Su, Y.-T.; Wang, G.-W. FeCl3-Mediated cyclization of [60]fullerene with N-benzhydryl sulfonamides under high-speed vibration milling conditions. Org. Lett. 2013, 15, 3408–3411. [Google Scholar] [CrossRef]
- Zhao, Y.; Rocha, S.V.; Swager, T.M. Mechanochemical synthesis of extended iptycenes. J. Am. Chem. Soc. 2016, 138, 13834–13837. [Google Scholar] [CrossRef]
- Turberg, M.; Ardila-Fierro, K.J.; Bolm, C.; Hernández, J.G. Altering copper-catalyzed A3 couplings by mechanochemistry: One-pot synthesis of 1,4-diamino-2-butynes from aldehydes, amines, and calcium carbide. Angew. Chem. Int. Ed. 2018, 57, 10718–10722. [Google Scholar] [CrossRef]
- Seo, T.; Ishiyama, T.; Kubota, K.; Ito, H. Solid-state Suzuki–Miyaura cross-coupling reactions: Olefin-accelerated C–C coupling using mechanochemistry. Chem. Sci. 2019, 10, 8202–8210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, K.; Pang, Y.; Miura, A.; Ito, H. Redox reactions of small organic molecules using ball milling and piezoelectric materials. Science 2019, 366, 1500–1504. [Google Scholar] [CrossRef] [PubMed]
- Pisanò, G.; Cazin, C.S.J. Mechanochemical synthesis of Cu(I)-N-heterocyclic carbene complexes. Green Chem. 2020, 22, 5253–5256. [Google Scholar] [CrossRef]
- Pang, Y.; Lee, J.W.; Kubota, K.; Ito, H. Solid-state radical C–H trifluoromethylation reactions using ball milling and piezoelectric materials. Angew. Chem. Int. Ed. 2020, 59, 22570–22576. [Google Scholar] [CrossRef]
- Das, D.; Bhosle, A.A.; Panjikar, P.C.; Chatterjee, A.; Banerjee, M. Mn(I)-catalyzed mechanochemical C–H bond activation: C-2 selective alkenylation of indoles. ACS Sustain. Chem. Eng. 2020, 8, 19105–19116. [Google Scholar] [CrossRef]
- Pisanò, G.; Cazin, C.S.J. General mechanochemical synthetic protocol to late transition metal-NHC (N-heterocyclic carbene) complexes. ACS Sustain. Chem. Eng. 2021, 9, 9625–9631. [Google Scholar] [CrossRef]
- Li, L.; Wang, G.-W. Mechanochemical solvent-free synthesis of indenones from aromatic carboxylic acids and alkynes. J. Org. Chem. 2021, 86, 14102–14112. [Google Scholar] [CrossRef]
- Ni, S.; Hribersek, M.; Baddigam, S.K.; Ingner, F.J.L.; Orthaber, A.; Gates, P.J.; Pilarski, L.T. Mechanochemical solvent-free catalytic C–H methylation. Angew. Chem. Int. Ed. 2021, 60, 6660–6666. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhu, C.; Middleton, K.R.; Todora, A.D.; Kastern, B.J.; Maskaev, A.V.; Zhdankin, V.V. Hypoiodite mediated synthesis of isoxazolines from aldoximes and alkenes using catalytic KI and Oxone as the terminal oxidant. Chem. Commun. 2013, 49, 4800–4802. [Google Scholar] [CrossRef]
- Yoshimura, A.; Middleton, K.R.; Todora, A.D.; Kastern, B.J.; Koski, S.R.; Maskaev, A.V.; Zhdankin, V.V. Hypervalent iodine catalyzed generation of nitrile oxides from oximes and their cycloaddition with alkenes or alkynes. Org. Lett. 2013, 15, 4010–4013. [Google Scholar] [CrossRef]
- Zhao, G.; Liang, L.; Wen, C.H.E.; Tong, R. In situ generation of nitrile oxides from NaCl-Oxone oxidation of various aldoximes and their 1,3-dipolar cycloaddition. Org. Lett. 2019, 21, 315–319. [Google Scholar] [CrossRef]
- Chen, K.; Niu, C.; Wang, G.-W. Reaction of aldoximes with sodium chloride and Oxone under ball-milling conditions. Molecules 2020, 25, 3719. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, G.-W. Direct oxidative amidation of aldehydes with anilines under mechanical milling conditions. J. Org. Chem. 2008, 73, 2955–2958. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-W.; Gao, J. Solvent-free bromination reactions with sodium bromide and oxone promoted by mechanical milling. Green Chem. 2012, 14, 1125–1131. [Google Scholar] [CrossRef]
- Schmidt, R.; Stolle, A.; Ondruschka, B. Aromatic substitution in ball mills: Formation of aryl chlorides and bromides using potassium peroxomonosulfate and NaX. Green Chem. 2012, 14, 1673–1679. [Google Scholar] [CrossRef]
- Friščić, T.; Jones, W. Recent advances in understanding the mechanism of cocrystal formation via grinding. Cryst. Growth Des. 2009, 9, 1621–1637. [Google Scholar] [CrossRef]
- Štrukil, V.; Margetić, D.; Igrc, M.D.; Eckert-Maksić, M.; Friščić, T. Desymmetrisation of aromatic diamines and synthesis of non-symmetrical thiourea derivatives by click-mechanochemistry. Chem. Commun. 2012, 48, 9705–9707. [Google Scholar] [CrossRef]
- Tan, D.; Štrukil, V.; Mottillo, C.; Friščić, T. Mechanosynthesis of pharmaceutically relevant sulfonyl-(thio)ureas. Chem. Commun. 2014, 50, 5248–5250. [Google Scholar] [CrossRef]
- Jiang, Z.-J.; Li, Z.-H.; Yu, J.-B.; Su, W.-K. Liquid-assisted grinding accelerating: Suzuki-Miyaura reaction of aryl chlorides under high-speed ball-milling conditions. J. Org. Chem. 2016, 81, 10049–10055. [Google Scholar] [CrossRef]
- Li, H.-G.; Wang, G.-W. Liquid-assisted one-pot mechanosynthesis and properties of neutral donor−acceptor [2]rotaxanes. J. Org. Chem. 2017, 82, 6341–6348. [Google Scholar] [CrossRef]
- Xua, W.; Wang, J.; Liu, C.; Chen, C.-L. Experimental and computational studies of the isomerization between Z and E isomers of benzaldoximes. J. Chin. Chem. Soc. 2004, 51, 1259–1266. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Nambu, H.; Okamoto, M. Deoxygenations of isocyanates and diphenylketene by tertiary phosphites. J. Org. Chem. 1962, 27, 3651–3654. [Google Scholar] [CrossRef]
- McIntosh, M.L.; Naffziger, M.R.; Ashburn, B.O.; Zakharovb, L.N.; Carter, R.G. Highly regioselective nitrile oxide dipolar cycloadditions with ortho-nitrophenyl alkynes. Org. Biomol. Chem. 2012, 10, 9204–9213. [Google Scholar] [CrossRef]
- Hwang, K.-J.; Park, Y.C.; Kim, H.J.; Lee, J.H. Synthesis and antifungal activities of furoxan derivatives designed as novel fungicide. Biosci. Biotechnol. Biochem. 1998, 62, 1693–1697. [Google Scholar] [CrossRef]
- Ovchinnikov, I.V.; Strelenko, Y.A.; Popov, N.A.; Finogenov, A.O.; Makhova, N.N. A study of the reaction mechanism of 3-nitro-4-R-furoxans formation by nitrosation of dipotassium salts of 1-hydroxyimino-2,2-dinitro-1-R-ethanes. Russ. Chem. Bull. Int. Ed. 2011, 60, 855–860. [Google Scholar] [CrossRef]
- Kadama, K.S.; Gandhi, T.; Gupte, A.; Gangopadhyay, A.K.; Sharma, R. Alkyl nitrites: Novel reagents for one-pot synthesis of 3,5-disubstituted isoxazoles from aldoximes and alkynes. Synthesis 2016, 48, 3996–4008. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.A. Organic synthesis-past, present and future. Chem. Ind. 1992, 23, 903–906. [Google Scholar]
- Sheldon, R.A. The E Factor: Fifteen years on. Green Chem. 2007, 9, 1273–1283. [Google Scholar] [CrossRef]
- Sheldon, R.A. The E factor 25 years on: The rise of green chemistry and sustainability. Green Chem. 2017, 19, 18–43. [Google Scholar] [CrossRef]
- Roschangar, F.; Sheldon, R.A.; Senanayake, C.H. Overcoming barriers to green chemistry in the pharmaceutical industry—The Green Aspiration Level™ concept. Green Chem. 2014, 17, 752–768. [Google Scholar] [CrossRef]
- Trost, B. The Atom Economy—A Search for Synthetic Efficiency. Science 1991, 254, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Curzons, A.D.; Mortimer, D.N.; Constable, D.J.C.; Cunningham, V.L. So you think your process is green, how do you know?—Using principles of sustainability to determine what is green—A corporate perspective. Green Chem. 2001, 3, 1–6. [Google Scholar] [CrossRef]
Scheme 1.
Comparison of different pathways in our previous and current work.

Scheme 2.
Scope of aldoximes (1a–r) a,b. a Unless otherwise stated, the reactions were performed in a stainless-steel jar (5 mL) with (1a–r) (0.2 mmol), NaCl (0.2 mmol), Oxone (0.2 mmol), NEt3 (0.2 mmol) together with four stainless-steel balls (5 mm in diameter) using a Retsch MM400 mixer mill at 30 Hz for 30 min. b Isolated yields based on (1a–r). c NaCl (0.8 mmol), Oxone (0.2 mmol), Na2CO3 (0.4 mmol). d Reaction time was 60 min. e NaCl (0.4 mmol), Oxone (0.6 mmol), NaOtBu (0.2 mmol). f NaCl (0.4 mmol), Oxone (0.2 mmol), Na2CO3 (0.4 mmol). g NaCl (0.4 mmol), Oxone (0.2 mmol), Na2CO3 (0.6 mmol).
Scheme 2.
Scope of aldoximes (1a–r) a,b. a Unless otherwise stated, the reactions were performed in a stainless-steel jar (5 mL) with (1a–r) (0.2 mmol), NaCl (0.2 mmol), Oxone (0.2 mmol), NEt3 (0.2 mmol) together with four stainless-steel balls (5 mm in diameter) using a Retsch MM400 mixer mill at 30 Hz for 30 min. b Isolated yields based on (1a–r). c NaCl (0.8 mmol), Oxone (0.2 mmol), Na2CO3 (0.4 mmol). d Reaction time was 60 min. e NaCl (0.4 mmol), Oxone (0.6 mmol), NaOtBu (0.2 mmol). f NaCl (0.4 mmol), Oxone (0.2 mmol), Na2CO3 (0.4 mmol). g NaCl (0.4 mmol), Oxone (0.2 mmol), Na2CO3 (0.6 mmol).

Scheme 3.
Formation of nitrile oxides from 1s and 1t under ball-milling conditions.

Scheme 4.
Control experiments.

Scheme 5.
Proposed mechanism for the formation of 2.

Scheme 6.
Mechanosynthesis of 2a from a mixture of 1a and 1a′.

Scheme 7.
The deoxygenation reaction of 2a.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of the reaction conditions a.

| Entry | NaCl (Equiv.) | Oxone (Equiv.) | Base (Equiv.) | Yield of 2a (%) b |
|---|---|---|---|---|
| 1 | 1.0 | 1.0 | Na2CO3 (1.0) | 8 |
| 2 | 1.0 | 1.0 | NaOtBu (1.0) | trace |
| 3 | 1.0 | 1.0 | NaOAc (1.0) | trace |
| 4 | 1.0 | 1.0 | NaHCO3 (1.0) | trace |
| 5 | 1.0 | 1.0 | K2CO3 (1.0) | 36 |
| 6 | 1.0 | 1.0 | Cs2CO3 (1.0) | 7 |
| 7 | 1.0 | 1.0 | DMAP (1.0) | 0 |
| 8 | 1.0 | 1.0 | DBU (1.0) | 0 |
| 9 | 1.0 | 1.0 | DABCO (1.0) | 0 |
| 10 | 1.0 | 1.0 | NEt3(1.0) | 79 |
| 11 | 1.0 | 1.0 | NEt3 (1.25) | 69 |
| 12 | 1.0 | 1.0 | NEt3 (1.5) | 53 |
| 13 | 1.0 | 1.0 | NEt3 (0.75) | 62 |
| 14 | 1.0 | 1.0 | NEt3 (0.5) | 38 |
| 15 c | 1.0 | 1.0 | NEt3 (1.0) | trace |
| 16 d | 1.0 | 1.0 | NEt3 (1.0) | 47 |
| 17 e | 1.0 | 1.0 | NEt3 (1.0) | 78 |
| 18 | 1.5 | 1.0 | NEt3 (1.0) | 79 |
| 19 | 1.0 | 1.5 | NEt3 (1.0) | 77 |
| 20 f,g | 1.0 | 1.0 | NEt3 (1.0) | 50 |
| 21 f,h | 1.0 | 1.0 | NEt3 (1.0) | 55 |
| 22 f,i | 1.0 | 1.0 | NEt3 (1.0) | 51 |
| 23 f,j | 1.0 | 1.0 | NEt3 (1.0) | 71 |
a Unless otherwise stated, the reactions were performed in a stainless-steel jar (5 mL) with 1a (0.2 mmol), NaCl (1.0 equiv.), Oxone (1.0 equiv.) and base (1.0 equiv.) together with four stainless-steel balls (5 mm in diameter) using a Retsch MM400 mixer mill at 30 Hz for 30 min. b Isolated yield based on 1a. c Magnetic stirring for 2 h instead of ball milling. d Reaction time was 15 min. e Reaction time was 40 min. f A liquid (22 µL, η = 0.17 µL/mg) was added as a LAG agent. g EtOH was added. h DCM was added. i EtOAc was added. j CH3CN was added.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fang, R.-K.; Chen, K.; Niu, C.; Wang, G.-W. Mechanochemical Dimerization of Aldoximes to Furoxans. Molecules 2022, 27, 2604. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082604
AMA Style
Fang R-K, Chen K, Niu C, Wang G-W. Mechanochemical Dimerization of Aldoximes to Furoxans. Molecules. 2022; 27(8):2604. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082604
Chicago/Turabian StyleFang, Run-Kai, Kuan Chen, Chuang Niu, and Guan-Wu Wang. 2022. "Mechanochemical Dimerization of Aldoximes to Furoxans" Molecules 27, no. 8: 2604. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082604