A QSAR Toxicity Study of a Series of Alkaloids with the Lycoctonine Skeleton

1
Institute of Chemistry of Plant Substances, Academy of Sciences of the Republic of Uzbekistan,
Kh. Abdullaev Str., 77, Tashkent, 700170, Uzbekistan
2
Chemistry Department, National University of Uzbekistan named after Mirzo Ulugbek, Vuzgorodok
Tashkent, 700174, Uzbekistan
*
Author to whom correspondence should be addressed.
Molecules 2004, 9(12), 1194-1207; https://0-doi-org.brum.beds.ac.uk/10.3390/91201194
Submission received: 26 April 2004
/
Accepted: 10 August 2004
/
Published: 31 December 2004
Abstract
:A QSAR toxicity analysis has been performed for a series of 19 alkaloids with the lycoctonine skeleton. GA-MLRA (Genetic Algorithm combined with Multiple Linear Regression Analysis) technique was applied for the generation of two types of QSARs: first, models containing exclusively 3D-descriptors and second, models consisting of physicochemical descriptors. As expected, 3D-descriptor QSARs have better statistical fits. Physicochemical-descriptor containing models, that are in a good agreement with the mode of toxic action exerted by the alkaloids studied, have also been identified and discussed. In particular, TPSA (Topological Polar Surface Area) and nC=O (number of –C(O)– fragments) parameters give the best statistically significant mono- and bidescriptor models (when combined with lipophilicity, MlogP) confirming the importance of H-bonding capability of the alkaloids for binding at the receptor site.
Introduction
Diterpene alkaloids isolated from the Aconitum and Delphinium plant species have been used mainly for preparation of a wide spectrum of poisons as well as medicinals for many centuries [1]. Owing to their structure diversity, these alkaloids can be divided into a number of subgroups represented by molecules with the lycoctonine, heteratisine and napelline diterpene skeletons [2]. Extensive biological and pharmacological studies performed have shown that Aconitum and Delphinium alkaloids affect substantially the cardiac and central nervous systems. Thus, at present, a number of diterpene alkaloids have been identified that exert analgesic, anti-inflammatory, antiepileptiform and cardiovascular effects [3,4].
Nevertheless, the practical application of Aconitum and Delphinium alkaloids in medicine is limited by their high toxicity. For the last few decades, these alkaloids have been the target of considerable interest by medicinal chemists who have put great effort into identifying structure-activity and/or structure-toxicity relationships. Thus, in the paper of Ameri et al. [1] Aconitum alkaloids are subdivided into three main groups: group number one consists of highly toxic alkaloids with two ester bonds ─OC(O)─ ; monoester derivatives of lappaconitine-like alkaloids of lower toxicity represent the second group. The most striking fact about these two groups is that they are antagonists when their affect on the voltage-dependent sodium channel is considered. Thus, group one compounds have been found to activate sodium channels, whereas alkaloids of group two block passive diffusion of sodium ions. The third group is comprised of the Aconitum alkaloids exerting the lowest toxicity due to the absence of any ester side chains. With no effect on the neuronal system, the alkaloids of this last group are still found to exhibit antiarrhythmic activity.
The high toxicity of various species of Delphinium plants is attributed to the norditerpenoid alkaloids present. These plants still continue to be the main cause of extensive cattle poisoning resulting in substantial losses for the cattle-breeding industry [5,6]. A number of investigations have been performed on systematisation and taxonomic classification of Delphinium alkaloids known at present. Thus, according to the classification reported by Panter et al. [5], Delphinium species alkaloids can be divided also into three general types: (I) N-(methylsuccinyl) anthranoyllycoctonine (MSAL)-type alkaloids with the highest toxicity; (II) Lycoctonine-type alkaloids of moderate toxicity; and (III) 7,8-methylenedioxyllycoctonine (MDL)-type with low toxicity.
Having screened the literature, one might conclude that structure-activity investigations of Aconitum and Delphinium alkaloids have been carried out basically using conventional approaches. Likewise, toxicity of the alkaloids has been investigated by modifying certain functional groups. Further, a careful literature search has revealed that investigation of diterpene alkaloids by means of QSAR approach has received very little attention and only recently has an attempt been made to perform a QSAR analysis of the analgesic properties for twelve Aconitum alkaloids [7].
QSAR [8,9,10,11,12] is a powerful lead-compound optimisation technique, which quantitatively relates variations in biological activity to changes in molecular properties (descriptors). In other words, it attempts to link activity data with descriptors chosen via identification of the “rules” that can be further used to guide chemical synthesis when new chemical entities are developed. Recently new programs have emerged allowing more than thousand descriptors to be generated for the single molecule. Therefore, the search for successful combinations of mutually inter-related descriptors becomes an issue even for small series of compounds. For the last few years the Genetic Algorithm (GA) method, together with Multiple Linear Regression Analysis (MLRA) has become a valuable approach for deriving and validating QSARs. GA is an optimisation algorithm based on the mechanisms of Darwinian evolution that uses random mutation, crossover and selection procedures to breed better models or solutions from an originally random starting population or sample [13,14].
In this paper we present the results of a QSAR toxicity study carried out for 19 alkaloids with the lycoctonine skeleton. Structure –toxicity relationships are discussed in terms of the QSAR models built and the descriptors chosen.
Materials and Methods
Training Compounds
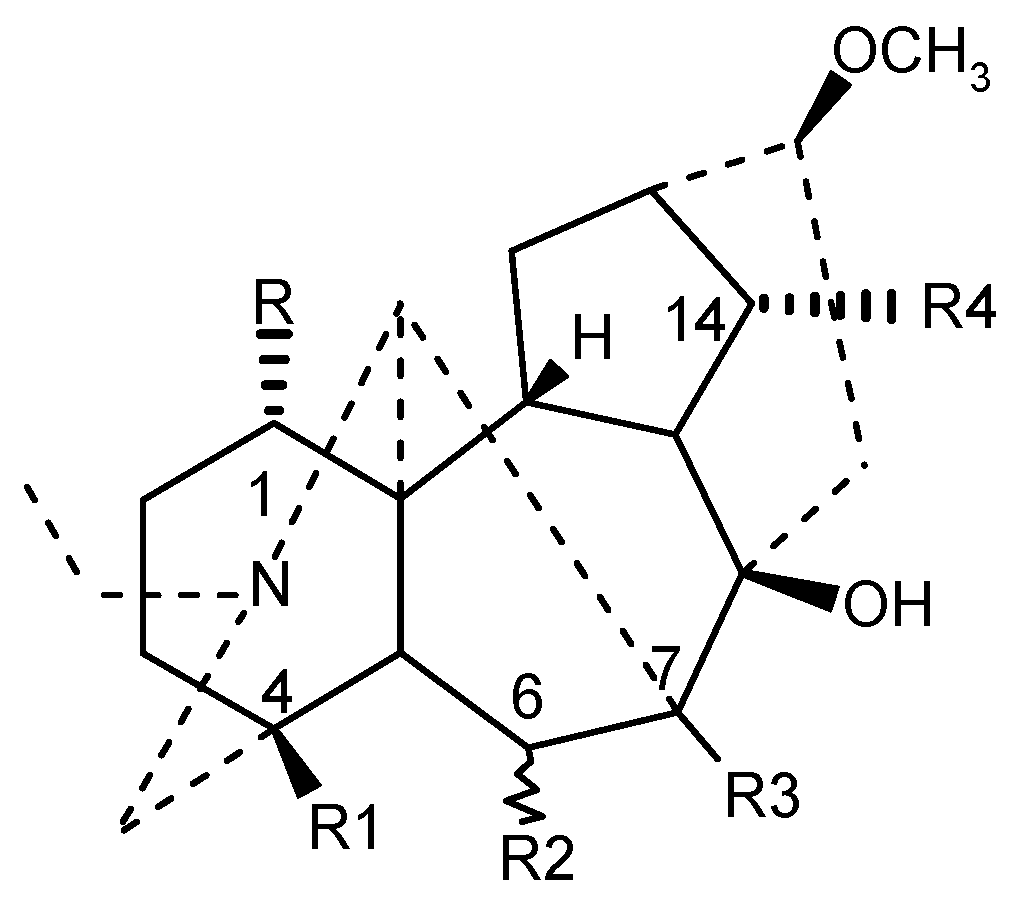
The structures of the 19 diterpenoids chosen for this work and their functional groups are shown in Figure 1.
Compounds are subdivided into two general groups in accordance with the functionality natureat C4. Thus the first group alkaloids contain ─CH3, ─CH2OCH3, and ─CH2OH residues, whereas the second type of alkaloids has a benzoylester side chain. Toxicity data used in this study is taken from the reference [15] suggesting compounds of the second group to be more toxic than the first group alkaloids. This is in a good agreement with structure-toxicity investigations reported in two recent reviews [1,5]. All original LD50 toxicity data (mg/kg) has been converted to molar –logLD50 response variables.
Figure 1.
Diterpene alkaloids with variations at C-1, C-4, C-6, C-7 and C-14 of lycoctonine ring.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | R1 | R2 | R3 | R4 |
|---|---|---|---|---|---|
| Neoline | OH | CH2OCH3 | αOCH3 | H | OH |
| Isotalatisidine | OH | CH2OCH3 | H | H | OH |
| Кarakoline | OH | CH3 | H | H | OH |
| Delsoline | OH | CH2OCH3 | βOCH3 | OH | OCH3 |
| Kondelfine | OH | CH2OCH3 | H | H | OCOCH3 |
| 14-Acetylvirescenine | OH | CH2OCH3 | H | OH | OCOCH3 |
| Gigactonine | OH | CH2OH | βOCH3 | OH | OCH3 |
| Lycoctonine | OCH3 | CH2OH | βOCH3 | OH | OCH3 |
| Delectinine | OCH3 | CH2OH | βOCH3 | OH | OH |

| Compound | R | R1 | Compound | R | R1 |
|---|---|---|---|---|---|
| Lycaconitine | OCH3 |  | Delectine | OH | NH2 |
| Nudicauline | OCOCH3 |  | О- Acetyldelectine | OCOCH3 | NH2 |
| Меthyllycaconitine | OCH3 | | N- Acetyldelectine | OH | NHCOCH3 |
| Anthranoyllycoctonine | OCH3 | NH2 | N-О-Diacetyldelectine | OCOCH3 | NHCOCH3 |
| Puberaconitine | OCH3 | NHCO(CH2)2COOH | Ajacine | OCH3 | NHCOCH3 |
Molecular Modelling
The molecular modelling studies (molecular mechanics and semi-empirical calculations) were carried out using the Hyperchem 6.01 software package [16]. The Molecular Mechanics (MM+) force field was applied for preliminary structure optimisation and study of the conformational behaviour of each alkaloid. Molecular mechanics has been shown to produce more realistic geometry values for the majority of organic molecules owing to the fact of being highly parameterised [17]. The next step was a re-optimisation of the MM+ optimised structures by applying AM1 semi-empirical method. Quantum mechanical method has been used in order to obtain an accurate charge distribution and quantum-chemical descriptors for each compound in the series.
Molecular Descriptors
Descriptors are normally calculated for molecules after a low-energy conformation has been found and optimised using any standard optimisation technique, e.g. molecular mechanics, ab initio, DFT or semi-empirical methods. The molecular descriptors used in this study have been calculated applying the DRAGON program [18]. The program contains scripts for generating 1497 descriptors of different types including: constitutional, topological, RDF, GETAWAY, functional groups, WHIM, Randic, 3D-Morse etc [19]. A set of additional quantum-chemical descriptors (energy of heat of formation, dipole moment, atomic charges, energy of highest occupied orbital HOMO, energy of lowest unoccupied orbital LUMO, HOMO-LUMO energy gap etc.) has also been obtained for the each molecule after geometry optimisation procedure.
Statistical Methods
Preliminary models selection was performed by means of GA-MLRA technique as implemented in the BuildQSAR [20] program. As mentioned before, this approach allows selection of the models with the following characteristics: high quadratic correlation coefficient R2, low standard deviation S and the least number of descriptors involved. Next, the NCSS98 [21] professional software package was applied for detailed statistical analysis of the models obtained. Thus, the high Fisher coefficient F, non-collinear descriptors, and the significance level P variable served as additional selection parameters. A final set of QSARs was identified by applying the “leave-one-out” technique with its predicting ability being evaluated and confirmed by cross validation coefficient Q2 based on predictive error sum of squares (SPRESS).
Results and Discussion
As mentioned above, a QSAR analysis has been performed for 19 alkaloids with the lycoctonine skeleton (Figure 1) aiming at establishing a structure-toxicity relationship. For the sake of simplicity we have divided the models into two general groups in accordance with nature of the descriptors involved: group one, comprised of 3D descriptors generated by DRAGON, and group two, containing physicochemical descriptors only. While constructing the models, great care was taken in order to avoid inclusion of highly collinear descriptors. The correlation matrix for the 3D together with physicochemical descriptors used in this study is given in Table 1. The table includes only those variables that have comprised the most populated models selected by the variable selection Genetic Algorithm method.
3D Descriptor Containing Models
One of the main advantages of these descriptors is the unambiguity regarding the 3D arrangement of atoms. There are also other properties making them flexible and therefore popular descriptors to be used. One of them is independence from the molecular size resulting in applicability to the large datasets with great structural variance. Another important property of 3D descriptors is their invariance against translation and rotation of the molecule. Such atom-based descriptors are also suggested to be applied for the collection of exotic chemicals, since there is a greater chance of physicochemical descriptors giving misleading information [22].
For the model generation we have chosen 3D descriptors of the following type: RDF, 3D-Morse, WHIM, GETAWAY, and Randic, available within DRAGON. Several runs of GA-MLRA variable selection technique implemented in BuildQSAR program have resulted in models containing mainly RDF, and 3D-Morse type descriptors. Values of 3D descriptors together with the toxicity data are indicated in Table 2. 3D-Morse descriptors were obtained on the basis of the molecular transform equation used in electron diffraction [23]. RDF code is based on the radial distribution function of an ensemble with N atoms, i.e. probability distribution of finding atom on a sphere with radius r [24].
| DiMo | lgP | E | HOMO | LUMO | GAP | MW | TPSA | MlogP | nHDon | nHAcc | nC=O | MR | Mor28e | RDF020u | Mor07p | |
| DiMo | 1 | .38 | .428 | .406 | .514 | .533 | .635 | .565 | .007 | .092 | .618 | .517 | .643 | .022 | .452 | .433 |
| lgP | 1 | .604 | .599 | .402 | .482 | .504 | .372 | .21 | .638 | .606 | .237 | .494 | .259 | .862 | .234 | |
| E | 1 | .487 | .554 | .607 | .784 | .761 | .044 | .228 | .882 | .669 | .737 | .035 | .594 | .571 | ||
| HOMO | 1 | .411 | .512 | .594 | .482 | .054 | .353 | .62 | .332 | .594 | .018 | .654 | .449 | |||
| LUMO | 1 | .985 | .857 | .811 | .118 | .092 | .792 | .788 | .884 | .034 | .397 | .728 | ||||
| GAP | 1 | .897 | .832 | .068 | .139 | .847 | .78 | .921 | .042 | .473 | .752 | |||||
| MW | 1 | .959 | .017 | .11 | .977 | .88 | .995 | .007 | .552 | .875 | ||||||
| TPSA | 1 | .044 | .035 | .93 | .952 | .941 | .001 | .4 | .909 | |||||||
| MlogP | 1 | .333 | 0 | .117 | .028 | .126 | .173 | .09 | ||||||||
| nHDon | 1 | .175 | .006 | .11 | .331 | .516 | .008 | |||||||||
| nHAcc | 1 | .833 | .958 | .021 | .622 | .8 | ||||||||||
| nC=O | 1 | .861 | .000 | .236 | .878 | |||||||||||
| MR | 1 | .006 | .555 | .87 | ||||||||||||
| Mor28e | 1 | .119 | .015 | |||||||||||||
| RDF020u | 1 | .283 | ||||||||||||||
| Mor07p | 1 |
DiMo – Dipole moment, lgP – Lipophilicity (octanol-water partition coefficient, HyperChem code), Е – Heat of formation, HOMO – Energy of Highest Occupied Molecular Orbital, LUMO – Energy of Lowest Unoccupied Molecular Orbital, GAP – HOMO-LUMO energy gap, MW – Molecular Weight, ТPSA – Topological Polar Surface Area, MlogP – Moriguchi octanol-water partition coefficient (DRAGON code), nHDon – number of donor atoms for H-bonds (with N and O atoms), nHAcc – number of acceptor atoms (N, O, F) Hy- hydrophilic factor, nC=O – number of C=O fragments, MR – Ghose-Grippen Molar Refractivity, 3D-Descriptors: Mor28e, RDF020u, Mor07p
| Alkaloid | LD50 mg/kg* |  | RDF020u | Mor28e | Mor07p | TPSA | nC=O | MlogP | MW |
| Neoline | 69.0 | 3.80 | 18.075 | -1.417 | 3.436 | 30.93 | 0 | 1.473 | 437.64 |
| Isotalatisidine | 40.1 | 4.00 | 16.387 | -1.184 | 3.438 | 21.7 | 0 | 2.021 | 407.61 |
| Кarakoline | 51.5 | 3.86 | 13.17 | -1.383 | 3.164 | 12.47 | 0 | 2.578 | 377.58 |
| Delsoline | 175.0 | 3.43 | 21.23 | -0.985 | 3.516 | 40.16 | 0 | 0.932 | 467.67 |
| Kondelfinee | 18.5 | 4.39 | 15.798 | -1.076 | 3.723 | 48 | 1 | 2.348 | 449.65 |
| 14-Acetylvirescenine | 18.0 | 4.41 | 16.249 | -0.84 | 3.749 | 48 | 1 | 1.599 | 465.65 |
| Lycaconotine | 2.6 | 5.41 | 20.806 | -1.257 | 6.025 | 103.84 | 3 | 1.673 | 668.86 |
| Nudicauline | 1.8 | 5.60 | 19.957 | -1.241 | 6.337 | 120.91 | 4 | 2.046 | 710.9 |
| Gigactonine | 88.0 | 3.71 | 19.164 | -0.754 | 3.463 | 30.93 | 0 | 0.725 | 453.64 |
| Lycoctonine | 170.0 | 3.44 | 20.081 | -1.149 | 3.458 | 40.16 | 0 | 0.932 | 467.67 |
| Delectinine | 130.0 | 3.54 | 20.173 | -0.903 | 3.228 | 30.93 | 0 | 0.725 | 453.64 |
| Methyllycaconitine | 3.9 | 5.24 | 21.178 | -1.236 | 6.248 | 103.84 | 3 | 1.85 | 682.89 |
| Anthranoyllycoctonine | 20.1 | 4.46 | 22.304 | -1.138 | 4.786 | 66.46 | 1 | 1.741 | 586.8 |
| Puberaconitine | 22.5 | 4.48 | 23.449 | -1.247 | 4.999 | 100.6 | 3 | 1.368 | 686.88 |
| Delectine | 35.8 | 4.20 | 22.1 | -0.898 | 4.25 | 57.23 | 1 | 1.553 | 572.77 |
| О-Acetyldelectine | 15.5 | 4.60 | 21.765 | -1.011 | 5.08 | 83.53 | 2 | 1.929 | 614.81 |
| N- Acetyldelectine | 25.3 | 4.39 | 22.521 | -0.641 | 4.522 | 74.3 | 2 | 1.523 | 614.81 |
| N-О-Diacetyldelectine | 12.5 | 4.71 | 21.3 | -0.833 | 4.98 | 100.6 | 3 | 1.9 | 656.85 |
| Ajacine | 9.0 | 4.84 | 22.423 | -0.683 | 4.733 | 83.53 | 2 | 1.706 | 628.84 |
* LD50 values have been taken from reference [15]
The following best QSAR models have been identified:
3D monodescriptor model (A):

n=19; r=0.93; r2adj=0.85 s=0.25; F=104.33; Q2=0.83; SPRESS=0.27; P=0.000000
3D bidescriptor model (B):

n=19; r=0.96; r2adj= 0.92; s=0.19; F=97.51; Q2=0.89; SPRESS=0.22; P=0.000000
3D tridescriptor model (C):

n=19; r=0.98; r2adj=0.94; s=0.154; F=98.39; Q2=0.92; SPRESS=0.20; P=0.000000
The statistical significance of each model is evaluated by the correlation coefficient r, standard error s, adjusted r-squared r2adj, F-test value, significance level of the model P, leave-one-out cross-validation coefficient Q2 and predictive error sum of squares SPRESS. As can be seen, all three models have a high significance level P and the statistical fit of each model improves as the additional descriptors are included. Interestingly, these models cannot be further improved by deleting from the series the compound which deviates the most from the average. This can be explained by the fact that 3D descriptors take into account 3D structure of each molecule of the series. Though being non-mechanistically relevant, the models obtained show high flexibility and the predicting ability of 3D descriptors, which is confirmed by the close values of r2adj and Q2.
Physicochemical descriptor containing models.
A number of physicochemical descriptors used in this study are shown in Table 1. Two descriptors, such as TPSA (Topological Polar Surface Area) [25] and nC=O (descriptor indicating the number of –C(O)─ fragments present in the molecule) have been identified by the GA-MLRA model search as the best correlated with the toxicity data (refer to Table 2 for their values). TPSA descriptor is described as a polar part of the molecule associated with the oxygen, nitrogen, sulfur atoms and also hydrogens connected to these heteroatoms. It has been reported to be one of the best predictivity descriptors to build a QSAR model for the drugs affecting Central Nervous System (CNS) [26]. Thus, TPSA has demonstrated a strong correlation with drug absorption: decrease of the surface of polar area results in increase of blood-brain permeation of CNS drug molecules [27] and improved intestinal epithelial permeability of orally administrated medicinal agents [28]. A larger topological surface area was also proposed as a feature favoring HERG K+ channel blockers binding [29]. The QSAR model containing TPSA and generated in this study is as follows:
PhysChem monodescriptor model (A):

n=19; r=0.88; r2adj=0.75; s=0.318; F=56.00; Q2=0.70; SPRESS=0.36; P=0.000001
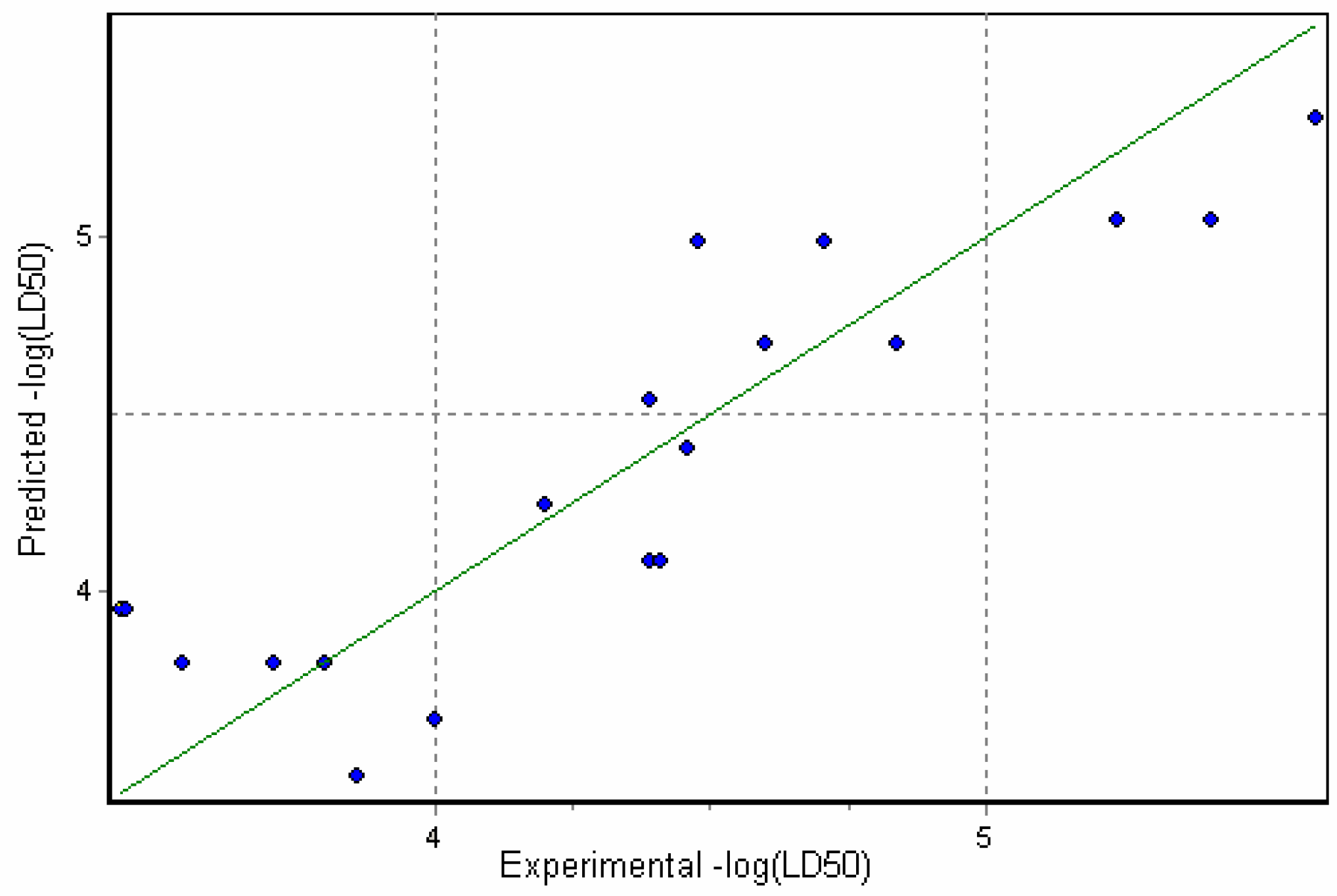
The graphical view of the model is shown in Figure 2. As can be seen from this model, the toxicity of the alkaloids increases with the number of heteroatoms in the molecule. This contrasts with the model obtained for other CNS-drugs and confirms the difference in mechanisms of action between the two types of CNS affecting drugs. Thus, lycoctonine alkaloids have been reported to affect CNS by binding to the sodium ion channel, i.e. the membrane protein, leading to conformational changes and subsequently to functional disruption of the channel, whereas low molecular weight CNS drugs induce toxicity at a certain concentration and therefore their ability to pass through biological membranes (e.g. blood-brain barrier) plays a key role in the mode of their toxic action.
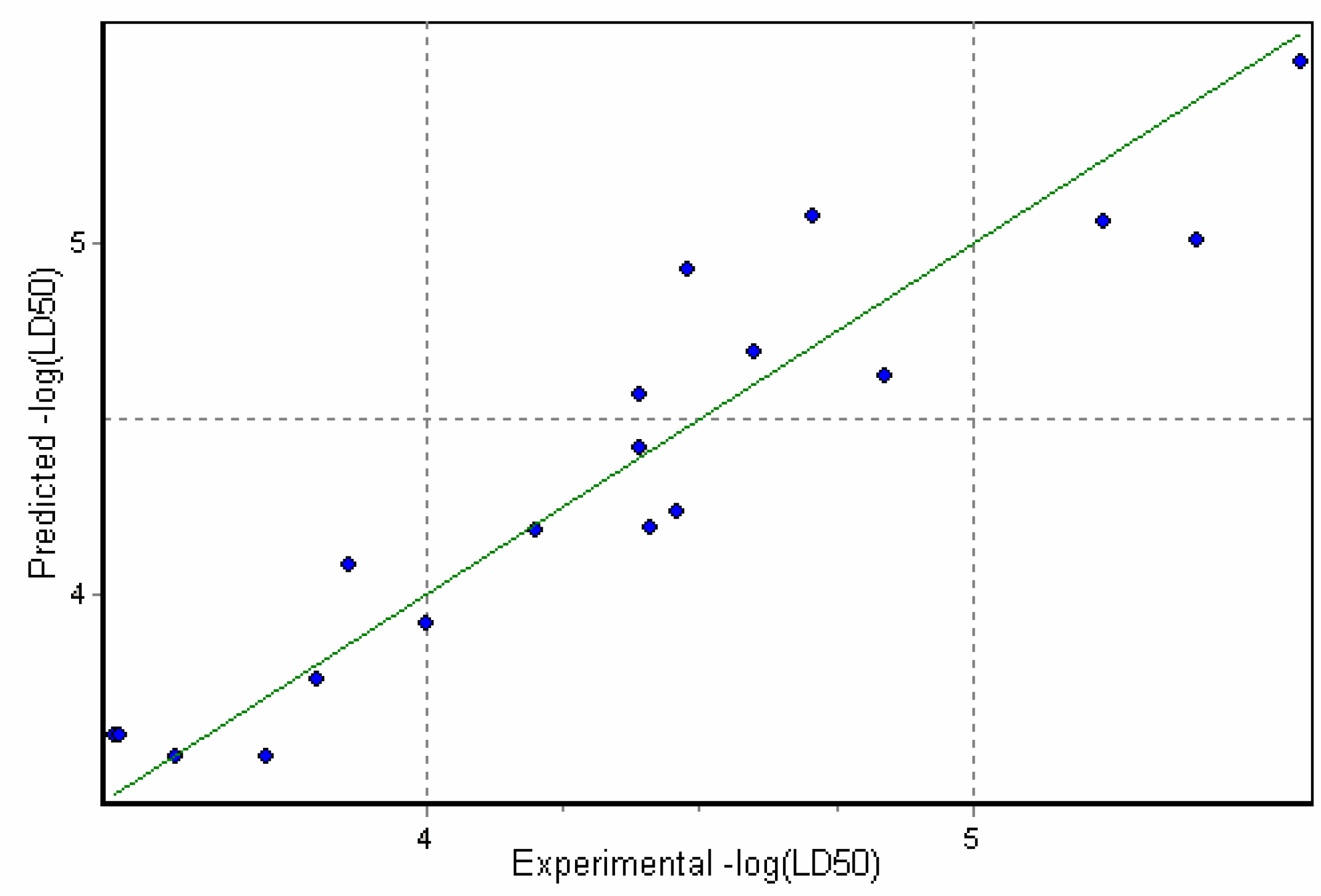
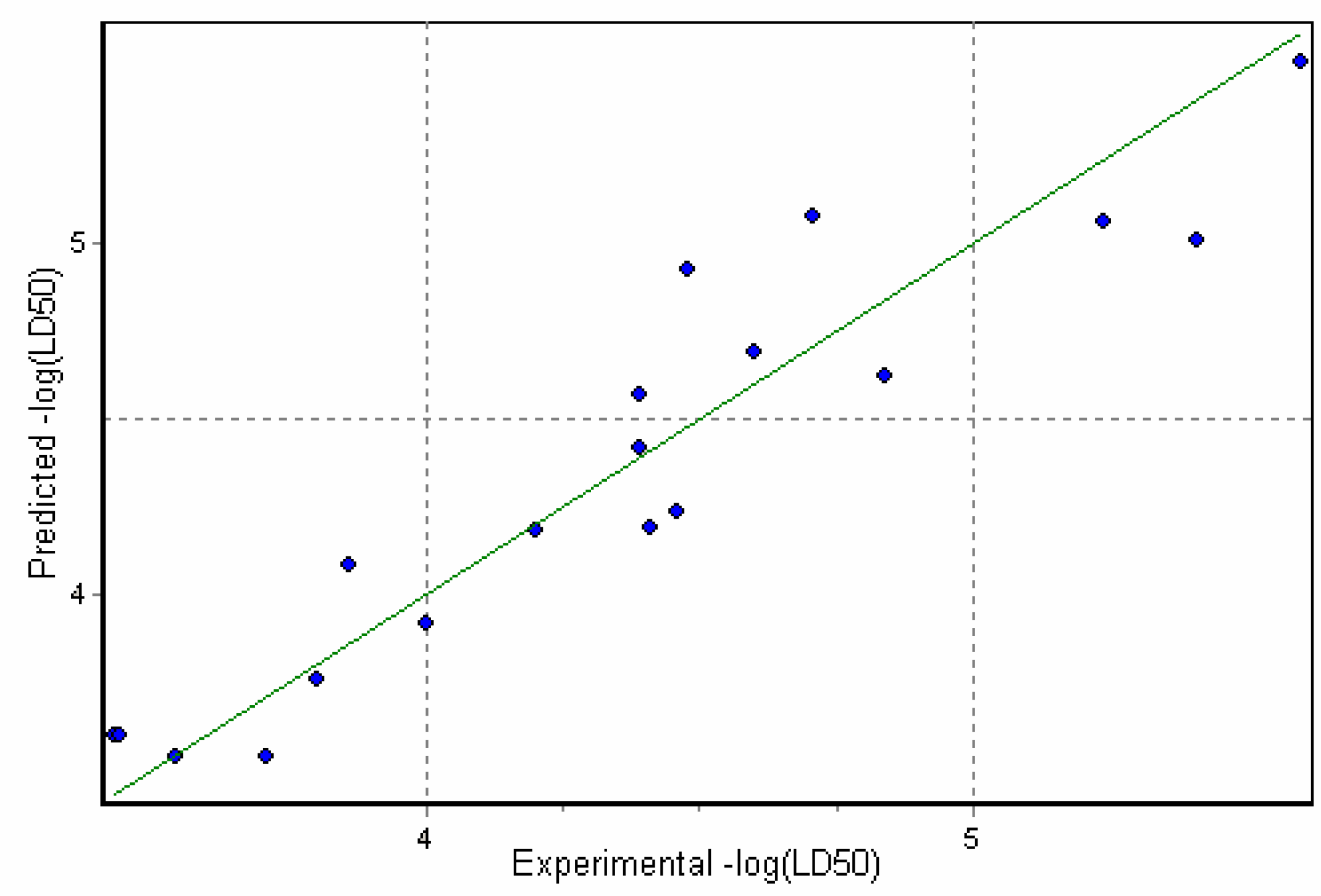
Figure 2.
Correlation of Predicted versus Experimental toxicities for model A (Eqn.4)
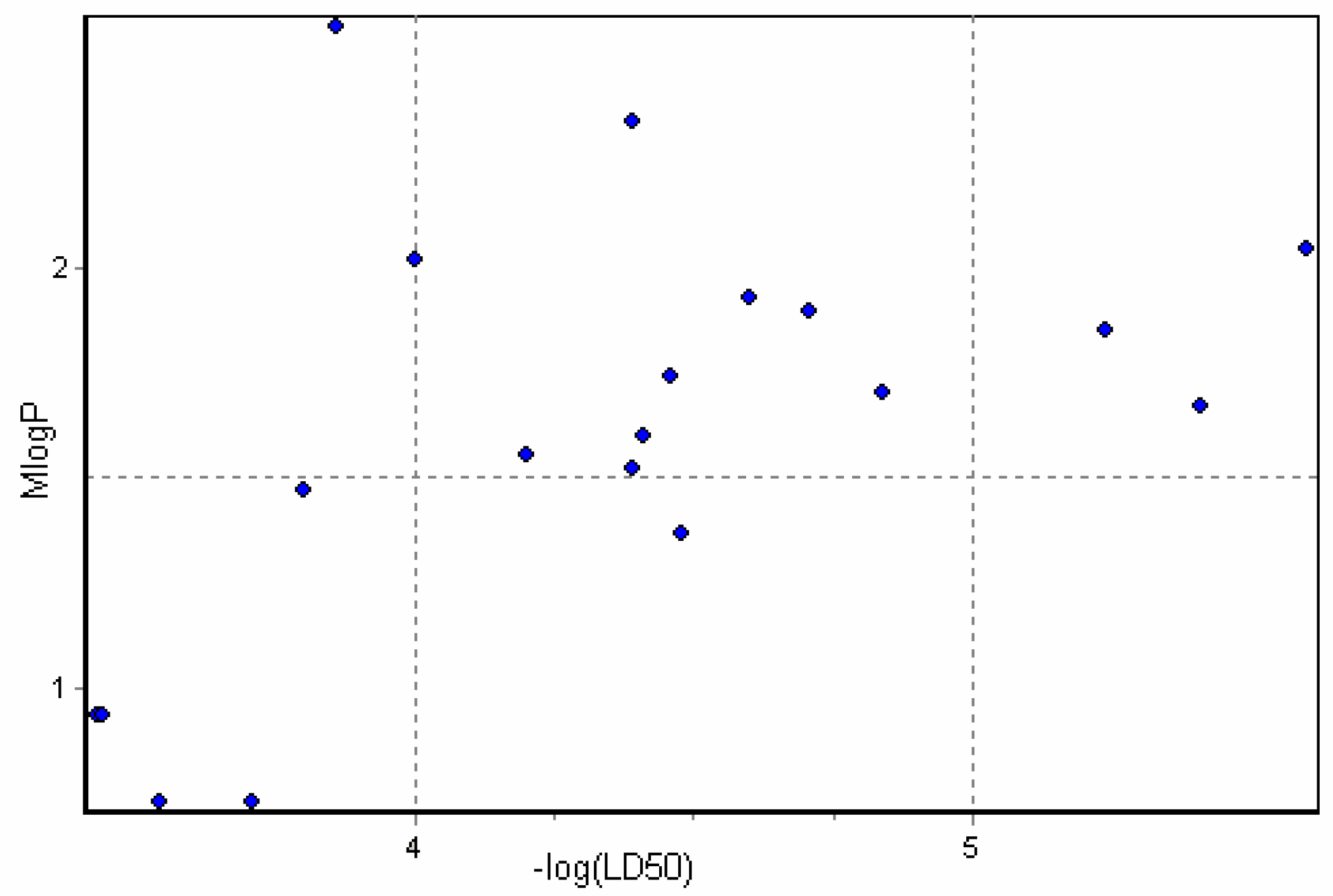
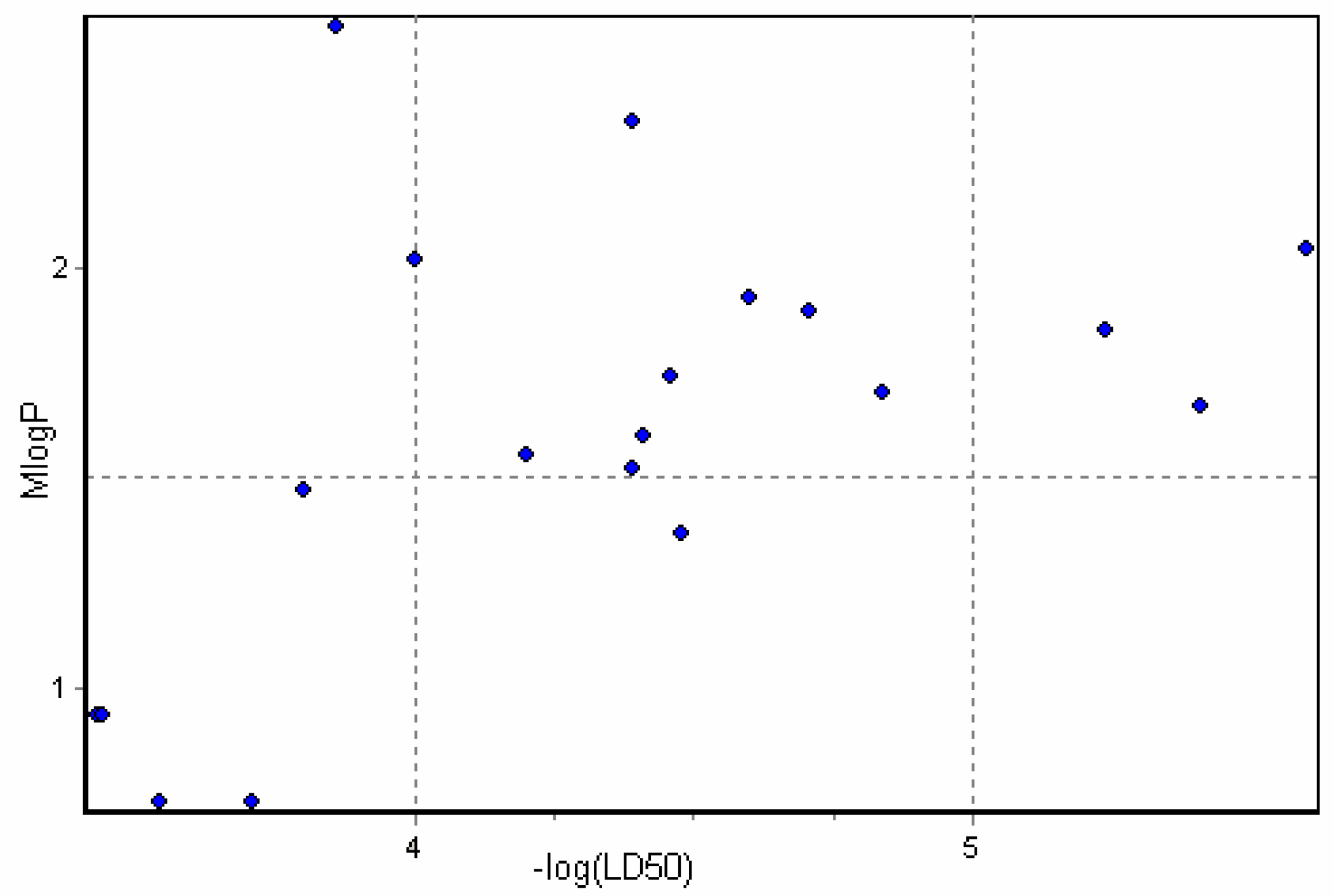
Furthermore, as reported elsewhere [30], no correlation should be observed for octanol-water coefficient and toxicity for the specific interactions of the toxicant with the receptor site. This is in a good agreement with our results as no substantial correlation has been observed for LogP/MlogP and toxicity data (for example, see Figure 3 with MlogP plotted against toxicity data). Having taken into account that TPSA describes H-bonding capability of the molecules, one can conclude that high values of this descriptor can be attributed to the alkaloids of higher toxicity due to the stronger binding at the receptor site.
Figure 3.
MlogP plotted versus Toxicity data
Next, a monodescriptor model is represented by nC=O fragment descriptor, which also can be considered as a H-bonding type of physicochemical descriptors (Figure 4):
PhysChem monodescriptor model (B):

n=19; r=0.91; r2adj=0.83; s=0.27; F=86.30; Q2=0.79; SPRESS=0.30; P=0.000000
Figure 4 below shows a 2D plot for nC=O and toxicity:
Figure 4.
nC=O plotted versus Toxicity data
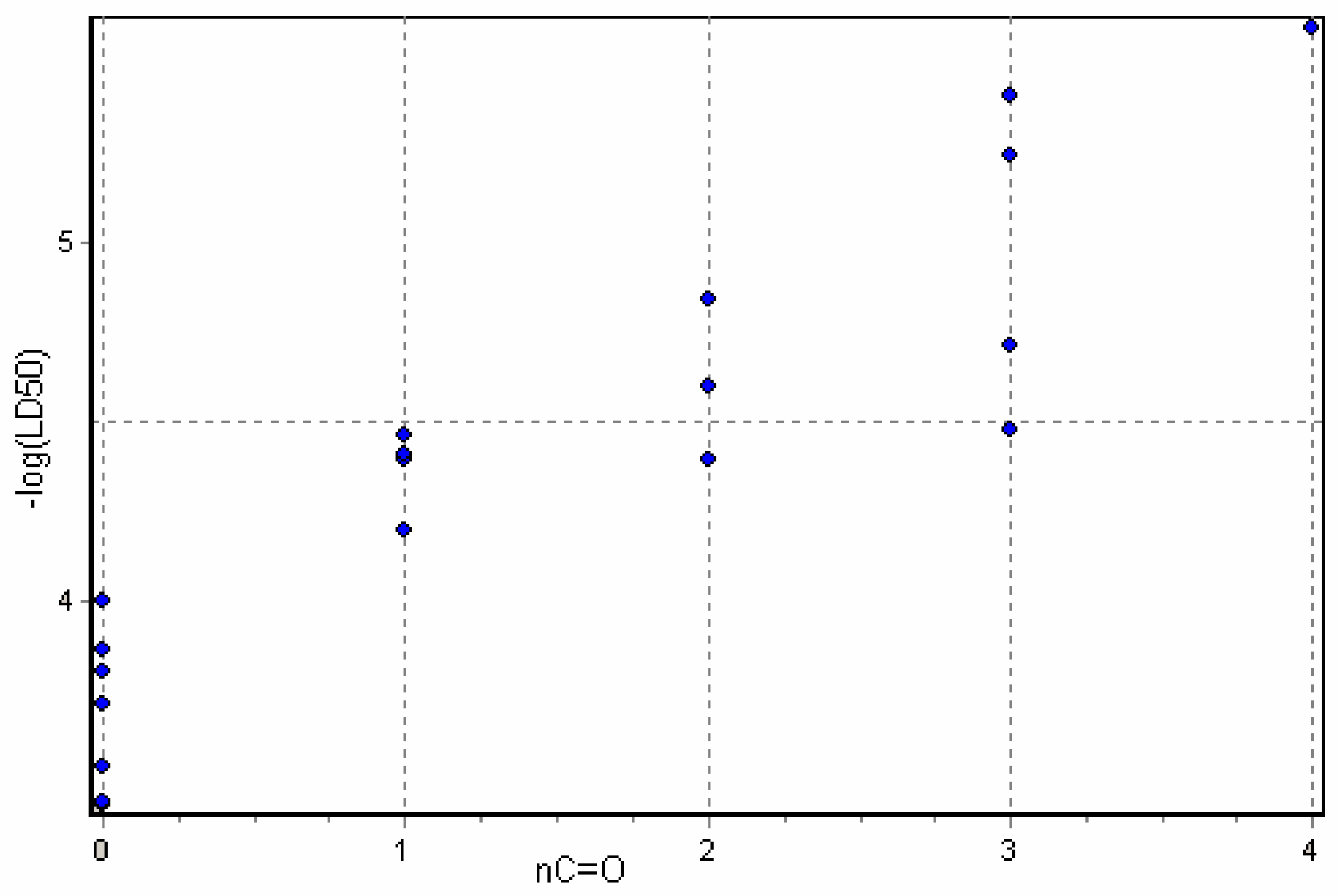
According to Figure 4, the toxicity of alkaloids can be improved as the number of C=O fragments decreases. Thus, the least toxic are alkaloids with no C=O fragments, and the most toxic one contains four such fragments.
Development of mechanistically based models requires an accurate determination of the most relevant descriptors controlling the endpoints of interest. Even at the first glance, it becomes clear that the nC=O fragment descriptor is responsible for the receptor-ligand interaction strength. The spatial orientation of –C(O)─ makes them the most favored “targets” – donors of electron lone pairs promoting attachment and fixation of the ligand molecule in 3D receptor domain. It is worth to note, that MlogP descriptor has been identified as the best to combine with both TPSA and nC=O descriptors while generating two-descriptor QSAR models. MlogP (Moriguchi octanol-water partition coefficient) [19] is a popular and traditional descriptor used in QSAR model building. It describes one of the most important properties of any compound– its lipophilicity which indicates the ability to penetrate lipid-rich zones from aqueous solutions. The later becomes a very important feature of any drug that is administrated orally and supposed to pass gastrointestinal epithelium. Thus in accordance with the reference [28], medicinal agents with PSA (general case of TPSA) varying in the range from 61Ǻ to 140 Ǻ and with LogP (general case of MlogP) less than 5 are proposed to be well absorbed. The following two-descriptor models obtained also indicate lipophilicity being directly related to toxicity for the given range of MlogP values when it combined with H-bonding descriptors:
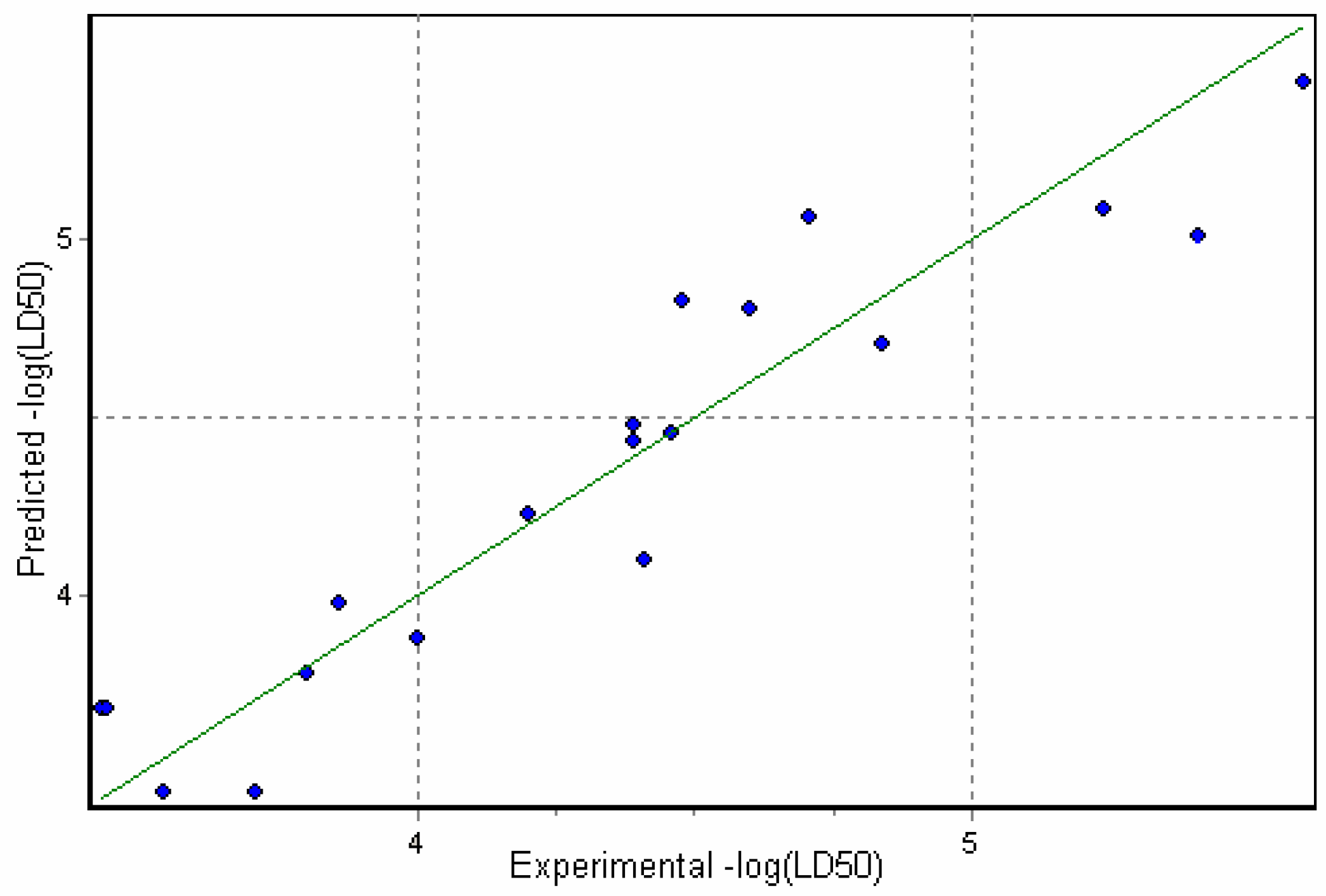
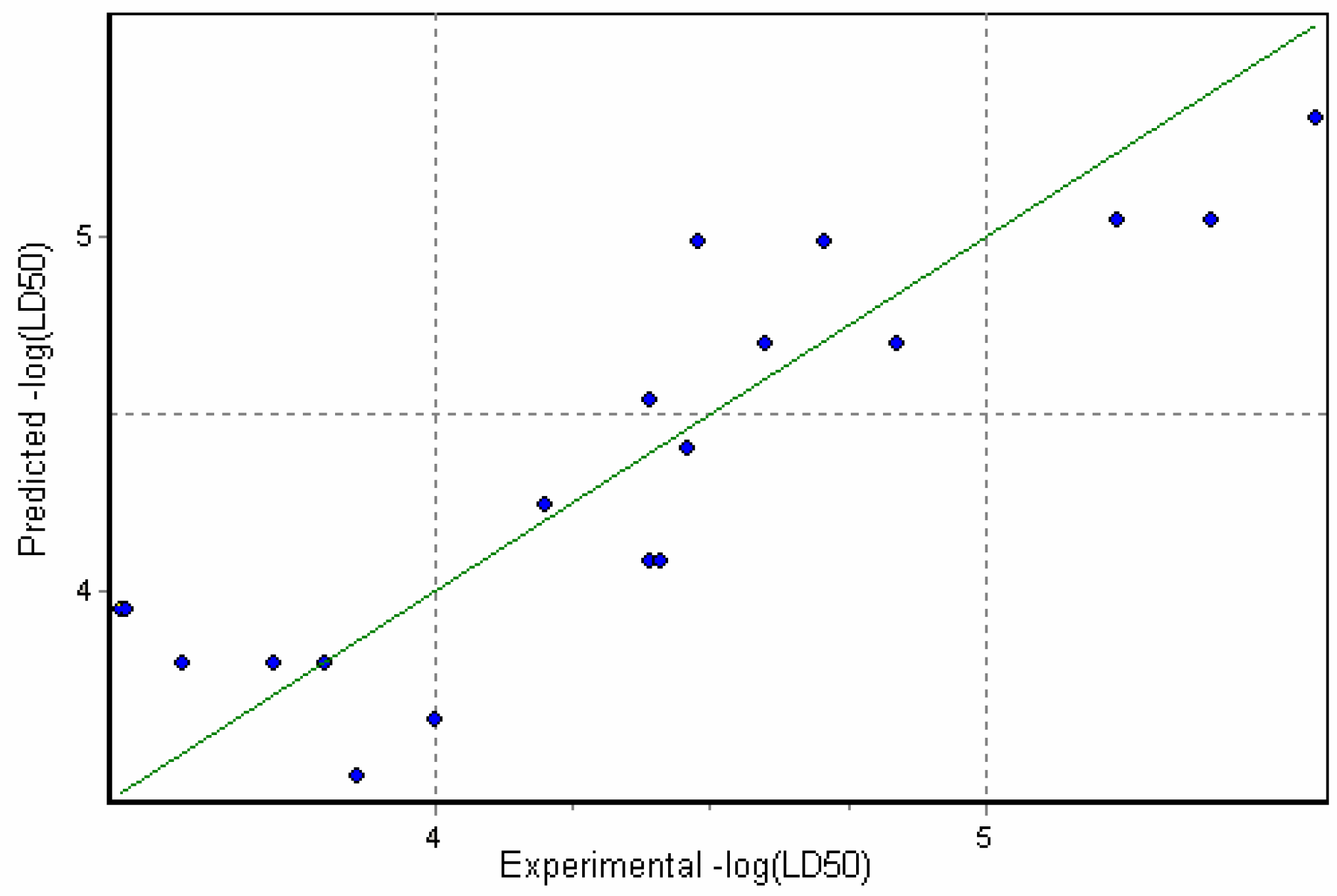
PhysChem bidescriptor model (C) (Figure 5):

n=19; r=0.94; r2adj=0.87; s=0.23; F=61.79; Q2=0.84; SPRESS=0.27; P=0.000000
Figure 5.
Correlation of Predicted versus Experimental toxicities for model C (Eqn.6)

PhysChem bidescriptor model (D) (Figure 6):

n=19; r=0.94; r2adj=0.87; s=0.23; F=61.06; Q2=0.83; SPRESS=0.28; P+0.000000
Figure 6.
Correlation of Predicted versus Experimental toxicities for model D (Eqn.7)
Despite the fact that nHAcc (number of acceptor atoms N, O, F) is collinear with TPSA, we have applied the latter descriptor as it gives the better models and describes both electron-donor and electron-acceptor properties of the molecule. Next, as can be seen from Table 1, two lipophilicity descriptors have been calculated for the alkaloids: MlogP (DRAGON program code) and LogP (HyperChem program code). However, the second octanol-water partition coefficient has not been considered in this study for two reasons: firstly, it has higher collinearity with the descriptor MW (Molecular Weight) and therefore should be avoided; secondly, it results in less significant models when combined with either TPSA or nC=O descriptors. GA-MLRA QSARs generation procedure has resulted in a number of statistically equivalent combinations of TPSA and nC=O with the other physicochemical descriptors. However, these models are not presented in this paper as the authors consider them being unable to reflect a true relationship between toxicity and physicochemical properties. It is also interesting to note, that both descriptors chosen (TPSA and nC=O) are highly collinear with the descriptor MW that results in statistically equal two-descriptor model when combined with MlogP (Eqn. 8):
PhysChem bidescriptor model (E):

n=19; r=0.92; r2adj=0.83; s=0.27; F=44.20; Q2=0.78; SPRESS=0.32; P=0.000000
However, the QSAR model built does not have a real value or meaning as it cannot explain the toxicity mechanism of action of the alkaloids studied (Eqn. 8). As shown by this model, alkaloids of higher molecular weight are more toxic. Nevertheless it is important to clarify the real cause of the phenomena observed. Thus, according to classical theory, an increase in MW due to the bulky part of the molecule (alkyl groups or aromatic rings) should result in a lipophilicity increase, which is not observed with the present series of compounds. Also, as can be seen from the descriptor correlation table (Table 1), the MW descriptor is not collinear with MlogP but with nHAcc, TPSA, and nC=O. Therefore, it can be concluded that the major factor influencing the toxicity of alkaloids is the number of electron donor (H-bonding) atoms or substituents, rather than lipophilicity.
In the recently reported QSAR analysis of 12 Aconitum alkaloids [7], the authors have identified a number of structural characteristics, chemical manipulation of which might result in both an increase of analgesic potency and a limitation of toxicity. Mono-parameter equations consisting of reactivity index, heat of formation, total, electronic and steric energies, molecular weight and core-core repulsion were selected as statistically significant ones. Two-descriptor models have not been proposed because of the considerable interrelation of the variables studied. For the same reason, the authors of the present paper are not presenting the QSAR models containing three descriptors due to a good chance of getting two highly collinear parameters in one equation.
Keeping in mind that realistically r2 values may be as low as the range of 0.6 - 0.7 [22], all the QSAR models obtained in this study are of satisfactory statistical fit with acceptable correlation coefficients, and with great significance level of each equation. The ability of each model to make predictions has been evaluated by obtaining cross-validation coefficients. Close values of both adjusted for the degrees of freedom correlation coefficients and cross-validation coefficients confirm a good predictivity of QSARs, especially for the ones containing two descriptors.
Conclusions
To summarize, 19 Aconitum and Delphinium alkaloids with the lycoctonine skeleton have been analyzed applying a QSAR approach. The best models selected and studied in this paper are the ones containing 3D descriptors (RDF020u, Mor28e, Mor07p) and physicochemical descriptors (ТPSA, MlogP, nC=O). The major difference between the two types of models is the better statistical fit of the 3D-descriptor containing models. It is worth mentioning that the exclusion of the deviant compound does not lead to the notable model improvement in both 3D- and physicochemical descriptors containing models. QSARs consisting of physicochemical descriptors confirm the mode of toxic action of lycoctonine-type alkaloids that interact with the sodium ion channel. Thus, a correlation observed between TPSA (determined as a Van der Waals area of electron donor and electron acceptor atoms) and toxicity demonstrates an importance of H-bonding capability of each alkaloid for binding to the bioreceptor. The descriptor most related to toxicity is the number of nC=O fragments in each molecule, which also describes the H-bonding formation capacity of the molecule. It has also been shown that toxicity increases with the number of atoms participating in hydrogen bonding and not due to the increase of bulky part of the molecules. This confirms experimental results suggesting Aconitum and Delphinium alkaloids interact with specific receptor sites in the sodium ion channel and therefore, hydrogen bonds playing an important role in their binding processes.
Acknowledgments
This work was supported by a grant from Science and Technology Committee of Republic of Uzbekistan (F № 3.8). Author M.A.T. would like to thank Chemistry School of Birmingham University (United Kingdom) for providing informational support. Both authors also would like to thank Dr. E.Y. Cheung for proofreading the manuscript and helpful comments.
References and Notes
- Ameri, A. Prog. Neurobiol. 1998, 56, 211.
- Dzhakhangirov, F. N.; Sultankhodzhaev, M. N.; Tashkhodzhaev, B.; Salimov, B. T. Chem. Nat. Comp. 1997, 33, 190. [CrossRef]
- Zhu, D. Y.; Bai, D.L.; Tang, X.C. Drug Dev. Res. 1996, 39, 147.
- Heubach, J.F.; Schule, A. Planta Med. 1998, 64, 22.
- Panter, K.E.; Manners, G.D.; Stegelmeier, B.L.; Lee, S.; Gardner, D.R.; Ralphs, M.H.; Pfister, J.A.; James, L.F. Biochem. Syst. Ecol. 2002, 30, 113. [CrossRef]
- Siemion, R.S.; Raisbek, M.F.; Waggoner, J.W.; Tidwell, M.A.; Sanchez, D.A. Vet. Human Toxicol. 1992, 34, 206.
- Bello-Ramirez, A.M.; Buendia-Orozco, J.; Nava-Ocampo, A.A. Fundam. Clin. Pharmacol. 2003, 17, 575.
- Fujita, T. QSAR and Drug Design; New Developments and Applications; Elsevier: Amsterdam, (The Netherlands), 1995. [Google Scholar]
- Lee, D. L.; Kollman, P. A.; Marsh, F. J.; Wolff, M. E. J. Med. Chem. 1977, 20, 1139. [CrossRef]
- Kubinyi, H. 3D QSAR in Drug Design: Theory, Methods and Applications; ESCOM: Amsterdam, (The Netherlands), 1993. [Google Scholar]
- Zhou, Y.-X.; Xu, L.; Wu, Y.-P.; Liu, B.-L. J. Chem. Intell. Lab. Sys. 1999, 45, 95. [CrossRef]
- Hou, T. J.; Wang, J. M.; Liao, N.; Xu, X. J. J. Chem. Inf. Comput. Sci. 1999, 39, 775. [CrossRef]
- Davis, L. Handbook of Genetic Algorithms; Van Nostrand Reinhold: New York (USA), 1991. [Google Scholar]
- Devillers, J. Genetic Algorithms in Molecular Modeling; Academic Press Ltd.: London, (U.K.), 1996. [Google Scholar]
- Dzhakhangirov, F. N.; Salimov, B. T.; Bessonova, I. A.; Sultankhodzhaev, M. N. Chem. Nat.Comp. 1995, 31, 708. [CrossRef]
- HyperChem 6.01 (HyperCube, Inc), 2000
- Young, D. Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems; John-Wiley & Sons: New York (USA), 2001. [Google Scholar]
- Todeschini, R.; Consonni, V. DRAGON software for the Calculation of Molecular Descriptors, Web version 3.0 for Windows.
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Mannhold, R., Kubinyi, H., Timmerman, H., Eds.; Wiley-VCH: Weinheim, New York, 2000. [Google Scholar]
- de Oliveira, D.B.; Gaudio, A.C. Quant. Struct.-Act. Relat. 2000, 19, 599.
- Statistical analysis software NCSS98, trial version, 1998
- Cronin, M.T.D.; Schultz, T. W. J. Mol. Struct. (Theochem) 2003, 622, 39. [CrossRef]
- Schuur, J.H.; Selzer, P.; Gasteiger, J. J. Chem. Inf. Comput. Sci. 1996, 36, 334. [CrossRef]
- Hemmer, M.C.; Steinhauer, V.; Gasteiger, J. Vibrat. Spectr. 1999, 19, 151.
- Ertl, P.; Rohde, B.; Selzer, P. J. Med. Chem. 2000, 43, 3714. [CrossRef]
- Kelder, J.; Grootenhuis, P.D.J.; Bayada, D.M.; Delbressine, L.P.C.; Ploemen, J-P. Pharm. Res. 1999, 16, 1514. [CrossRef] [PubMed]
- Clark, D.E. J. Pharm. Sci. 1999, 88, 815. [CrossRef]
- Clark, D.E. J. Pharm. Sci. 1999, 88, 807. [CrossRef]
- Aronov, A. M.; Goldman, B. B. Bioorg. Med. Chem. 2004, 12, 2307.
- Kutzenko, S.A. Osnovy Toxikologii (Fundamentals of Toxicology) Sankt-Petersburg. 2002. (electronic version at www.medline.ru/monograf/toxicology).
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Turabekova, M.A.; Rasulev, B.F. A QSAR Toxicity Study of a Series of Alkaloids with the Lycoctonine Skeleton. Molecules 2004, 9, 1194-1207. https://0-doi-org.brum.beds.ac.uk/10.3390/91201194
AMA Style
Turabekova MA, Rasulev BF. A QSAR Toxicity Study of a Series of Alkaloids with the Lycoctonine Skeleton. Molecules. 2004; 9(12):1194-1207. https://0-doi-org.brum.beds.ac.uk/10.3390/91201194
Chicago/Turabian StyleTurabekova, Malakhat A., and Bakhtiyor F. Rasulev. 2004. "A QSAR Toxicity Study of a Series of Alkaloids with the Lycoctonine Skeleton" Molecules 9, no. 12: 1194-1207. https://0-doi-org.brum.beds.ac.uk/10.3390/91201194