Molecular Motions in Functional Self-Assembled Nanostructures


Abstract
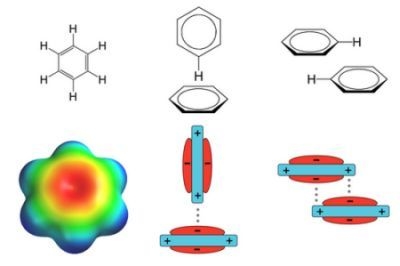
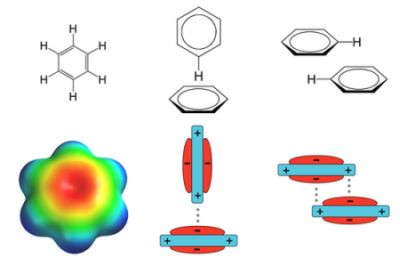
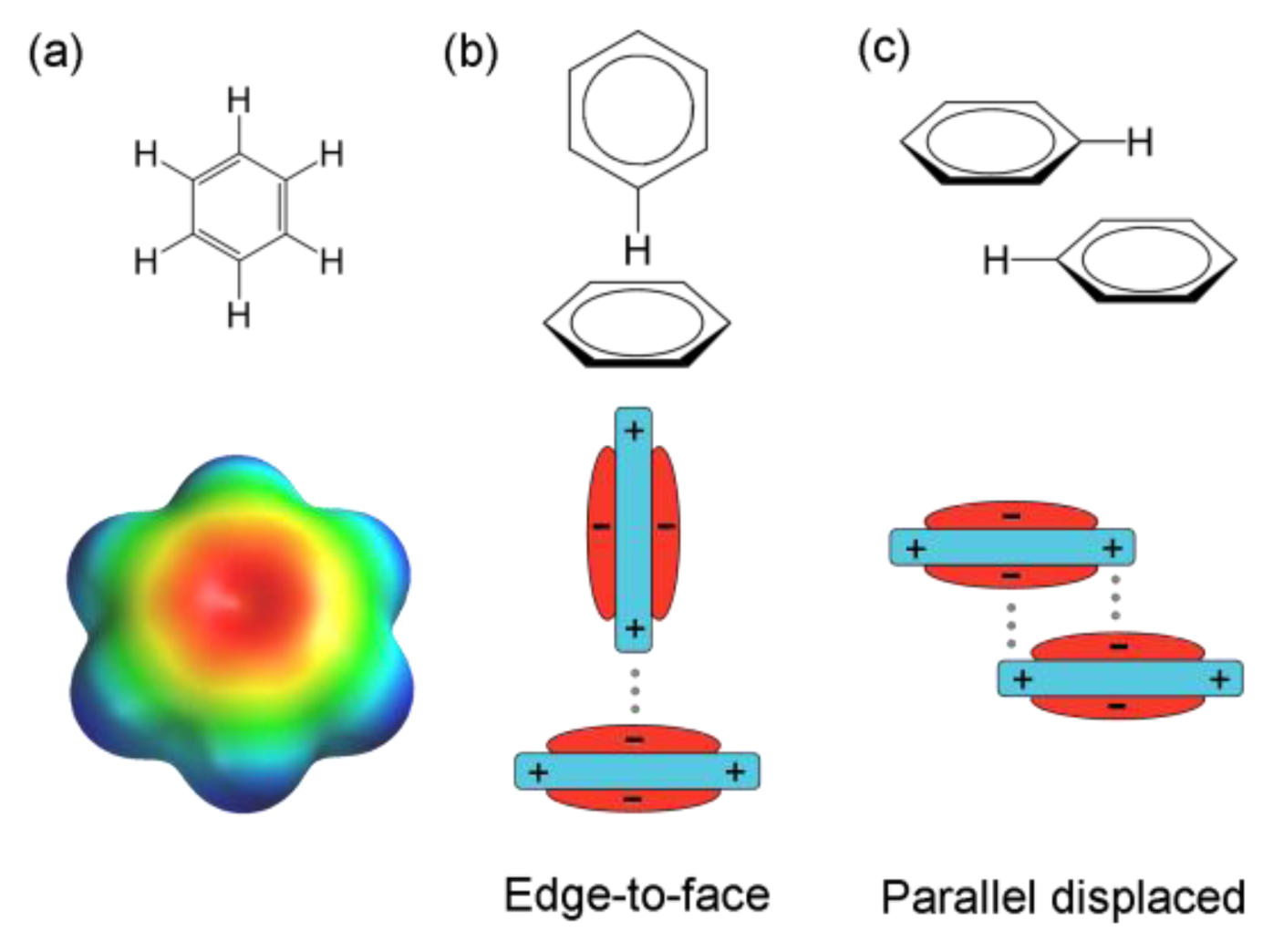
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Flexibility of Bonding in Self-Assembled Systems
2.1. Weak Interactions
2.2. Strong Bonding
3. Flexibility of Organic Nanostructures
3.1. Functional DNA Nanostructures
3.2. Stimuli-Responsive Polymer Systems
4. Flexibility of Organic-Inorganic Nanolayers
4.1. Functional Coatings
4.2. Stimuli-Responsive Nanolayers
5. Flexibility in Metal-Organic Frameworks
5.1. Gas Pressure Induced Breathing of MOFs
5.2. Exchange of Guest Molecules
5.3. External Stimuli
6. Conclusion
Acknowledgments
References
- Palmer, L.C.; Stupp, S.I. Molecular self-assembly into one-dimensional nanostructures. Acc. Chem. Res 2008, 41, 1674–1684. [Google Scholar]
- Capito, R.M.; Azevedo, H.S.; Velichko, Y.S.; Mata, A.; Stupp, S.I. Self-assembly of large and small molecules into hierarchically ordered sacs and membranes. Science 2008, 319, 1812–1816. [Google Scholar]
- Zubarev, E.R.; Pralle, M.U.; Sone, E.D.; Stupp, S.I. Self-assembly of dendron rodcoil molecules into nanoribbons. J. Am. Chem. Soc 2001, 123, 4105–4106. [Google Scholar]
- Jiyun, C.H. Guided molecular self-assembly: A review of recent efforts. Smart Mater. Struct 2003, 12, 264. [Google Scholar]
- Amigoni, S.; Taffin de Givenchy, E.; Dufay, M.; Guittard, F. Covalent layer-by-layer assembled superhydrophobic organic-inorganic hybrid films. Langmuir 2009, 25, 11073–11077. [Google Scholar]
- Minari, T.; Liu, C.; Kano, M.; Tsukagoshi, K. Controlled self-assembly of organic semiconductors for solution-based fabrication of organic field-effect transistors. Adv. Mater 2012, 24, 299–306. [Google Scholar]
- Halik, M.; Hirsch, A. The potential of molecular self-assembled monolayers in organic electronic devices. Adv. Mater 2011, 23, 2689–2695. [Google Scholar]
- Xie, G.; Sun, P.; Yan, X.; Du, X.; Jiang, Y. Fabrication of methane gas sensor by layer-by-layer self-assembly of polyaniline/pdo ultra thin films on quartz crystal microbalance. Sens. Actuators B 2010, 145, 373–377. [Google Scholar]
- Bussetti, G.; Violante, A.; Yivlialin, R.; Cirilli, S.; Bonanni, B.; Chiaradia, P.; Goletti, C.; Tortora, L.; Paolesse, R.; Martinelli, E.; et al. Site-sensitive gas sensing and analyte discrimination in langmuir–blodgett porphyrin films. J. Phys. Chem. C 2011, 115, 8189–8194. [Google Scholar]
- Li, Y.-C.; Mannen, S.; Morgan, A.B.; Chang, S.; Yang, Y.-H.; Condon, B.; Grunlan, J.C. Intumescent all-polymer multilayer nanocoating capable of extinguishing flame on fabric. Adv. Mater 2011, 23, 3926–3931. [Google Scholar]
- Zhang, S. Fabrication of novel biomaterials through molecular self-assembly. Nat. Biotech 2003, 21, 1171–1178. [Google Scholar]
- Bishop, K.J.M.; Wilmer, C.E.; Soh, S.; Grzybowski, B.A. Nanoscale forces and their uses in self-assembly. Small 2009, 5, 1600–1630. [Google Scholar]
- Claessens, C.G.; Stoddart, J.F. π–π interactions in self-assembly. J. Phys. Org. Chem 1997, 10, 254–272. [Google Scholar]
- Krische, M.; Lehn, J.-M. The Utilization Of Persistent H-Bonding Motifs in the Self-Assembly of Supramolecular Architectures. In Molecular Self-Assembly Organic Versus Inorganic Approaches; Fuiita, M, Ed.; Springer: Berlin/Heidelberg, Germnay, 2000; Volume 96, pp. 3–29. [Google Scholar]
- Sherrington, D.C.; Taskinen, K.A. Self-assembly in synthetic macromolecular systems multiple hydrogen bonding interactions. Chem. Soc. Rev 2001, 30, 83–93. [Google Scholar]
- Whitesides, G.M.; Boncheva, M. Beyond molecules: Self-assembly of mesoscopic and macroscopic components. Proc. Natl. Acad. Sci. USA 2002, 99, 4769–4774. [Google Scholar]
- Ferrighi, L.; Pan, Y.; Grönbeck, H.; Hammer, B. Study of alkylthiolate self-assembled monolayers on Au(111) using a semilocal meta-gga density functional. J. Phys. Chem. C 2012, 116, 7374–7379. [Google Scholar]
- Vericat, C.; Vela, M.E.; Benitez, G.; Carro, P.; Salvarezza, R.C. Self-assembled monolayers of thiols and dithiols on gold: New challenges for a well-known system. Chem. Soc. Rev 2010, 39, 1805–1834. [Google Scholar]
- Ni, L.; Chemtob, A.; Croutxe-Barghorn, C.; Brendle, J.; Vidal, L.; Rigolet, S. Photoinduced synthesis and ordering of lamellar n-alkylsiloxane films. J. Mater. Chem 2012, 22, 643–652. [Google Scholar]
- Lerouge, F.; Cerveau, G.; Corriu, R.J.P. Supramolecular self-organization in non-crystalline hybrid organic-inorganic nanomaterials induced by van der waals interactions. New J. Chem 2006, 30, 1364–1376. [Google Scholar]
- Jiang, J.; Lima, O.V.; Pei, Y.; Zeng, X.C.; Tan, L.; Forsythe, E. Dipole-induced, thermally stable lamellar structure by polar aromatic silane. J. Am. Chem. Soc 2009, 131, 900–901. [Google Scholar]
- Dhotel, A.; Delbreilh, L.; Youssef, B.; Jiang, J.; Coquerel, G.; Saiter, J.-M.; Tan, L. Thermal growth of organic supramolecular crystals with screw dislocations. J. Therm. Anal. Calorim. 2012. [Google Scholar] [CrossRef]
- Gearba, R.I.; Lehmann, M.; Levin, J.; Ivanov, D.A.; Koch, M.H.J.; Barberá, J.; Debije, M.G.; Piris, J.; Geerts, Y.H. Tailoring discotic mesophases: Columnar order enforced with hydrogen bonds. Adv. Mater 2003, 15, 1614–1618. [Google Scholar]
- Deng, R.; Liu, S.; Li, J.; Liao, Y.; Tao, J.; Zhu, J. Mesoporous block copolymer nanoparticles with tailored structures by hydrogen-bonding-assisted self-assembly. Adv. Mater 2012, 24, 1889–1893. [Google Scholar]
- Kimizuka, N.; Kawasaki, T.; Kunitake, T. Self-organization of bilayer membranes from amphiphilic networks of complementary hydrogen bonds. J. Am. Chem. Soc 1993, 115, 4387–4388. [Google Scholar]
- Aakeroy, C.B.; Seddon, K.R. The hydrogen bond and crystal engineering. Chem. Soc. Rev 1993, 22, 397–407. [Google Scholar]
- Cordier, P.; Tournilhac, F.; Soulie-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451, 977–980. [Google Scholar]
- Schmidt, D.J.; Hammond, P.T. Electrochemically erasable hydrogen-bonded thin films. Chem. Comm 2010, 46, 7358–7360. [Google Scholar]
- Rose, G.D.; Fleming, P.J.; Banavar, J.R.; Maritan, A. A backbone-based theory of protein folding. Proc. Natl. Acad. Sci. USA 2006, 103, 16623–16633. [Google Scholar]
- Deechongkit, S.; Nguyen, H.; Powers, E.T.; Dawson, P.E.; Gruebele, M.; Kelly, J.W. Context-dependent contributions of backbone hydrogen bonding to [beta]-sheet folding energetics. Nature 2004, 430, 101–105. [Google Scholar]
- Mignon, P.; Loverix, S.; Steyaert, J.; Geerlings, P. Influence of the π–π interaction on the hydrogen bonding capacity of stacked DNA/rna bases. Nucleic Acids Res 2005, 33, 1779–1789. [Google Scholar]
- Yakovchuk, P.; Protozanova, E.; Frank-Kamenetskii, M.D. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res 2006, 34, 564–574. [Google Scholar]
- Kool, E.T. Hydrogen bonding, base stacking, and steric effects in DNA replication. Annu. Rev. Biophys. Biomol. Struct 2001, 30, 1–22. [Google Scholar]
- Van Oss, C.J.; Good, R.J.; Chaudhury, M.K. The role of van der waals forces and hydrogen bonds in “hydrophobic interactions” between biopolymers and low energy surfaces. J. Colloid Interface Sci 1986, 111, 378–390. [Google Scholar]
- Silverstein, T.P. The real reason why oil and water don’t mix. J. Chem. Educ 1998, 75, 116. [Google Scholar]
- Priimagi, A.; Cavallo, G.; Forni, A.; Gorynsztejn-Leben, M.; Kaivola, M.; Metrangolo, P.; Milani, R.; Shishido, A.; Pilati, T.; Resnati, G.; et al. Halogen bonding versus hydrogen bonding in driving self-assembly and performance of light-responsive supramolecular polymers. Adv. Funct. Mater. 2012, 22, 2572–2579. [Google Scholar]
- Hunter, C.A.; Sanders, J.K.M. The nature of .pi.-.pi. interactions. J. Am. Chem. Soc 1990, 112, 5525–5534. [Google Scholar]
- Eric, V.; Anslyn, D.A.D. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, USA, 2005. [Google Scholar]
- Salonen, L.M.; Ellermann, M.; Diederich, F. Aromatic rings in chemical and biological recognition: Energetics and structures. Angew. Chem. Int. Ed 2011, 50, 4808–4842. [Google Scholar]
- Martinez, C.R.; Iverson, B.L. Rethinking the term “pi-stacking”. Chem. Sci 2012, 3, 2191–2201. [Google Scholar]
- Tiekink, E.R.T.; Zukerman-Schpector, J. The Importance Of Pi-Interactions In Crystal Engineering: Frontiers in Crystal Engineering, 2nd Ed. ed; Wiley: Chichester, UK, 2012. [Google Scholar]
- Grimme, S. Do special noncovalent π–π stacking interactions really exist? Angew. Chem. Int. Ed 2008, 47, 3430–3434. [Google Scholar]
- Beaujuge, P.M.; Fréchet, J.M.J. Molecular design and ordering effects in π-functional materials for transistor and solar cell applications. J. Am. Chem. Soc 2011, 133, 20009–20029. [Google Scholar]
- Jiang, J.; Lima, O.V.; Pei, Y.; Jiang, Z.; Chen, Z.; Yu, C.; Wang, J.; Zeng, X.C.; Forsythe, E.; Tan, L. Self-assembled nanolayers of conjugated silane with π–π interlocking. ACS Nano 2010, 4, 3773–3780. [Google Scholar]
- Desiraju, G.R.; Gavezzotti, A. From molecular to crystal structure; polynuclear aromatic hydrocarbons. J. Chem. Soc. Chem. Commun. 1989, 621–623. [Google Scholar]
- Pluth, M.D.; Raymond, K.N. Reversible guest exchange mechanisms in supramolecular host-guest assemblies. Chem. Soc. Rev 2007, 36, 161–171. [Google Scholar]
- Ferri, T.; Frasca, D.; Arias de Fuentes, O.; Santucci, R.; Frasconi, M. Spatially oriented and reversible surface assembly of single-walled carbon nanotubes: A strategy based on π–π interactions. Angew. Chem. Int. Ed 2011, 50, 7074–7078. [Google Scholar]
- Fasan, R.; Dias, R.L.A.; Moehle, K.; Zerbe, O.; Obrecht, D.; Mittl, P.R.E.; Grütter, M.G.; Robinson, J.A. Structure–activity studies in a family of β-hairpin protein epitope mimetic inhibitors of the p53–hdm2 protein–protein interaction. ChemBioChem 2006, 7, 515–526. [Google Scholar]
- Matta, C.F.; Castillo, N.; Boyd, R.J. Extended weak bonding interactions in DNA: π-stacking (base–base), base–backbone, and backbone–backbone interactions. J. Phys. Chem. B 2005, 110, 563–578. [Google Scholar]
- Altman, M.; Lee, P.; Rich, A.; Zhang, S. Conformational behavior of ionic self-complementary peptides. Protein Sci 2000, 9, 1095–1105. [Google Scholar]
- Zhao, X. Design of self-assembling surfactant-like peptides and their applications. Curr. Opin. Colloid Interface Sci 2009, 14, 340–348. [Google Scholar]
- Antonietti, M.; Burger, C.; Effing, J. Mesomorphous polyelectrolyte-surfactant complexes. Adv. Mater 1995, 7, 751–753. [Google Scholar]
- Franke, D.; Vos, M.; Antonietti, M.; Sommerdijk, N.A.J.M.; Faul, C.F.J. Induced supramolecular chirality in nanostructured materials: Ionic self-assembly of perylene-chiral surfactant complexes. Chem. Mater 2006, 18, 1839–1847. [Google Scholar]
- Antonietti, M.; Wenzel, A.; Thünemann, A. The “egg-carton” phase: A new morphology of complexes of polyelectrolytes with natural lipid mixtures. Langmuir 1996, 12, 2111–2114. [Google Scholar]
- Decher, G.; Hong, J.D.; Schmitt, J. Buildup of ultrathin multilayer films by a self-assembly process: III. Consecutively alternating adsorption of anionic and cationic polyelectrolytes on charged surfaces. Thin Solid Films 1992, 210–211, 831–835. [Google Scholar]
- Tauk, L.; Schroder, A.P.; Decher, G.; Giuseppone, N. Hierarchical functional gradients of ph-responsive self-assembled monolayers using dynamic covalent chemistry on surfaces. Nat. Chem 2009, 1, 739–739. [Google Scholar]
- Minkenberg, C.B.; Florusse, L.; Eelkema, R.; Koper, G.J.M.; van Esch, J.H. Triggered self-assembly of simple dynamic covalent surfactants. J. Am. Chem. Soc 2009, 131, 11274–11275. [Google Scholar]
- Liu, X.; Jiang, M. Optical switching of self-assembly: Micellization and micelle–hollow-sphere transition of hydrogen-bonded polymers. Angew. Chem. Int. Ed 2006, 45, 3846–3850. [Google Scholar]
- Yang, Y.; Yue, L.; Li, H.; Maher, E.; Li, Y.; Wang, Y.; Wu, L.; Yam, V.W.-W. Photo-responsive self-assembly of an azobenzene-ended surfactant-encapsulated polyoxometalate complex for modulating catalytic reactions. Small 2012, 8, 3105–3110. [Google Scholar]
- Liu, N.; Chen, Z.; Dunphy, D.R.; Jiang, Y.-B.; Assink, R.A.; Brinker, C.J. Photoresponsive nanocomposite formed by self-assembly of an azobenzene-modified silane. Angew. Chem. Int. Ed 2003, 42, 1731–1734. [Google Scholar]
- Lim, H.S.; Han, J.T.; Kwak, D.; Jin, M.; Cho, K. Photoreversibly switchable superhydrophobic surface with erasable and rewritable pattern. J. Am. Chem. Soc 2006, 128, 14458–14459. [Google Scholar]
- Gianneschi, N.C.; Masar, M.S.; Mirkin, C.A. Development of a coordination chemistry-based approach for functional supramolecular structures. Acc. Chem. Res 2005, 38, 825–837. [Google Scholar]
- Yoshizawa, M.; Fujita, M. Development of unique chemical phenomena within nanometer-sized, self-assembled coordination hosts. Bull. Chem. Soc. Jpn 2010, 83, 609–618. [Google Scholar]
- Burnett, B.J.; Choe, W. Sequential self-assembly in metal-organic frameworks. Dalton Trans 2012, 41, 3889–3894. [Google Scholar]
- Chakrabarty, R.; Mukherjee, P.S.; Stang, P.J. Supramolecular coordination: Self-assembly of finite two- and three-dimensional ensembles. Chem. Rev 2011, 111, 6810–6918. [Google Scholar]
- Swiegers, G.F.; Malefetse, T.J. Classification of coordination polygons and polyhedra according to their mode of self-assembly. 2. Review of the literature. Coord. Chem. Rev 2002, 225, 91–121. [Google Scholar]
- O’Mahony, C.T.; Farrell, R.A.; Goshal, T.; Holmes, J.D.; Morris, M.A. The thermodynamics of defect formation in self-assembled systems. In Thermodynamics—Systems in Equilibrium and Non-Equilibrium; Moreno-Piraján, J.C., Ed.; InTech: Rijeka, Croatia, 2011; pp. 279–306. [Google Scholar]
- Nakamura, I.; Shi, A.-C. Study of entropy-driven self-assembly of rigid macromolecules. Phys. Rev. E 2009, 80, 021112. [Google Scholar]
- Patra, N.; Wang, B.; Král, P. Nanodroplet activated and guided folding of graphene nanostructures. Nano Lett 2009, 9, 3766–3771. [Google Scholar]
- Bellido, E.P.; Seminario, J.M. Molecular dynamics simulations of folding of supported graphene. J. Phys. Chem. C 2010, 114, 22472–22477. [Google Scholar]
- Chen, Y.; Guo, F.; Jachak, A.; Kim, S.-P.; Datta, D.; Liu, J.; Kulaots, I.; Vaslet, C.; Jang, H.D.; Huang, J.; et al. Aerosol synthesis of cargo-filled graphene nanosacks. Nano Lett. 2012, 12, 1996–2002. [Google Scholar]
- Park, S.H.; Yan, H.; Reif, J.H.; LaBean, T.H.; Finkelstein, G. Electronic nanostructures templated on self-assembled DNA scaffolds. Nanotechnology 2004, 15, S525–S527. [Google Scholar]
- Rinker, S.; Ke, Y.G.; Liu, Y.; Chhabra, R.; Yan, H. Self-assembled DNA nanostructures for distance-dependent multivalent ligand-protein binding. Nat. Nanotechnol 2008, 3, 418–422. [Google Scholar]
- Alivisatos, A.P.; Johnsson, K.P.; Peng, X.G.; Wilson, T.E.; Loweth, C.J.; Bruchez, M.P.; Schultz, P.G. Organization of “nanocrystal molecules” using DNA. Nature 1996, 382, 609–611. [Google Scholar]
- Seeman, N.C. DNA in a material world. Nature 2003, 421, 427–431. [Google Scholar]
- Hamley, I.W. Nanostructure fabrication using block copolymers. Nanotechnology 2003, 14, R39–R54. [Google Scholar]
- Fasolka, M.J.; Mayes, A.M. Block copolymer thin films: Physics and applications. Annu. Rev. Mater. Res 2001, 31, 323–355. [Google Scholar]
- Morikawa, Y.; Nagano, S.; Watanabe, K.; Kamata, K.; Iyoda, T.; Seki, T. Optical alignment and patterning of nanoscale microdomains in a block copolymer thin film. Adv. Mater 2006, 18, 883–886. [Google Scholar]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar]
- Douglas, S.M.; Marblestone, A.H.; Teerapittayanon, S.; Vazquez, A.; Church, G.M.; Shih, W.M. Rapid prototyping of 3D DNA-origami shapes with cadnano. Nucleic Acids Res 2009, 37, 5001–5006. [Google Scholar]
- Douglas, S.M.; Dietz, H.; Liedl, T.; Hogberg, B.; Graf, F.; Shih, W.M. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 2009, 459, 414–418. [Google Scholar]
- Dietz, H.; Douglas, S.M.; Shih, W.M. Folding DNA into twisted and curved nanoscale shapes. Science 2009, 325, 725–730. [Google Scholar]
- Goodman, R.P.; Schaap, I.A.T.; Tardin, C.F.; Erben, C.M.; Berry, R.M.; Schmidt, C.F.; Turberfield, A.J. Rapid chiral assembly of rigid DNA building blocks for molecular nanofabrication. Science 2005, 310, 1661–1665. [Google Scholar]
- Seeman, N.C. An overview of structural DNA nanotechnology. Mol. Biotechnol 2007, 37, 246–257. [Google Scholar]
- Bath, J.; Turberfield, A.J. DNA nanomachines. Nat. Nanotechnol 2007, 2, 275–284. [Google Scholar]
- Goodman, R.P.; Heilemann, M.; Doose, S.; Erben, C.M.; Kapanidis, A.N.; Turberfield, A.J. Reconfigurable, braced, three-dimensional DNA nanostructures. Nat. Nanotechnol 2008, 3, 93–96. [Google Scholar]
- Zhang, D.Y.; Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem 2011, 3, 103–113. [Google Scholar]
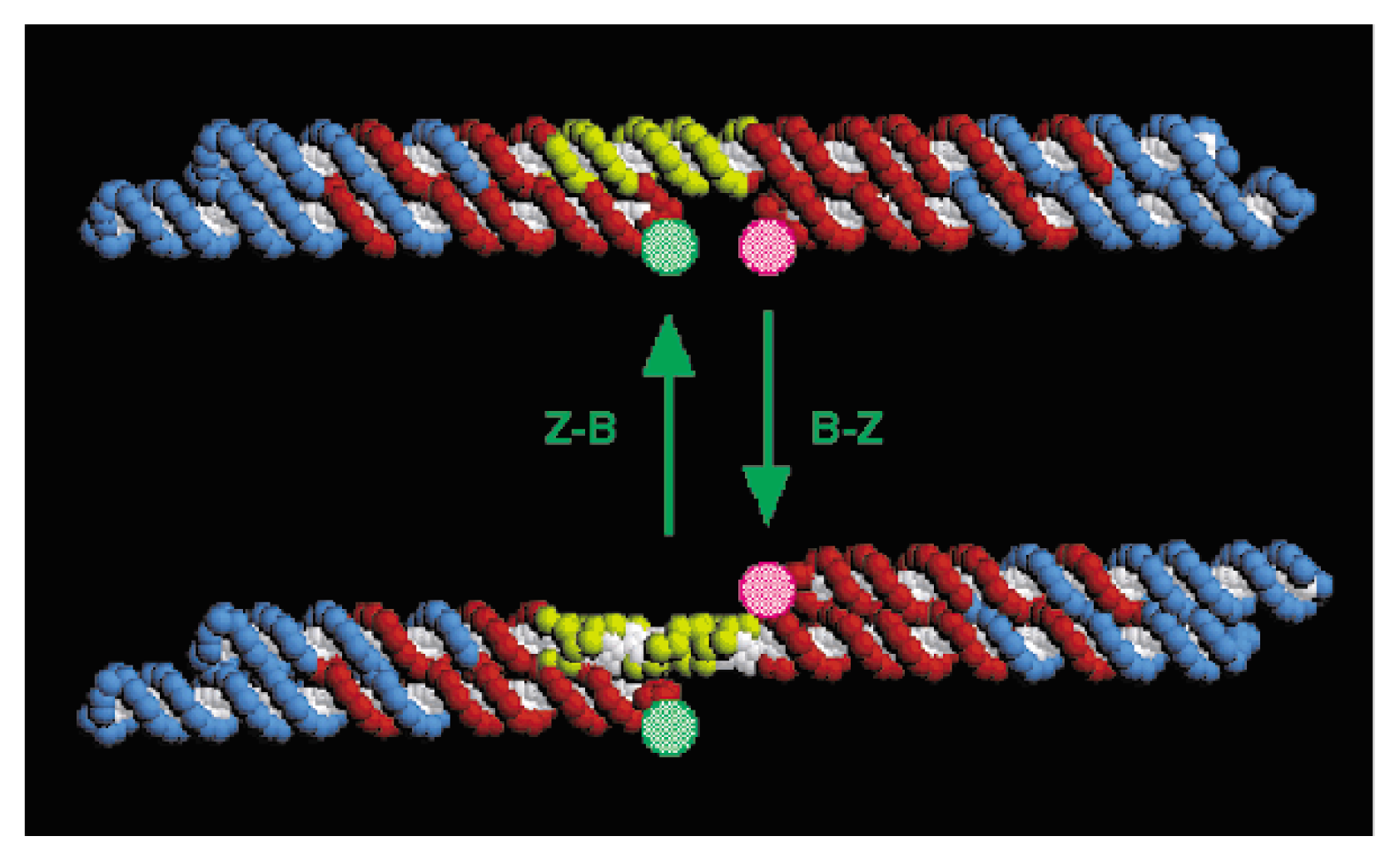
- Mao, C.D.; Sun, W.Q.; Shen, Z.Y.; Seeman, N.C. A nanomechanical device based on the B-Z transition of DNA. Nature 1999, 397, 144–146. [Google Scholar]
- Yurke, B.; Turberfield, A.J.; Mills, A.P.; Simmel, F.C.; Neumann, J.L. A DNA-fuelled molecular machine made of DNA. Nature 2000, 406, 605–608. [Google Scholar]
- Wang, S.T.; Liu, H.J.; Liu, D.S.; Ma, X.Y.; Fang, X.H.; Jiang, L. Enthalpy-driven three-state switching of a superhydrophilic/superhydrophobic surface. Angew. Chem. Int. Ed 2007, 46, 3915–3917. [Google Scholar]
- Yan, H.; Zhang, X.P.; Shen, Z.Y.; Seeman, N.C. A robust DNA mechanical device controlled by hybridization topology. Nature 2002, 415, 62–65. [Google Scholar]
- Feng, L.P.; Park, S.H.; Reif, J.H.; Yan, H. A two-state DNA lattice switched by DNA nanoactuator. Angew. Chem. Int. Ed 2003, 42, 4342–4346. [Google Scholar]
- Robinson, B.H.; Seeman, N.C. The design of a biochip—A self-assembling molecular-scale memory device. Protein Eng 1987, 1, 295–300. [Google Scholar]
- Keren, K.; Krueger, M.; Gilad, R.; Ben-Yoseph, G.; Sivan, U.; Braun, E. Sequence-specific molecular lithography on single DNA molecules. Science 2002, 297, 72–75. [Google Scholar]
- Heilemann, M.; Tinnefeld, P.; Mosteiro, G.S.; Garcia-Parajo, M.; van Hulst, N.F.; Sauer, M. Multistep energy transfer in single molecular photonic wires. J. Am. Chem. Soc 2004, 126, 6514–6515. [Google Scholar]
- Douglas, S.M.; Bachelet, I.; Church, G.M. A logic-gated nanorobot for targeted transport of molecular payloads. Science 2012, 335, 831–834. [Google Scholar]
- Andersen, E.S.; Dong, M.; Nielsen, M.M.; Jahn, K.; Subramani, R.; Mamdouh, W.; Golas, M.M.; Sander, B.; Stark, H.; Oliveira, C.L.P.; et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature 2009, 459, 73–76. [Google Scholar]
- Von Delius, M.; Leigh, D.A. Walking molecules. Chem. Soc. Rev 2011, 40, 3656–3676. [Google Scholar]
- Sherman, W.B.; Seeman, N.C. A precisely controlled DNA biped walking device. Nano Lett 2004, 4, 1203–1207. [Google Scholar]
- He, Y.; Liu, D.R. Autonomous multistep organic synthesis in a single isothermal solution mediated by a DNA walker. Nat. Nanotechnol 2010, 5, 778–782. [Google Scholar]
- Tian, Y.; He, Y.; Chen, Y.; Yin, P.; Mao, C.D. Molecular devices—A dnazyme that walks processively and autonomously along a one-dimensional track. Angew. Chem. Int. Ed 2005, 44, 4355–4358. [Google Scholar]
- Bath, J.; Green, S.J.; Turberfield, A.J. A free-running DNA motor powered by a nicking enzyme. Angew. Chem. Int. Ed 2005, 44, 4358–4361. [Google Scholar]
- Lund, K.; Manzo, A.J.; Dabby, N.; Michelotti, N.; Johnson-Buck, A.; Nangreave, J.; Taylor, S.; Pei, R.J.; Stojanovic, M.N.; Walter, N.G.; et al. Molecular robots guided by prescriptive landscapes. Nature 2010, 465, 206–210. [Google Scholar]
- Wang, C.Y.; Tao, Y.; Song, G.T.; Ren, J.S.; Qu, X.G. Speeding up a bidirectional DNA walking device. Langmuir 2012, 28, 14829–14837. [Google Scholar]
- Liu, D.S.; Balasubramanian, S. A proton-fuelled DNA nanomachine. Angew. Chem. Int. Ed 2003, 42, 5734–5736. [Google Scholar]
- Shu, W.M.; Liu, D.S.; Watari, M.; Riener, C.K.; Strunz, T.; Welland, M.E.; Balasubramanian, S.; McKendry, R.A. DNA molecular motor driven micromechanical cantilever arrays. J. Am. Chem. Soc 2005, 127, 17054–17060. [Google Scholar]
- Liu, D.S.; Bruckbauer, A.; Abell, C.; Balasubramanian, S.; Kang, D.J.; Klenerman, D.; Zhou, D.J. A reversible ph-driven DNA nanoswitch array. J. Am. Chem. Soc 2006, 128, 2067–2071. [Google Scholar]
- Arora, H.; Du, P.; Tan, K.W.; Hyun, J.K.; Grazul, J.; Xin, H.L.; Muller, D.A.; Thompson, M.O.; Wiesner, U. Block copolymer self-assembly-directed single-crystal homo- and heteroepitaxial nanostructures. Science 2010, 330, 214–219. [Google Scholar]
- Bates, F.S.; Fredrickson, G.H. Block copolymer thermodynamics—theory and experiment. Annu. Rev. Phys. Chem 1990, 41, 525–557. [Google Scholar]
- Cui, H.G.; Chen, Z.Y.; Zhong, S.; Wooley, K.L.; Pochan, D.J. Block copolymer assembly via kinetic control. Science 2007, 317, 647–650. [Google Scholar]
- Stoykovich, M.P.; Muller, M.; Kim, S.O.; Solak, H.H.; Edwards, E.W.; de Pablo, J.J.; Nealey, P.F. Directed assembly of block copolymer blends into nonregular device-oriented structures. Science 2005, 308, 1442–1446. [Google Scholar]
- Zhao, D.Y.; Feng, J.L.; Huo, Q.S.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 1998, 279, 548–552. [Google Scholar]
- Zhao, D.Y.; Huo, Q.S.; Feng, J.L.; Chmelka, B.F.; Stucky, G.D. Nonionic triblock and star diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J. Am. Chem. Soc 1998, 120, 6024–6036. [Google Scholar]
- Nie, Z.H.; Fava, D.; Kumacheva, E.; Zou, S.; Walker, G.C.; Rubinstein, M. Self-assembly of metal-polymer analogues of amphiphilic triblock copolymers. Nat. Mater 2007, 6, 609–614. [Google Scholar]
- Mertoglu, M.; Garnier, S.; Laschewsky, A.; Skrabania, K.; Storsberg, J. Stimuli responsive amphiphilic block copolymers for aqueous media synthesised via reversible addition fragmentation chain transfer polymerisation (raft). Polymer 2005, 46, 7726–7740. [Google Scholar]
- Nykanen, A.; Nuopponen, M.; Laukkanen, A.; Hirvonen, S.P.; Rytela, M.; Turunen, O.; Tenhu, H.; Mezzenga, R.; Ikkala, O.; Ruokolainen, J. Phase behavior and temperature-responsive molecular filters based on self-assembly of polystyrene-block-poly(n-isopropylacrylamide)-block-polystyrene. Macromolecules 2007, 40, 5827–5834. [Google Scholar]
- Tokarev, I.; Minko, S. Stimuli-responsive hydrogel thin films. Soft Matter 2009, 5, 511–524. [Google Scholar]
- Kumar, S.; Dory, Y.L.; Lepage, M.; Zhao, Y. Surface-grafted stimuli-responsive block copolymer brushes for the thermo-, photo- and ph-sensitive release of dye molecules. Macromolecules 2011, 44, 7385–7393. [Google Scholar]
- Stuart, M.A.C.; Huck, W.T.S.; Genzer, J.; Muller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar]
- Chu, L.Y.; Li, Y.; Zhu, J.H.; Chen, W.M. Negatively thermoresponsive membranes with functional gates driven by zipper-type hydrogen-bonding interactions. Angew. Chem. Int. Ed 2005, 44, 2124–2127. [Google Scholar]
- Lee, D.; Nolte, A.J.; Kunz, A.L.; Rubner, M.F.; Cohen, R.E. Ph-induced hysteretic gating of track-etched polycarbonate membranes: Swelling/deswelling behavior of polyelectrolyte multilayers in confined geometry. J. Am. Chem. Soc 2006, 128, 8521–8529. [Google Scholar]
- Song, Y.; Nair, R.; Zou, M.; Wang, Y. Superhydrophobic surfaces produced by applying a self-assembled monolayer to silicon micro/nano-textured surfaces. Nano Res 2009, 2, 143–150. [Google Scholar]
- Li, J.; Liu, X.; Ye, Y.; Zhou, H.; Chen, J. Fabrication of superhydrophobic cuo surfaces with tunable water adhesion. J. Phys. Chem. C 2011, 115, 4726–4729. [Google Scholar]
- Vuillaume, D.; Boulas, C.; Collet, J.; Davidovits, J.V.; Rondelez, F. Organic insulating films of nanometer thicknesses. Appl. Phys. Lett 1996, 69, 1646–1648. [Google Scholar]
- Fontaine, P.; Goguenheim, D.; Deresmes, D.; Vuillaume, D.; Garet, M.; Rondelez, F. Octadecyltrichlorosilane monolayers as ultrathin gate insulating films in metal-insulator-semiconductor devices. Appl. Phys. Lett 1993, 62, 2256–2258. [Google Scholar]
- Liu, J.; Hennek, J.W.; Buchholz, D.B.; Ha, Y.; Xie, S.; Dravid, V.P.; Chang, R.P.H.; Facchetti, A.; Marks, T.J. Reinforced self-assembled nanodielectrics for high-performance transparent thin film transistors. Adv. Mater 2011, 23, 992–997. [Google Scholar]
- Ha, Y.; Emery, J.D.; Bedzyk, M.J.; Usta, H.; Facchetti, A.; Marks, T.J. Solution-deposited organic–inorganic hybrid multilayer gate dielectrics. Design, synthesis, microstructures, and electrical properties with thin-film transistors. J. Am. Chem. Soc 2011, 133, 10239–10250. [Google Scholar]
- Schlitz, R.A.; Ha, Y.; Marks, T.J.; Lauhon, L.J. Quantitative statistical analysis of dielectric breakdown in zirconia-based self-assembled nanodielectrics. ACS Nano 2012, 6, 4452–4460. [Google Scholar]
- Boulas, C.; Davidovits, J.V.; Rondelez, F.; Vuillaume, D. Suppression of charge carrier tunneling through organic self-assembled monolayers. Phy. Rev. Lett 1996, 76, 4797–4800. [Google Scholar]
- Lee, H.S.; Kim, D.H.; Cho, J.H.; Hwang, M.; Jang, Y.; Cho, K. Effect of the phase states of self-assembled monolayers on pentacene growth and thin-film transistor characteristics. J. Am. Chem. Soc 2008, 130, 10556–10564. [Google Scholar]
- Walter, S.R.; Youn, J.; Emery, J.D.; Kewalramani, S.; Hennek, J.W.; Bedzyk, M.J.; Facchetti, A.; Marks, T.J.; Geiger, F.M. In-situ probe of gate dielectric-semiconductor interfacial order in organic transistors: Origin and control of large performance sensitivities. J. Am. Chem. Soc 2012, 134, 11726–11733. [Google Scholar]
- Dhotel, A.; Li, H.; Fernandez-Ballester, L.; Delbreilh, L.; Youssef, B.; Zeng, X.C.; Tan, L. Supramolecular nanolayer reconfiguration after molecular intercalation. J. Phys. Chem. C 2011, 115, 10351–10356. [Google Scholar]
- Yu, C.; Chen, Z.; Li, H.; Turner, J.; Zeng, X.C.; Jin, Z.; Jiang, J.; Youssef, B.; Tan, L. Molecularly intercalated nanoflakes: A supramolecular composite for strong energy absorption. Adv. Mater 2010, 22, 4457–4461. [Google Scholar]
- Morin, J.-F.; Shirai, Y.; Tour, J.M. En route to a motorized nanocar. Org. Lett 2006, 8, 1713–1716. [Google Scholar]
- Michl, J.; Sykes, E.C.H. Molecular rotors and motors: Recent advances and future challenges. ACS Nano 2009, 3, 1042–1048. [Google Scholar]
- Kottas, G.S.; Clarke, L.I.; Horinek, D.; Michl, J. Artificial molecular rotors. Chem. Rev 2005, 105, 1281–1376. [Google Scholar]
- Vogelsberg, C.S.; Bracco, S.; Beretta, M.; Comotti, A.; Sozzani, P.; Garcia-Garibay, M.A. Dynamics of molecular rotors confined in two dimensions: Transition from a 2D rotational glass to a 2D rotational fluid in a periodic mesoporous organosilica. J. Phys. Chem. B 2012, 116, 1623–1632. [Google Scholar]
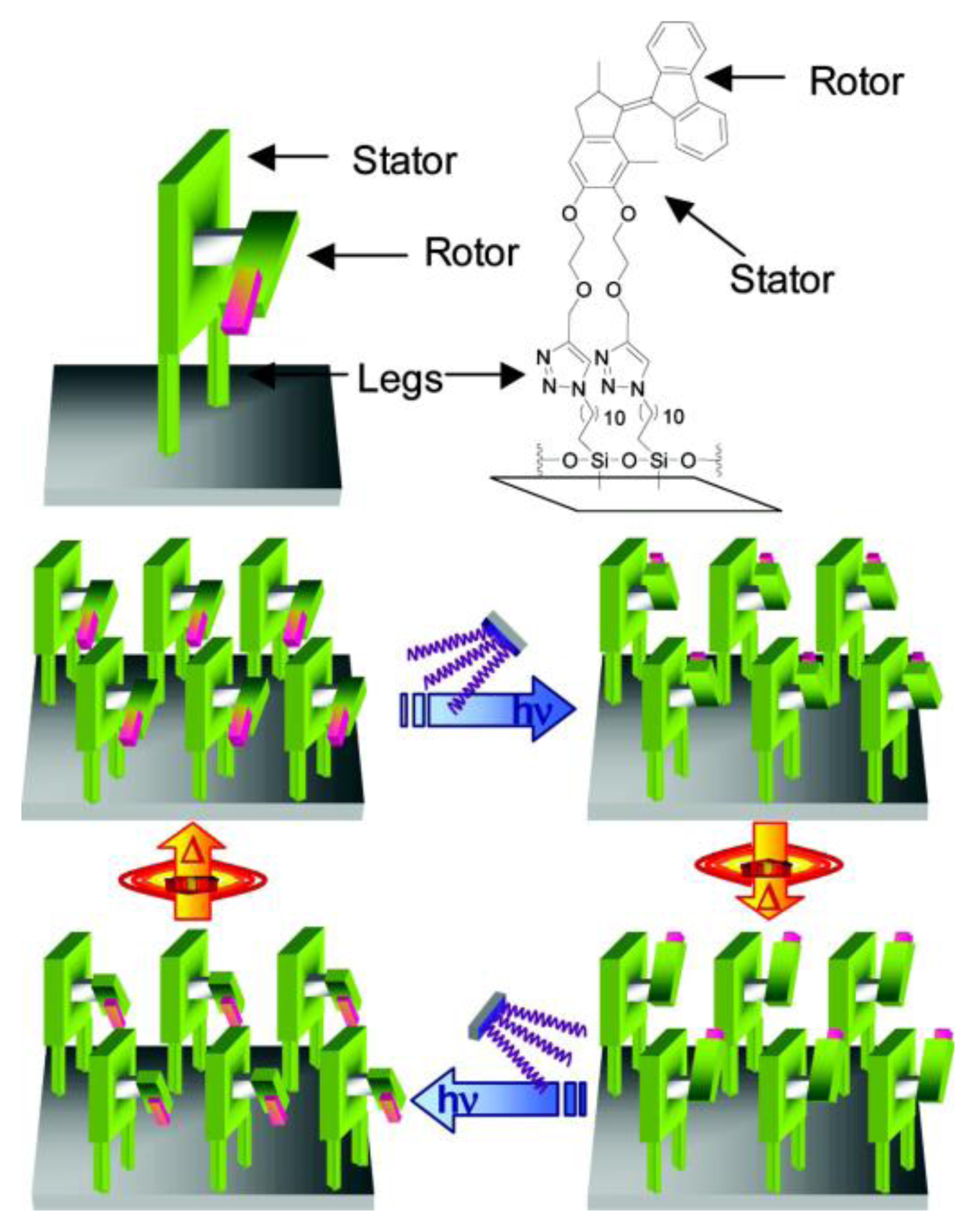
- Carroll, G.T.; London, G.; Landaluce, T.F.; Rudolf, P.; Feringa, B.L. Adhesion of photon-driven molecular motors to surfaces via 1,3-dipolar cycloadditions: Effect of interfacial interactions on molecular motion. ACS Nano 2011, 5, 622–630. [Google Scholar]
- Arcos-Ramos, R.; Rodríguez-Molina, B.; Romero, M.; Méndez-Stivalet, J.M.; Ochoa, M.E.; Ramírez-Montes, P.I.; Santillan, R.; Garcia-Garibay, M.A.; Farfán, N. Synthesis and evaluation of molecular rotors with large and bulky tert-butyldiphenylsilyloxy-substituted trityl stators. J. Org. Chem 2012, 77, 6887–6894. [Google Scholar]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev 2012, 41, 1809–1825. [Google Scholar]
- Beharry, A.A.; Woolley, G.A. Azobenzene photoswitches for biomolecules. Chem. Soc. Rev 2011, 40, 4422–4437. [Google Scholar]
- Yager, K.G.; Barrett, C.J. Azobenzene Polymers for Photonic Applications. In Smart Light-Responsive Materials; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2008; pp. 1–46. [Google Scholar]
- Wang, Y.; Cheng, H.-P. Electronic and transport properties of azobenzene monolayer junctions as molecular switches. Phys. Rev. B 2012, 86, 035444. [Google Scholar]
- Lee, S.; Kang, H.S.; Park, J.-K. Directional photofluidization lithography: Micro/nanostructural evolution by photofluidic motions of azobenzene materials. Adv. Mater 2012, 24, 2069–2103. [Google Scholar]
- Hu, J.; Meng, H.; Li, G.; Samuel, I.I. A review of stimuli-responsive polymers for smart textile applications. Smart Mater. Struct 2012, 21, 053001. [Google Scholar]
- Yuan, Q.; Zhang, Y.; Chen, T.; Lu, D.; Zhao, Z.; Zhang, X.; Li, Z.; Yan, C.-H.; Tan, W. Photon-manipulated drug release from a mesoporous nanocontainer controlled by azobenzene-modified nucleic acid. ACS Nano 2012, 6, 6337–6344. [Google Scholar]
- Wang, Y.; Xu, H.; Zhang, X. Tuning the amphiphilicity of building blocks: Controlled self-assembly and disassembly for functional supramolecular materials. Adv. Mater 2009, 21, 2849–2864. [Google Scholar]
- Wang, Y.; Han, P.; Xu, H.; Wang, Z.; Zhang, X.; Kabanov, A.V. Photocontrolled self-assembly and disassembly of block ionomer complex vesicles: A facile approach toward supramolecular polymer nanocontainers. Langmuir 2009, 26, 709–715. [Google Scholar]
- Mativetsky, J.M.; Pace, G.; Elbing, M.; Rampi, M.A.; Mayor, M.; Samorì, P. Azobenzenes as light-controlled molecular electronic switches in nanoscale metal–molecule–metal junctions. J. Am. Chem. Soc 2008, 130, 9192–9193. [Google Scholar]
- Crivillers, N.; Orgiu, E.; Reinders, F.; Mayor, M.; Samorì, P. Optical modulation of the charge injection in an organic field-effect transistor based on photochromic self-assembled-monolayer-functionalized electrodes. Adv. Mater 2011, 23, 1447–1452. [Google Scholar]
- Song, H.; Reed, M.A.; Lee, T. Single molecule electronic devices. Adv. Mater 2011, 23, 1583–1608. [Google Scholar]
- Demirel, G.B.; Dilsiz, N.; Cakmak, M.; Caykara, T. Molecular design of photoswitchable surfaces with controllable wettability. J. Mater. Chem 2011, 21, 3189–3196. [Google Scholar]
- Wan, P.; Jiang, Y.; Wang, Y.; Wang, Z.; Zhang, X. Tuning surface wettability through photocontrolled reversible molecular shuttle. Chem. Comm. 2008, 5710–5712. [Google Scholar]
- Evans, S.D.; Johnson, S.R.; Ringsdorf, H.; Williams, L.M.; Wolf, H. Photoswitching of azobenzene derivatives formed on planar and colloidal gold surfaces. Langmuir 1998, 14, 6436–6440. [Google Scholar]
- Wang, S.-Y.; Huang, D.-C.; Tao, Y.-T. Self-assembled azobenzenethiol monolayer on electrode surfaces: Effect of photo-switching on the surface and electrical property. J. Chin. Chem. Soc 2012, 59, 9–17. [Google Scholar]
- Wagner, S.; Leyssner, F.; Kordel, C.; Zarwell, S.; Schmidt, R.; Weinelt, M.; Ruck-Braun, K.; Wolf, M.; Tegeder, P. Reversible photoisomerization of an azobenzene-functionalized self-assembled monolayer probed by sum-frequency generation vibrational spectroscopy. Phys. Chem. Chem. Phys 2009, 11, 6242–6248. [Google Scholar]
- Takamatsu, D.; Yamakoshi, Y.; Fukui, K. Photoswitching behavior of a novel single molecular tip for noncontact atomic force microscopy designed for chemical identification. J. Phys. Chem. B 2006, 110, 1968–1970. [Google Scholar]
- Silien, C.; Räisänen, M.T.; Buck, M. A supramolecular network as sacrificial mask for the generation of a nanopatterned binary self-assembled monolayer. Small 2010, 6, 391–394. [Google Scholar]
- Madueno, R.; Raisanen, M.T.; Silien, C.; Buck, M. Functionalizing hydrogen-bonded surface networks with self-assembled monolayers. Nature 2008, 454, 618–621. [Google Scholar]
- Han, M.; Honda, T.; Ishikawa, D.; Ito, E.; Hara, M.; Norikane, Y. Realization of highly photoresponsive azobenzene-functionalized monolayers. J. Mater. Chem 2011, 21, 4696–4702. [Google Scholar]
- Elbing, M.; Błaszczyk, A.; von Hänisch, C.; Mayor, M.; Ferri, V.; Grave, C.; Rampi, M.A.; Pace, G.; Samorì, P.; Shaporenko, A.; et al. Single component self-assembled monolayers of aromatic azo-biphenyl: Influence of the packing tightness on the sam structure and light-induced molecular movements. Adv. Funct. Mater 2008, 18, 2972–2983. [Google Scholar]
- Lavalle, P.; Voegel, J.-C.; Vautier, D.; Senger, B.; Schaaf, P.; Ball, V. Dynamic aspects of films prepared by a sequential deposition of species: Perspectives for smart and responsive materials. Adv. Mater 2011, 23, 1191–1221. [Google Scholar]
- Ariga, K.; Ji, Q.; Hill, J.P.; Bando, Y.; Aono, M. Forming nanomaterials as layered functional structures toward materials nanoarchitectonics. NPG Asia Mater 2012, 4, e17. [Google Scholar]
- Schmidt, D.J.; Moskowitz, J.S.; Hammond, P.T. Electrically triggered release of a small molecule drug from a polyelectrolyte multilayer coating. Chem. Mater 2010, 22, 6416–6425. [Google Scholar]
- Ganta, S.; Devalapally, H.; Shahiwala, A.; Amiji, M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Controlled Release 2008, 126, 187–204. [Google Scholar]
- Wang, F.; Wang, J.; Zhai, Y.; Li, G.; Li, D.; Dong, S. Layer-by-layer assembly of biologically inert inorganic ions/DNA multilayer films for tunable DNA release by chelation. J. Control. Release 2008, 132, 65–73. [Google Scholar]
- Zhuk, A.; Mirza, R.; Sukhishvili, S. Multiresponsive clay-containing layer-by-layer films. ACS Nano 2011, 5, 8790–8799. [Google Scholar]
- Katagiri, K.; Koumoto, K.; Iseya, S.; Sakai, M.; Matsuda, A.; Caruso, F. Tunable UV-responsive organic–inorganic hybrid capsules. Chem. Mater 2008, 21, 195–197. [Google Scholar]
- Zhu, Z.; Senses, E.; Akcora, P.; Sukhishvili, S.A. Programmable light-controlled shape changes in layered polymer nanocomposites. ACS Nano 2012, 6, 3152–3162. [Google Scholar]
- Farha, O.K.; Hupp, J.T. Rational design, synthesis, purification, and activation of metal-organic framework materials. Acc. Chem. Res 2010, 43, 1166–1175. [Google Scholar]
- Rosi, N.L.; Eckert, J.; Eddaoudi, M.; Vodak, D.T.; Kim, J.; O’Keeffe, M.; Yaghi, O.M. Hydrogen storage in microporous metal-organic frameworks. Science 2003, 300, 1127–1129. [Google Scholar]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar]
- Kitaura, R.; Kitagawa, S.; Kubota, Y.; Kobayashi, T.C.; Kindo, K.; Mita, Y.; Matsuo, A.; Kobayashi, M.; Chang, H.C.; Ozawa, T.C.; et al. Formation of a one-dimensional array of oxygen in a microporous metal-organic solid. Science 2002, 298, 2358–2361. [Google Scholar]
- Ferey, G.; Mellot-Draznieks, C.; Serre, C.; Millange, F.; Dutour, J.; Surble, S.; Margiolaki, I. A chromium terephthalate-based solid with unusually large pore volumes and surface area. Science 2005, 309, 2040–2042. [Google Scholar]
- Wang, Z.Q.; Cohen, S.M. Postsynthetic modification of metal-organic frameworks. Chem. Soc. Rev 2009, 38, 1315–1329. [Google Scholar]
- Cohen, S.M. Postsynthetic methods for the functionalization of metal-organic frameworks. Chem. Rev 2012, 112, 970–1000. [Google Scholar]
- Deng, H.X.; Doonan, C.J.; Furukawa, H.; Ferreira, R.B.; Towne, J.; Knobler, C.B.; Wang, B.; Yaghi, O.M. Multiple functional groups of varying ratios in metal-organic frameworks. Science 2010, 327, 846–850. [Google Scholar]
- Wiers, B.M.; Foo, M.L.; Balsara, N.P.; Long, J.R. A solid lithium electrolyte via addition of lithium isopropoxide to a metal-organic framework with open metal sites. J. Am. Chem. Soc 2011, 133, 14522–14525. [Google Scholar]
- Kitagawa, S.; Uemura, K. Dynamic porous properties of coordination polymers inspired by hydrogen bonds. Chem. Soc. Rev 2005, 34, 109–119. [Google Scholar]
- Serre, C.; Mellot-Draznieks, C.; Surble, S.; Audebrand, N.; Filinchuk, Y.; Ferey, G. Role of solvent-host interactions that lead to very large swelling of hybrid frameworks. Science 2007, 315, 1828–1831. [Google Scholar]
- Llewellyn, P.L.; Maurin, G.; Devic, T.; Loera-Serna, S.; Rosenbach, N.; Serre, C.; Bourrelly, S.; Horcajada, P.; Filinchuk, Y.; Ferey, G. Prediction of the conditions for breathing of metal organic framework materials using a combination of X-ray powder diffraction, microcalorimetry, and molecular simulation. J. Am. Chem. Soc 2008, 130, 12808–12814. [Google Scholar]
- Ferey, G.; Serre, C. Large breathing effects in three-dimensional porous hybrid matter: Facts, analyses, rules and consequences. Chem. Soc. Rev 2009, 38, 1380–1399. [Google Scholar]
- Henke, S.; Schneemann, A.; Wutscher, A.; Fischer, R.A. Directing the breathing behavior of pillared-layered metal organic frameworks via a systematic library of functionalized linkers bearing flexible substituents. J. Am. Chem. Soc 2012, 134, 9464–9474. [Google Scholar]
- Horike, S.; Shimomura, S.; Kitagawa, S. Soft porous crystals. Nat. Chem 2009, 1, 695–704. [Google Scholar]
- Li, J.R.; Sculley, J.; Zhou, H.C. Metal-organic frameworks for separations. Chem. Rev 2012, 112, 869–932. [Google Scholar]
- Kreno, L.E.; Leong, K.; Farha, O.K.; Allendorf, M.; Van Duyne, R.P.; Hupp, J.T. Metal-organic framework materials as chemical sensors. Chem. Rev 2012, 112, 1105–1125. [Google Scholar]
- Alberti, G.; Murcia-Mascaros, S.; Vivani, R. Pillared derivatives of gamma-zirconium phosphate containing nonrigid alkyl chain pillars. J. Am. Chem. Soc 1998, 120, 9291–9295. [Google Scholar]
- Maspoch, D.; Ruiz-Molina, D.; Wurst, K.; Domingo, N.; Cavallini, M.; Biscarini, F.; Tejada, J.; Rovira, C.; Veciana, J. A nanoporous molecular magnet with reversible solvent-induced mechanical and magnetic properties. Nat. Mater 2003, 2, 190–195. [Google Scholar]
- Ferey, G. Nanoporous materials—a selective magnetic sponge. Nat. Mater 2003, 2, 136–137. [Google Scholar]
- Ferey, G.; Serre, C.; Devic, T.; Maurin, G.; Jobic, H.; Llewellyn, P.L.; De Weireld, G.; Vimont, A.; Daturi, M.; Chang, J.S. Why hybrid porous solids capture greenhouse gases? Chem. Soc. Rev 2011, 40, 550–562. [Google Scholar]
- Sumida, K.; Rogow, D.L.; Mason, J.A.; McDonald, T.M.; Bloch, E.D.; Herm, Z.R.; Bae, T.H.; Long, J.R. Carbon dioxide capture in metal-organic frameworks. Chem. Rev 2012, 112, 724–781. [Google Scholar]
- Carlucci, L.; Ciani, G.; Moret, M.; Proserpio, D.M.; Rizzato, S. Polymeric layers catenated by ribbons of rings in a three-dimensional self-assembled architecture: A nanoporous network with spongelike behavior. Angew. Chem. Int. Ed 2000, 39, 1506–1510. [Google Scholar]
- Kitaura, R.; Seki, K.; Akiyama, G.; Kitagawa, S. Porous coordination-polymer crystals with gated channels specific for supercritical gases. Angew. Chem. Int. Ed 2003, 42, 428–431. [Google Scholar]
- Nelson, A.P.; Parrish, D.A.; Cambrea, L.R.; Baldwin, L.C.; Trivedi, N.J.; Mulfort, K.L.; Farha, O.K.; Hupp, J.T. Crystal to crystal guest exchange in a mixed ligand metal-organic framework. Cryst. Growth Des 2009, 9, 4588–4591. [Google Scholar]
- Liu, Y.; Her, J.H.; Dailly, A.; Ramirez-Cuesta, A.J.; Neumann, D.A.; Brown, C.M. Reversible structural transition in mil-53 with large temperature hysteresis. J. Am. Chem. Soc 2008, 130, 11813–11818. [Google Scholar]
- Ma, Q.T.; Yang, Q.Y.; Ghoufi, A.; Ferey, G.; Zhong, C.L.; Maurin, G. Guest dependent pressure behavior of the flexible mil-53(cr): A computational exploration. Dalton Trans 2012, 41, 3915–3919. [Google Scholar]
- Yot, P.G.; Ma, Q.T.; Haines, J.; Yang, Q.Y.; Ghoufi, A.; Devic, T.; Serre, C.; Dmitriev, V.; Ferey, G.; Zhong, C.L.; et al. Large breathing of the mof mil-47(v–iv) under mechanical pressure: A joint experimental-modelling exploration. Chem. Sci. 2012, 3, 1100–1104. [Google Scholar]
- Ramsahye, N.A.; Trung, T.K.; Bourrelly, S.; Yang, Q.Y.; Devic, T.; Maurin, G.; Horcajada, P.; Llewellyn, P.L.; Yot, P.; Serre, C.; et al. Influence of the organic ligand functionalization on the breathing of the porous iron terephthalate metal organic framework type material upon hydrocarbon adsorption. J. Phys. Chem. C 2011, 115, 18683–18695. [Google Scholar]
- Sapchenko, S.A.; Samsonenko, D.G.; Dybtsev, D.N.; Melgunov, M.S.; Fedin, V.P. Microporous sensor: Gas sorption, guest exchange and guest-dependant luminescence of metal-organic framework. Dalton Trans 2011, 40, 2196–2203. [Google Scholar]
- Kondo, M.; Furukawa, S.; Hirai, K.; Kitagawa, S. Coordinatively immobilized monolayers on porous coordination polymer crystals. Angew. Chem. Int. Ed 2010, 49, 5327–5330. [Google Scholar]













© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dhotel, A.; Chen, Z.; Delbreilh, L.; Youssef, B.; Saiter, J.-M.; Tan, L. Molecular Motions in Functional Self-Assembled Nanostructures. Int. J. Mol. Sci. 2013, 14, 2303-2333. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14022303
Dhotel A, Chen Z, Delbreilh L, Youssef B, Saiter J-M, Tan L. Molecular Motions in Functional Self-Assembled Nanostructures. International Journal of Molecular Sciences. 2013; 14(2):2303-2333. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14022303
Chicago/Turabian StyleDhotel, Alexandre, Ziguang Chen, Laurent Delbreilh, Boulos Youssef, Jean-Marc Saiter, and Li Tan. 2013. "Molecular Motions in Functional Self-Assembled Nanostructures" International Journal of Molecular Sciences 14, no. 2: 2303-2333. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14022303