Caffeic Acid Phenethyl Ester as a Potential Treatment for Advanced Prostate Cancer Targeting Akt Signaling


Abstract
:1. Introduction
2. Akt Signaling and Prostate Cancer
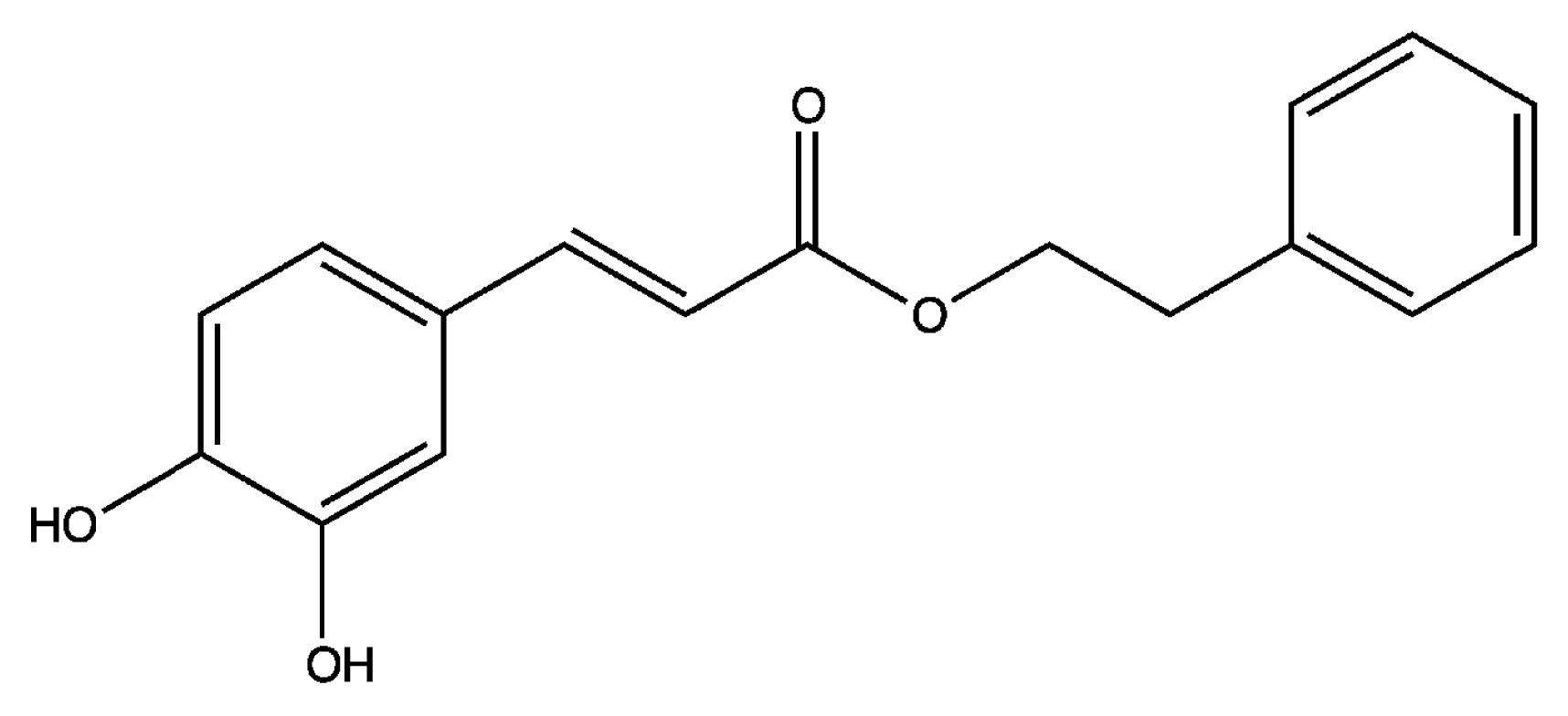
3. Caffeic Acid Phenethyl Ester (CAPE)
4. Anti-Cancer Effects of CAPE on Human Cancer Cell Lines
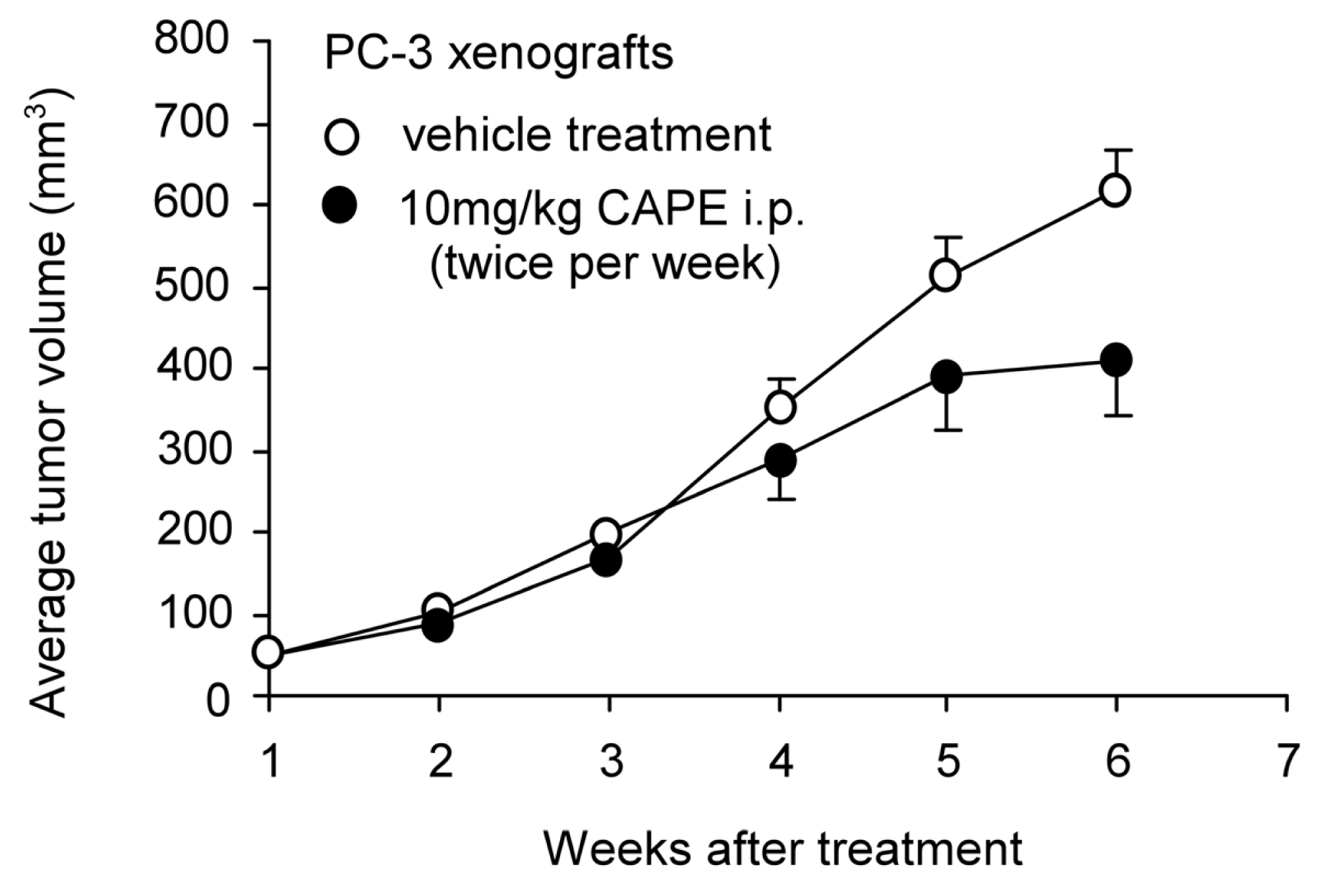
5. CAPE Suppresses Tumor Growth and Cancer Metastasis in Animal Models
6. Anticancer Effects of CAPE on Human Prostate Cancer Cells
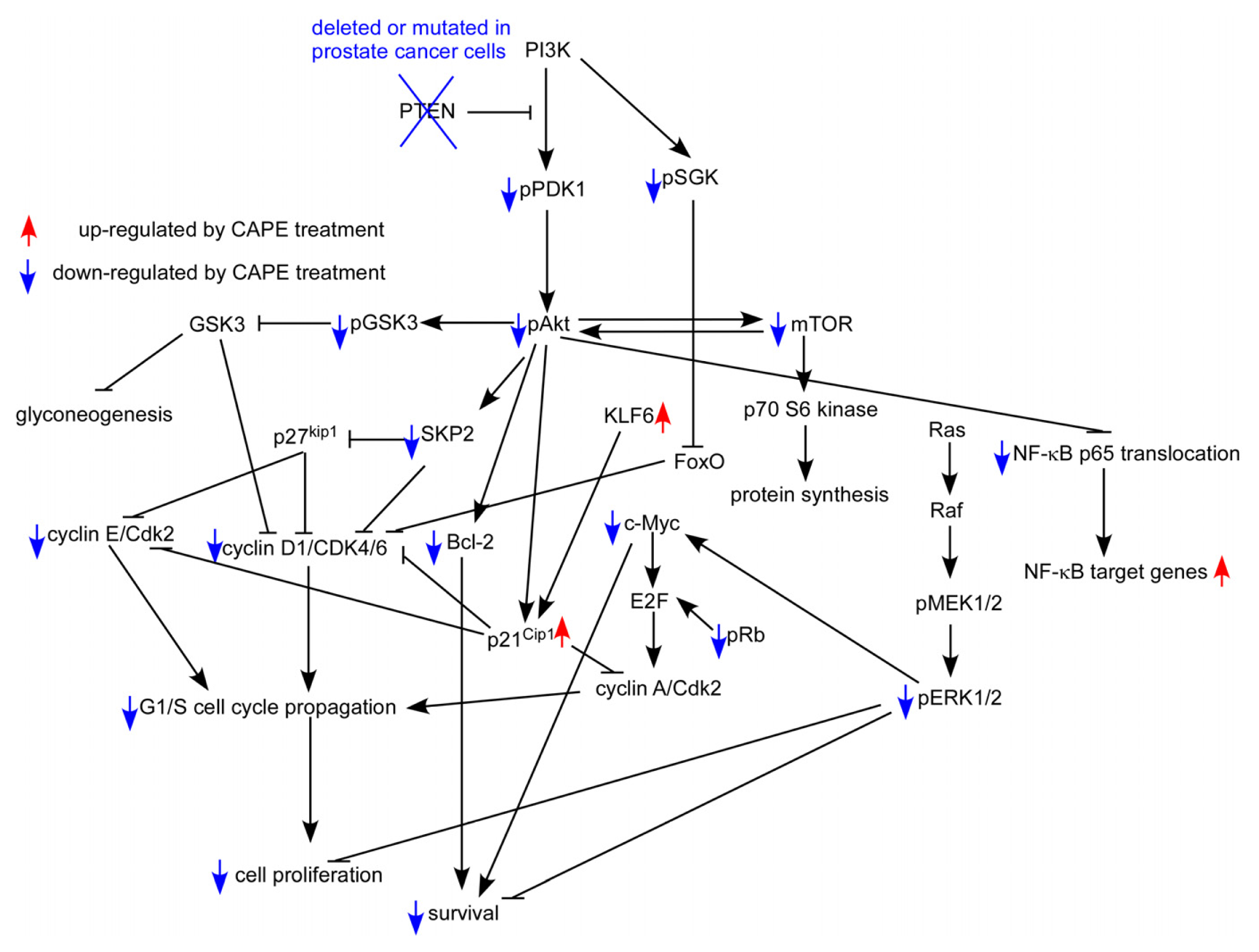
7. CAPE Inhibits Akt Signaling in Prostate Cancer Cells
8. Potential Clinical Application of CAPE
9. Conclusions
Acknowledgments
Conflict of Interest
References
- Karan, D.; Thrasher, J.B.; Lubaroff, D. Prostate cancer: Genes, environment, immunity and the use of immunotherapy. Prostate Cancer Prostatic Dis 2008, 11, 230–236. [Google Scholar]
- Kuriyama, M.; Wang, M.C.; Lee, C.I.; Papsidero, L.D.; Killian, C.S.; Inaji, H.; Slack, N.H.; Nishiura, T.; Murphy, G.P.; Chu, T.M. Use of human prostate-specific antigen in monitoring prostate cancer. Cancer Res 1981, 41, 3874–3876. [Google Scholar]
- Nadji, M.; Tabei, S.Z.; Castro, A.; Chu, T.M.; Murphy, G.P.; Wang, M.C.; Morales, A.R. Prostatic-Specific antigen: An immunohistologic marker for prostatic neoplasms. Cancer 1981, 48, 1229–1232. [Google Scholar]
- Riegman, P.H.; Vlietstra, R.J.; van der Korput, J.A.; Brinkmann, A.O.; Trapman, J. The promoter of the prostate-specific antigen gene contains a functional androgen responsive element. Mol. Endocrinol 1991, 5, 1921–1930. [Google Scholar]
- Wolf, D.A.; Schulz, P.; Fittler, F. Transcriptional regulation of prostate kallikrein-like genes by androgen. Mol. Endocrinol 1992, 6, 753–762. [Google Scholar]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar]
- Ricke, E.A.; Williams, K.; Lee, Y.F.; Couto, S.; Wang, Y.; Hayward, S.W.; Cunha, G.R.; Ricke, W.A. Androgen hormone action in prostatic carcinogenesis: Stromal androgen receptors mediate prostate cancer progression, malignant transformation and metastasis. Carcinogenesis 2012, 33, 1391–1398. [Google Scholar]
- Chuu, C.P.; Kokontis, J.M.; Hiipakka, R.A.; Fukuchi, J.; Lin, H.P.; Lin, C.Y.; Huo, C.; Su, L.C. Androgens as therapy for androgen receptor-positive castration-resistant prostate cancer. J. Biomed. Sci 2011, 18, 63. [Google Scholar]
- Chuu, C.P.; Hiipakka, R.A.; Fukuchi, J.; Kokontis, J.M.; Liao, S. Androgen causes growth suppression and reversion of androgen-independent prostate cancer xenografts to an androgen-stimulated phenotype in athymic mice. Cancer Res 2005, 65, 2082–2084. [Google Scholar]
- Chuu, C.P.; Hiipakka, R.A.; Kokontis, J.M.; Fukuchi, J.; Chen, R.Y.; Liao, S. Inhibition of tumor growth and progression of LNCaP prostate cancer cells in athymic mice by androgen and liver X receptor agonist. Cancer Res 2006, 66, 6482–6486. [Google Scholar]
- Chuu, C.P.; Kokontis, J.M.; Hiipakka, R.A.; Fukuchi, J.; Lin, H.P.; Lin, C.Y.; Huo, C.; Su, L.C.; Liao, S. Androgen suppresses proliferation of castration-resistant LNCaP 104-R2 prostate cancer cells through androgen receptor, Skp2, and c-Myc. Cancer Sci 2011, 102, 2022–2028. [Google Scholar]
- Wang, Q.; Li, W.; Liu, X.S.; Carroll, J.S.; Janne, O.A.; Keeton, E.K.; Chinnaiyan, A.M.; Pienta, K.J.; Brown, M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell 2007, 27, 380–392. [Google Scholar]
- Xu, Y.; Chen, S.Y.; Ross, K.N.; Balk, S.P. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res 2006, 66, 7783–7792. [Google Scholar]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009, 138, 245–256. [Google Scholar]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar]
- Cerveira, N.; Ribeiro, F.R.; Peixoto, A.; Costa, V.; Henrique, R.; Jeronimo, C.; Teixeira, M.R. TMPRSS2-ERG gene fusion causing ERG overexpression precedes chromosome copy number changes in prostate carcinomas and paired HGPIN lesions. Neoplasia 2006, 8, 826–832. [Google Scholar]
- Cai, C.; Wang, H.; Xu, Y.; Chen, S.; Balk, S.P. Reactivation of androgen receptor-regulated TMPRSS2:ERG gene expression in castration-resistant prostate cancer. Cancer Res 2009, 69, 6027–6032. [Google Scholar]
- Van der Kwast, T.H.; Schalken, J.; Ruizeveld de Winter, J.A.; van Vroonhoven, C.C.; Mulder, E.; Boersma, W.; Trapman, J. Androgen receptors in endocrine-therapy-resistant human prostate cancer. Int. J. Cancer 1991, 48, 189–193. [Google Scholar]
- Ruizeveld de Winter, J.A.; Janssen, P.J.; Sleddens, H.M.; Verleun-Mooijman, M.C.; Trapman, J.; Brinkmann, A.O.; Santerse, A.B.; Schroder, F.H.; van der Kwast, T.H. Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer. Am. J. Pathol 1994, 144, 735–746. [Google Scholar]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinanen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet 1995, 9, 401–406. [Google Scholar]
- Bubendorf, L.; Kononen, J.; Koivisto, P.; Schraml, P.; Moch, H.; Gasser, T.C.; Willi, N.; Mihatsch, M.J.; Sauter, G.; Kallioniemi, O.P. Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res 1999, 59, 803–806. [Google Scholar]
- Linja, M.J.; Savinainen, K.J.; Saramaki, O.R.; Tammela, T.L.; Vessella, R.L.; Visakorpi, T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res 2001, 61, 3550–3555. [Google Scholar]
- Ford, O.H., III; Gregory, C.W.; Kim, D.; Smitherman, A.B.; Mohler, J.L. Androgen receptor gene amplification and protein expression in recurrent prostate cancer. J. Urol. 2003, 170, 1817–1821. [Google Scholar]
- Holzbeierlein, J.; Lal, P.; LaTulippe, E.; Smith, A.; Satagopan, J.; Zhang, L.; Ryan, C.; Smith, S.; Scher, H.; Scardino, P.; et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am. J. Pathol 2004, 164, 217–227. [Google Scholar]
- Mohler, J.L.; Gregory, C.W.; Ford, O.H., III; Kim, D.; Weaver, C.M.; Petrusz, P.; Wilson, E.M.; French, F.S. The androgen axis in recurrent prostate cancer. Clin. Cancer Res. 2004, 10, 440–448. [Google Scholar]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res 2006, 66, 2815–2825. [Google Scholar]
- Koivisto, P.; Kononen, J.; Palmberg, C.; Tammela, T.; Hyytinen, E.; Isola, J.; Trapman, J.; Cleutjens, K.; Noordzij, A.; Visakorpi, T.; et al. Androgen receptor gene amplification: A possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res 1997, 57, 314–319. [Google Scholar]
- Brown, R.S.; Edwards, J.; Dogan, A.; Payne, H.; Harland, S.J.; Bartlett, J.M.; Masters, J.R. Amplification of the androgen receptor gene in bone metastases from hormone-refractory prostate cancer. J. Pathol 2002, 198, 237–244. [Google Scholar]
- Edwards, J.; Krishna, N.S.; Grigor, K.M.; Bartlett, J.M. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br. J. Cancer 2003, 89, 552–556. [Google Scholar]
- Sadar, M.D. Small molecule inhibitors targeting the “achilles’ heel” of androgen receptor activity. Cancer Res 2011, 71, 1208–1213. [Google Scholar]
- Bubendorf, L.; Schopfer, A.; Wagner, U.; Sauter, G.; Moch, H.; Willi, N.; Gasser, T.C.; Mihatsch, M.J. Metastatic patterns of prostate cancer: An autopsy study of 1,589 patients. Hum. Pathol 2000, 31, 578–583. [Google Scholar]
- Ibrahim, T.; Flamini, E.; Mercatali, L.; Sacanna, E.; Serra, P.; Amadori, D. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer 2010, 116, 1406–1418. [Google Scholar]
- Keller, E.T.; Zhang, J.; Cooper, C.R.; Smith, P.C.; McCauley, L.K.; Pienta, K.J.; Taichman, R.S. Prostate carcinoma skeletal metastases: Cross-Talk between tumor and bone. Cancer Metastasis Rev 2001, 20, 333–349. [Google Scholar]
- Huggins, C.; Stevens, R.; Hodges, C. Studies on prostatic cancer: II. The effects of castration on advanced carcinoma of the prostate gland. Arch. Surg 1941, 43, 15. [Google Scholar]
- Seruga, B.; Tannock, I.F. Intermittent androgen blockade should be regarded as standard therapy in prostate cancer. Nat. Clin. Pract. Oncol 2008, 5, 574–576. [Google Scholar]
- Hellerstedt, B.A.; Pienta, K.J. The current state of hormonal therapy for prostate cancer. CA Cancer J. Clin 2002, 52, 154–179. [Google Scholar]
- Gilligan, T.; Kantoff, P.W. Chemotherapy for prostate cancer. Urology 2002, 60, 94–100. [Google Scholar]
- Pinto, A.C.; Moreira, J.N.; Simoes, S. Liposomal imatinib-mitoxantrone combination: Formulation development and therapeutic evaluation in an animal model of prostate cancer. Prostate 2011, 71, 81–90. [Google Scholar]
- Coffer, P.J.; Jin, J.; Woodgett, J.R. Protein kinase B (c-Akt): A multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J 1998, 335, 1–13. [Google Scholar]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell. Cycle 2009, 8, 2502–2508. [Google Scholar]
- Cantley, L.C.; Neel, B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 4240–4245. [Google Scholar]
- Sarker, D.; Reid, A.H.; Yap, T.A.; de Bono, J.S. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin. Cancer Res 2009, 15, 4799–4805. [Google Scholar]
- Bedolla, R.; Prihoda, T.J.; Kreisberg, J.I.; Malik, S.N.; Krishnegowda, N.K.; Troyer, D.A.; Ghosh, P.M. Determining risk of biochemical recurrence in prostate cancer by immunohistochemical detection of PTEN expression and Akt activation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 2007, 13, 3860–3867. [Google Scholar]
- Squire, J.A. TMPRSS2-ERG and PTEN loss in prostate cancer. Nat. Genet 2009, 41, 509–510. [Google Scholar]
- King, J.C.; Xu, J.; Wongvipat, J.; Hieronymus, H.; Carver, B.S.; Leung, D.H.; Taylor, B.S.; Sander, C.; Cardiff, R.D.; Couto, S.S.; et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet 2009, 41, 524–526. [Google Scholar]
- Carver, B.S.; Tran, J.; Gopalan, A.; Chen, Z.; Shaikh, S.; Carracedo, A.; Alimonti, A.; Nardella, C.; Varmeh, S.; Scardino, P.T.; et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat. Genet 2009, 41, 619–624. [Google Scholar]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol 1997, 7, 261–269. [Google Scholar]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar]
- Hammarsten, P.; Cipriano, M.; Josefsson, A.; Stattin, P.; Egevad, L.; Granfors, T.; Fowler, C.J. Phospho-Akt immunoreactivity in prostate cancer: Relationship to disease severity and outcome, Ki67 and phosphorylated EGFR expression. PLoS One 2012, 7, e47994. [Google Scholar]
- Chuu, C.P.; Chen, R.Y.; Barkinge, J.L.; Ciaccio, M.F.; Jones, R.B. Systems-Level analysis of ErbB4 signaling in breast cancer: A laboratory to clinical perspective. Mol. Cancer Res 2008, 6, 885–891. [Google Scholar]
- Morgan, T.M.; Koreckij, T.D.; Corey, E. Targeted therapy for advanced prostate cancer: Inhibition of the PI3K/Akt/mTOR pathway. Curr. Cancer Drug Targets 2009, 9, 237–249. [Google Scholar]
- Dai, B.; Kong, Y.Y.; Ye, D.W.; Ma, C.G.; Zhou, X.; Yao, X.D. Activation of the mammalian target of rapamycin signalling pathway in prostate cancer and its association with patient clinicopathological characteristics. BJU Int 2009, 104, 1009–1016. [Google Scholar]
- Kreisberg, J.I.; Malik, S.N.; Prihoda, T.J.; Bedolla, R.G.; Troyer, D.A.; Kreisberg, S.; Ghosh, P.M. Phosphorylation of Akt (Ser473) is an excellent predictor of poor clinical outcome in prostate cancer. Cancer Res 2004, 64, 5232–5236. [Google Scholar]
- Sircar, K.; Yoshimoto, M.; Monzon, F.A.; Koumakpayi, I.H.; Katz, R.L.; Khanna, A.; Alvarez, K.; Chen, G.; Darnel, A.D.; Aprikian, A.G.; et al. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J. Pathol 2009, 218, 505–513. [Google Scholar]
- Wegiel, B.; Bjartell, A.; Culig, Z.; Persson, J.L. Interleukin-6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int. J. Cancer 2008, 122, 1521–1529. [Google Scholar]
- McCall, P.; Gemmell, L.K.; Mukherjee, R.; Bartlett, J.M.; Edwards, J. Phosphorylation of the androgen receptor is associated with reduced survival in hormone-refractory prostate cancer patients. Br. J. Cancer 2008, 98, 1094–1101. [Google Scholar]
- Shimizu, Y.; Segawa, T.; Inoue, T.; Shiraishi, T.; Yoshida, T.; Toda, Y.; Yamada, T.; Kinukawa, N.; Terada, N.; Kobayashi, T.; et al. Increased Akt and phosphorylated Akt expression are associated with malignant biological features of prostate cancer in Japanese men. BJU Int 2007, 100, 685–690. [Google Scholar]
- Ayala, G.; Thompson, T.; Yang, G.; Frolov, A.; Li, R.; Scardino, P.; Ohori, M.; Wheeler, T.; Harper, W. High levels of phosphorylated form of Akt-1 in prostate cancer and non-neoplastic prostate tissues are strong predictors of biochemical recurrence. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 2004, 10, 6572–6578. [Google Scholar]
- Orio, F., Jr; Terouanne, B.; Georget, V.; Lumbroso, S.; Avances, C.; Siatka, C.; Sultan, C. Potential action of IGF-1 and EGF on androgen receptor nuclear transfer and transactivation in normal and cancer human prostate cell lines. Mol. Cell. Endocrinol. 2002, 198, 105–114. [Google Scholar]
- Wang, Y.; Kreisberg, J.I.; Ghosh, P.M. Cross-Talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr. Cancer Drug Targets 2007, 7, 591–604. [Google Scholar]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res 1994, 54, 5474–5478. [Google Scholar]
- Fan, W.; Yanase, T.; Morinaga, H.; Okabe, T.; Nomura, M.; Daitoku, H.; Fukamizu, A.; Kato, S.; Takayanagi, R.; Nawata, H. Insulin-Like growth factor 1/insulin signaling activates androgen signaling through direct interactions of Foxo1 with androgen receptor. J. Biol. Chem 2007, 282, 7329–7338. [Google Scholar]
- Nelson, E.C.; Evans, C.P.; Mack, P.C.; Devere-White, R.W.; Lara, P.N., Jr. Inhibition of Akt pathways in the treatment of prostate cancer. Prostate Cancer Prostatic Dis. 2007, 10, 331–339. [Google Scholar]
- Bhimani, R.S.; Troll, W.; Grunberger, D.; Frenkel, K. Inhibition of oxidative stress in HeLa cells by chemopreventive agents. Cancer Res 1993, 53, 4528–4533. [Google Scholar]
- Natarajan, K.; Singh, S.; Burke, T.R., Jr; Grunberger, D.; Aggarwal, B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 1996, 93, 9090–9095. [Google Scholar]
- Nomura, M.; Kaji, A.; Ma, W.; Miyamoto, K.; Dong, Z. Suppression of cell transformation and induction of apoptosis by caffeic acid phenethyl ester. Mol. Carcinog 2001, 31, 83–89. [Google Scholar]
- Wu, J.; Omene, C.; Karkoszka, J.; Bosland, M.; Eckard, J.; Klein, C.B.; Frenkel, K. Caffeic acid phenethyl ester (CAPE), derived from a honeybee product propolis, exhibits a diversity of anti-tumor effects in pre-clinical models of human breast cancer. Cancer Lett 2011, 308, 43–53. [Google Scholar]
- Watabe, M.; Hishikawa, K.; Takayanagi, A.; Shimizu, N.; Nakaki, T. Caffeic acid phenethyl ester induces apoptosis by inhibition of NFkappaB and activation of Fas in human breast cancer MCF-7 cells. J. Biol. Chem 2004, 279, 6017–6026. [Google Scholar]
- Chuu, C.P.; Lin, H.P.; Ciaccio, M.F.; Kokontis, J.M.; Hause, R.J., Jr; Hiipakka, R.A.; Liao, S.; Jones, R.B. Caffeic acid phenethyl ester suppresses the proliferation of human prostate cancer cells through inhibition of p70S6K and Akt signaling networks. Cancer Prev. Res. (Phila) 2012, 5, 788–797. [Google Scholar]
- Lin, H.P.; Jiang, S.S.; Chuu, C.P. Caffeic acid phenethyl ester causes p21 induction, Akt Signaling reduction, and growth inhibition in PC-3 human prostate cancer cells. PLoS One 2012, 7, e31286. [Google Scholar]
- McEleny, K.; Coffey, R.; Morrissey, C.; Fitzpatrick, J.M.; Watson, R.W. Caffeic acid phenethyl ester-induced PC-3 cell apoptosis is caspase-dependent and mediated through the loss of inhibitors of apoptosis proteins. BJU Int 2004, 94, 402–406. [Google Scholar]
- Chen, M.F.; Wu, C.T.; Chen, Y.J.; Keng, P.C.; Chen, W.C. Cell killing and radiosensitization by caffeic acid phenethyl ester (CAPE) in lung cancer cells. J. Radiat. Res. (Tokyo) 2004, 45, 253–260. [Google Scholar]
- Lin, H.P.; Kuo, L.K.; Chuu, C.P. Combined treatment of curcumin and small molecule inhibitors suppresses proliferation of A549 and H1299 human non-small-cell lung cancer cells. Phytother. Res 2011, 26, 122–126. [Google Scholar]
- Lee, Y.T.; Don, M.J.; Hung, P.S.; Shen, Y.C.; Lo, Y.S.; Chang, K.W.; Chen, C.F.; Ho, L.K. Cytotoxicity of phenolic acid phenethyl esters on oral cancer cells. Cancer Lett 2005, 223, 19–25. [Google Scholar]
- Onori, P.; DeMorrow, S.; Gaudio, E.; Franchitto, A.; Mancinelli, R.; Venter, J.; Kopriva, S.; Ueno, Y.; Alvaro, D.; Savage, J.; et al. Caffeic acid phenethyl ester decreases cholangiocarcinoma growth by inhibition of NF-kappaB and induction of apoptosis. Int. J. Cancer J. Int. Cancer 2009, 125, 565–576. [Google Scholar]
- Hung, M.W.; Shiao, M.S.; Tsai, L.C.; Chang, G.G.; Chang, T.C. Apoptotic effect of caffeic acid phenethyl ester and its ester and amide analogues in human cervical cancer ME180 cells. Anticancer Res 2003, 23, 4773–4780. [Google Scholar]
- Usia, T.; Banskota, A.H.; Tezuka, Y.; Midorikawa, K.; Matsushige, K.; Kadota, S. Constituents of Chinese propolis and their antiproliferative activities. J. Nat. Prod 2002, 65, 673–676. [Google Scholar]
- Chen, Y.J.; Shiao, M.S.; Hsu, M.L.; Tsai, T.H.; Wang, S.Y. Effect of caffeic acid phenethyl ester, an antioxidant from propolis, on inducing apoptosis in human leukemic HL-60 cells. J. Agric. Food Chem 2001, 49, 5615–5619. [Google Scholar]
- Jin, U.H.; Song, K.H.; Motomura, M.; Suzuki, I.; Gu, Y.H.; Kang, Y.J.; Moon, T.C.; Kim, C.H. Caffeic acid phenethyl ester induces mitochondria-mediated apoptosis in human myeloid leukemia U937 cells. Mol. Cell Biochem 2008, 310, 43–48. [Google Scholar]
- Lee, Y.J.; Kuo, H.C.; Chu, C.Y.; Wang, C.J.; Lin, W.C.; Tseng, T.H. Involvement of tumor suppressor protein p53 and p38 MAPK in caffeic acid phenethyl ester-induced apoptosis of C6 glioma cells. Biochem. Pharmacol 2003, 66, 2281–2289. [Google Scholar]
- Su, Z.Z.; Lin, J.; Grunberger, D.; Fisher, P.B. Growth suppression and toxicity induced by caffeic acid phenethyl ester (CAPE) in type 5 adenovirus-transformed rat embryo cells correlate directly with transformation progression. Cancer Res 1994, 54, 1865–1870. [Google Scholar]
- Lin, Y.H.; Chiu, J.H.; Tseng, W.S.; Wong, T.T.; Chiou, S.H.; Yen, S.H. Antiproliferation and radiosensitization of caffeic acid phenethyl ester on human medulloblastoma cells. Cancer Chemother Pharmacol 2006, 57, 525–532. [Google Scholar]
- He, Y.J.; Liu, B.H.; Xiang, D.B.; Qiao, Z.Y.; Fu, T.; He, Y.H. Inhibitory effect of caffeic acid phenethyl ester on the growth of SW480 colorectal tumor cells involves beta-catenin associated signaling pathway down-regulation. World J. Gastroenterol 2006, 12, 4981–4985. [Google Scholar]
- Kuo, H.C.; Kuo, W.H.; Lee, Y.J.; Lin, W.L.; Chou, F.P.; Tseng, T.H. Inhibitory effect of caffeic acid phenethyl ester on the growth of C6 glioma cells in vitro and in vivo. Cancer Lett 2006, 234, 199–208. [Google Scholar]
- Wang, D.; Xiang, D.B.; He, Y.J.; Li, Z.P.; Wu, X.H.; Mou, J.H.; Xiao, H.L.; Zhang, Q.H. Effect of caffeic acid phenethyl ester on proliferation and apoptosis of colorectal cancer cells in vitro. World J. Gastroenterol 2005, 11, 4008–4012. [Google Scholar]
- Xiang, D.; Wang, D.; He, Y.; Xie, J.; Zhong, Z.; Li, Z. Caffeic acid phenethyl ester induces growth arrest and apoptosis of colon cancer cells via the beta-catenin/T-cell factor signaling. Anticancer Drugs 2006, 17, 753–762. [Google Scholar]
- Shigeoka, Y.; Igishi, T.; Matsumoto, S.; Nakanishi, H.; Kodani, M.; Yasuda, K.; Hitsuda, Y.; Shimizu, E. Sulindac sulfide and caffeic acid phenethyl ester suppress the motility of lung adenocarcinoma cells promoted by transforming growth factor-beta through Akt inhibition. J. Cancer Res. Clin. Oncol 2004, 130, 146–152. [Google Scholar]
- Weyant, M.J.; Carothers, A.M.; Bertagnolli, M.E.; Bertagnolli, M.M. Colon cancer chemopreventive drugs modulate integrin-mediated signaling pathways. Clin. Cancer Res 2000, 6, 949–956. [Google Scholar]
- Mahmoud, N.N.; Carothers, A.M.; Grunberger, D.; Bilinski, R.T.; Churchill, M.R.; Martucci, C.; Newmark, H.L.; Bertagnolli, M.M. Plant phenolics decrease intestinal tumors in an animal model of familial adenomatous polyposis. Carcinogenesis 2000, 21, 921–927. [Google Scholar]
- Nagaoka, T.; Banskota, A.H.; Tezuka, Y.; Harimaya, Y.; Koizumi, K.; Saiki, I.; Kadota, S. Inhibitory effects of caffeic acid phenethyl ester analogues on experimental lung metastasis of murine colon 26-L5 carcinoma cells. Biol. Pharm. Bull 2003, 26, 638–641. [Google Scholar]
- Borrelli, F.; Izzo, A.A.; Di Carlo, G.; Maffia, P.; Russo, A.; Maiello, F.M.; Capasso, F.; Mascolo, N. Effect of a propolis extract and caffeic acid phenethyl ester on formation of aberrant crypt foci and tumors in the rat colon. Fitoterapia 2002, 73, S38–S43. [Google Scholar]
- Carrasco-Legleu, C.E.; Sanchez-Perez, Y.; Marquez-Rosado, L.; Fattel-Fazenda, S.; Arce-Popoca, E.; Hernandez-Garcia, S.; Villa-Trevino, S. A single dose of caffeic acid phenethyl ester prevents initiation in a medium-term rat hepatocarcinogenesis model. World J. Gastroenterol 2006, 12, 6779–6785. [Google Scholar]
- Carrasco-Legleu, C.E.; Marquez-Rosado, L.; Fattel-Fazenda, S.; Arce-Popoca, E.; Perez-Carreon, J.I.; Villa-Trevino, S. Chemoprotective effect of caffeic acid phenethyl ester on promotion in a medium-term rat hepatocarcinogenesis assay. Int. J. Cancer 2004, 108, 488–492. [Google Scholar]
- Kudugunti, S.K.; Vad, N.M.; Ekogbo, E.; Moridani, M.Y. Efficacy of caffeic acid phenethyl ester (CAPE) in skin B16-F0 melanoma tumor bearing C57BL/6 mice. Investig. New Drugs 2011, 29, 52–62. [Google Scholar]
- Liao, H.F.; Chen, Y.Y.; Liu, J.J.; Hsu, M.L.; Shieh, H.J.; Liao, H.J.; Shieh, C.J.; Shiao, M.S.; Chen, Y.J. Inhibitory effect of caffeic acid phenethyl ester on angiogenesis, tumor invasion, and metastasis. J. Agric. Food Chem 2003, 51, 7907–7912. [Google Scholar]
- Orsolic, N.; Knezevic, A.H.; Sver, L.; Terzic, S.; Basic, I. Immunomodulatory and antimetastatic action of propolis and related polyphenolic compounds. J. Ethnopharmacol 2004, 94, 307–315. [Google Scholar]
- Chung, T.W.; Moon, S.K.; Chang, Y.C.; Ko, J.H.; Lee, Y.C.; Cho, G.; Kim, S.H.; Kim, J.G.; Kim, C.H. Novel and therapeutic effect of caffeic acid and caffeic acid phenyl ester on hepatocarcinoma cells: Complete regression of hepatoma growth and metastasis by dual mechanism. FASEB J 2004, 18, 1670–1681. [Google Scholar]
- Horoszewicz, J.S.; Leong, S.S.; Chu, T.M.; Wajsman, Z.L.; Friedman, M.; Papsidero, L.; Kim, U.; Chai, L.S.; Kakati, S.; Arya, S.K.; et al. The LNCaP cell line—A new model for studies on human prostatic carcinoma. Prog. Clin. Biol. Res 1980, 37, 115–132. [Google Scholar]
- Chuu, C.P.; Kokontis, J.M.; Hiipakka, R.A.; Liao, S. Modulation of liver X receptor signaling as novel therapy for prostate cancer. J. Biomed. Sci 2007, 14, 543–553. [Google Scholar]
- Stone, K.R.; Mickey, D.D.; Wunderli, H.; Mickey, G.H.; Paulson, D.F. Isolation of a human prostate carcinoma cell line (DU 145). Int. J. Cancer 1978, 21, 274–281. [Google Scholar]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest. Urol 1979, 17, 16–23. [Google Scholar]
- Kokontis, J.; Takakura, K.; Hay, N.; Liao, S. Increased androgen receptor activity and altered c-myc expression in prostate cancer cells after long-term androgen deprivation. Cancer Res 1994, 54, 1566–1573. [Google Scholar]
- Kokontis, J.M.; Hay, N.; Liao, S. Progression of LNCaP prostate tumor cells during androgen deprivation: Hormone-Independent growth, repression of proliferation by androgen, and role for p27Kip1 in androgen-induced cell cycle arrest. Mol. Endocrinol 1998, 12, 941–953. [Google Scholar]
- Kokontis, J.M.; Hsu, S.; Chuu, C.P.; Dang, M.; Fukuchi, J.; Hiipakka, R.A.; Liao, S. Role of androgen receptor in the progression of human prostate tumor cells to androgen independence and insensitivity. Prostate 2005, 65, 287–298. [Google Scholar]
- Hiipakka, R.A.; Zhang, H.Z.; Dai, W.; Dai, Q.; Liao, S. Structure-Activity relationships for inhibition of human 5alpha-reductases by polyphenols. Biochem. Pharmacol 2002, 63, 1165–1176. [Google Scholar]
- Wang, L.C.; Chu, K.H.; Liang, Y.C.; Lin, Y.L.; Chiang, B.L. Caffeic acid phenethyl ester inhibits nuclear factor-kappaB and protein kinase B signalling pathways and induces caspase-3 expression in primary human CD4+ T cells. Clin. Exp. Immunol 2010, 160, 223–232. [Google Scholar]
- Ho, H.C.; Hsu, S.L.; Ting, C.T.; Kuo, C.Y.; Yang, V.C. Caffeic acid phenethyl ester inhibits arterial smooth muscle cell proliferation and migration in vitro and in vivo using a local delivery system. Cell. Mol. Biol. (Noisy-le-grand) 2009, 55, OL1161–1167. [Google Scholar]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar]
- Liang, J.; Slingerland, J.M. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2003, 2, 339–345. [Google Scholar]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar]
- Brunet, A.; Park, J.; Tran, H.; Hu, L.S.; Hemmings, B.A.; Greenberg, M.E. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol 2001, 21, 952–965. [Google Scholar]
- Gu, Y.; Rosenblatt, J.; Morgan, D.O. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J 1992, 11, 3995–4005. [Google Scholar]
- Morrison, D.K.; Heidecker, G.; Rapp, U.R.; Copeland, T.D. Identification of the major phosphorylation sites of the Raf-1 kinase. J. Biol. Chem 1993, 268, 17309–17316. [Google Scholar]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell. Biol 2006, 7, 85–96. [Google Scholar]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997, 275, 661–665. [Google Scholar]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 1996, 15, 6541–6551. [Google Scholar]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol 2001, 3, 1009–1013. [Google Scholar]
- Duan, C.; Liimatta, M.B.; Bottum, O.L. Insulin-Like growth factor (IGF)-I regulates IGF-binding protein-5 gene expression through the phosphatidylinositol 3-kinase, protein kinase B/Akt, and p70 S6 kinase signaling pathway. J. Biol. Chem 1999, 274, 37147–37153. [Google Scholar]
- Celli, N.; Dragani, L.K.; Murzilli, S.; Pagliani, T.; Poggi, A. In vitro and in vivo stability of caffeic acid phenethyl ester, a bioactive compound of propolis. J. Agric. Food Chem 2007, 55, 3398–3407. [Google Scholar]
- Wang, X.; Pang, J.; Maffucci, J.A.; Pade, D.S.; Newman, R.A.; Kerwin, S.M.; Bowman, P.D.; Stavchansky, S. Pharmacokinetics of caffeic acid phenethyl ester and its catechol-ring fluorinated derivative following intravenous administration to rats. Biopharm. Drug Dispos 2009, 30, 221–228. [Google Scholar]
- Akyol, S.; Ginis, Z.; Armutcu, F.; Ozturk, G.; Yigitoglu, M.R.; Akyol, O. The potential usage of caffeic acid phenethyl ester (CAPE) against chemotherapy-induced and radiotherapy-induced toxicity. Cell. Biochem. Funct 2012, 30, 438–443. [Google Scholar]
- Yagmurca, M.; Erdogan, H.; Iraz, M.; Songur, A.; Ucar, M.; Fadillioglu, E. Caffeic acid phenethyl ester as a protective agent against doxorubicin nephrotoxicity in rats. Clin. Chim. Acta 2004, 348, 27–34. [Google Scholar]
- Fadillioglu, E.; Oztas, E.; Erdogan, H.; Yagmurca, M.; Sogut, S.; Ucar, M.; Irmak, M.K. Protective effects of caffeic acid phenethyl ester on doxorubicin-induced cardiotoxicity in rats. J. Appl. Toxicol 2004, 24, 47–52. [Google Scholar]
- Irmak, M.K.; Fadillioglu, E.; Sogut, S.; Erdogan, H.; Gulec, M.; Ozer, M.; Yagmurca, M.; Gozukara, M.E. Effects of caffeic acid phenethyl ester and alpha-tocopherol on reperfusion injury in rat brain. Cell. Biochem. Funct 2003, 21, 283–289. [Google Scholar]
- Iraz, M.; Ozerol, E.; Gulec, M.; Tasdemir, S.; Idiz, N.; Fadillioglu, E.; Naziroglu, M.; Akyol, O. Protective effect of caffeic acid phenethyl ester (CAPE) administration on cisplatin-induced oxidative damage to liver in rat. Cell. Biochem. Funct 2006, 24, 357–361. [Google Scholar]
- Yilmaz, H.R.; Sogut, S.; Ozyurt, B.; Ozugurlu, F.; Sahin, S.; Isik, B.; Uz, E.; Ozyurt, H. The activities of liver adenosine deaminase, xanthine oxidase, catalase, superoxide dismutase enzymes and the levels of malondialdehyde and nitric oxide after cisplatin toxicity in rats: protective effect of caffeic acid phenethyl ester. Toxicol. Ind. Health 2005, 21, 67–73. [Google Scholar]
- Oktem, F.; Yilmaz, H.R.; Ozguner, F.; Olgar, S.; Ayata, A.; Uzare, E.; Uz, E. Methotrexate-induced renal oxidative stress in rats: The role of a novel antioxidant caffeic acid phenethyl ester. Toxicol. Ind. Health 2006, 22, 241–247. [Google Scholar]
- Ozyurt, H.; Sogut, S.; Yildirim, Z.; Kart, L.; Iraz, M.; Armutcu, F.; Temel, I.; Ozen, S.; Uzun, A.; Akyol, O. Inhibitory effect of caffeic acid phenethyl ester on bleomycine-induced lung fibrosis in rats. Clin. Chim. Acta 2004, 339, 65–75. [Google Scholar]
- Albukhari, A.A.; Gashlan, H.M.; El-Beshbishy, H.A.; Nagy, A.A.; Abdel-Naim, A.B. Caffeic acid phenethyl ester protects against tamoxifen-induced hepatotoxicity in rats. Food Chem. Toxicol 2009, 47, 1689–1695. [Google Scholar]
- Yildiz, O.G.; Soyuer, S.; Saraymen, R.; Eroglu, C. Protective effects of caffeic acid phenethyl ester on radiation induced lung injury in rats. Clin. Invest. Med 2008, 31, E242–E247. [Google Scholar]
- Chen, Y.J.; Liao, H.F.; Tsai, T.H.; Wang, S.Y.; Shiao, M.S. Caffeic acid phenethyl ester preferentially sensitizes CT26 colorectal adenocarcinoma to ionizing radiation without affecting bone marrow radioresponse. Int. J. Radiat. Oncol. Biol. Phys 2005, 63, 1252–1261. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LNCaP 104-S | PC-3 | DU-145 | |
|---|---|---|---|
| AR | + | − | − |
| p53 | + | − | mutant |
| PTEN | − | − | + |
| androgen-dependency | dependent | independent | independent |
| androgen effect on cell proliferation | stimulation | no response | no response |
| differentiation | well | poorly | poorly |
| originality | lymph-node metastases | bone-metastases | brain-metastases |
| PSA production | + | − | − |
| EC50 to CAPE treatment (μM) | 0.68 | 18.65 | 9.54 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lin, H.-P.; Lin, C.-Y.; Liu, C.-C.; Su, L.-C.; Huo, C.; Kuo, Y.-Y.; Tseng, J.-C.; Hsu, J.-M.; Chen, C.-K.; Chuu, C.-P. Caffeic Acid Phenethyl Ester as a Potential Treatment for Advanced Prostate Cancer Targeting Akt Signaling. Int. J. Mol. Sci. 2013, 14, 5264-5283. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14035264
Lin H-P, Lin C-Y, Liu C-C, Su L-C, Huo C, Kuo Y-Y, Tseng J-C, Hsu J-M, Chen C-K, Chuu C-P. Caffeic Acid Phenethyl Ester as a Potential Treatment for Advanced Prostate Cancer Targeting Akt Signaling. International Journal of Molecular Sciences. 2013; 14(3):5264-5283. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14035264
Chicago/Turabian StyleLin, Hui-Ping, Ching-Yu Lin, Chun-Chieh Liu, Liang-Cheng Su, Chieh Huo, Ying-Yu Kuo, Jen-Chih Tseng, Jong-Ming Hsu, Chi-Kuan Chen, and Chih-Pin Chuu. 2013. "Caffeic Acid Phenethyl Ester as a Potential Treatment for Advanced Prostate Cancer Targeting Akt Signaling" International Journal of Molecular Sciences 14, no. 3: 5264-5283. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14035264