CentroidAlign-Web: A Fast and Accurate Multiple Aligner for Long Non-Coding RNAs

Abstract
:1. Introduction
- CentroidAlign-Web can accept long RNA sequences, such as rRNAs. In order to handle those RNA sequences, we have reduced the time complexity of CentroidAlign by integrating the Rfold algorithm [15] into it (see the next section for details).
- Users can (optionally) give the secondary structure(s) of input sequences, if this information is available. For example, secondary structures of long RNA sequences from HIV-1 [16], HCV (hepatitis C virus) [17] and lincRNA (the steroid receptor RNA activator (SRA)) [18] have been recently determined by combining experimental techniques with computational approaches. This secondary structure information is useful for estimating multiple alignments.
- CentroidAlign-Web has an interface in which users can specify a region of the human genome (hg18) from which to extract a multiple alignment, and re-align that region using CentroidAlign. Because recent studies have suggested that re-alignment of genome sequence alignments reveals new non-coding RNAs [19], this function will be useful.
2. Materials and Methods
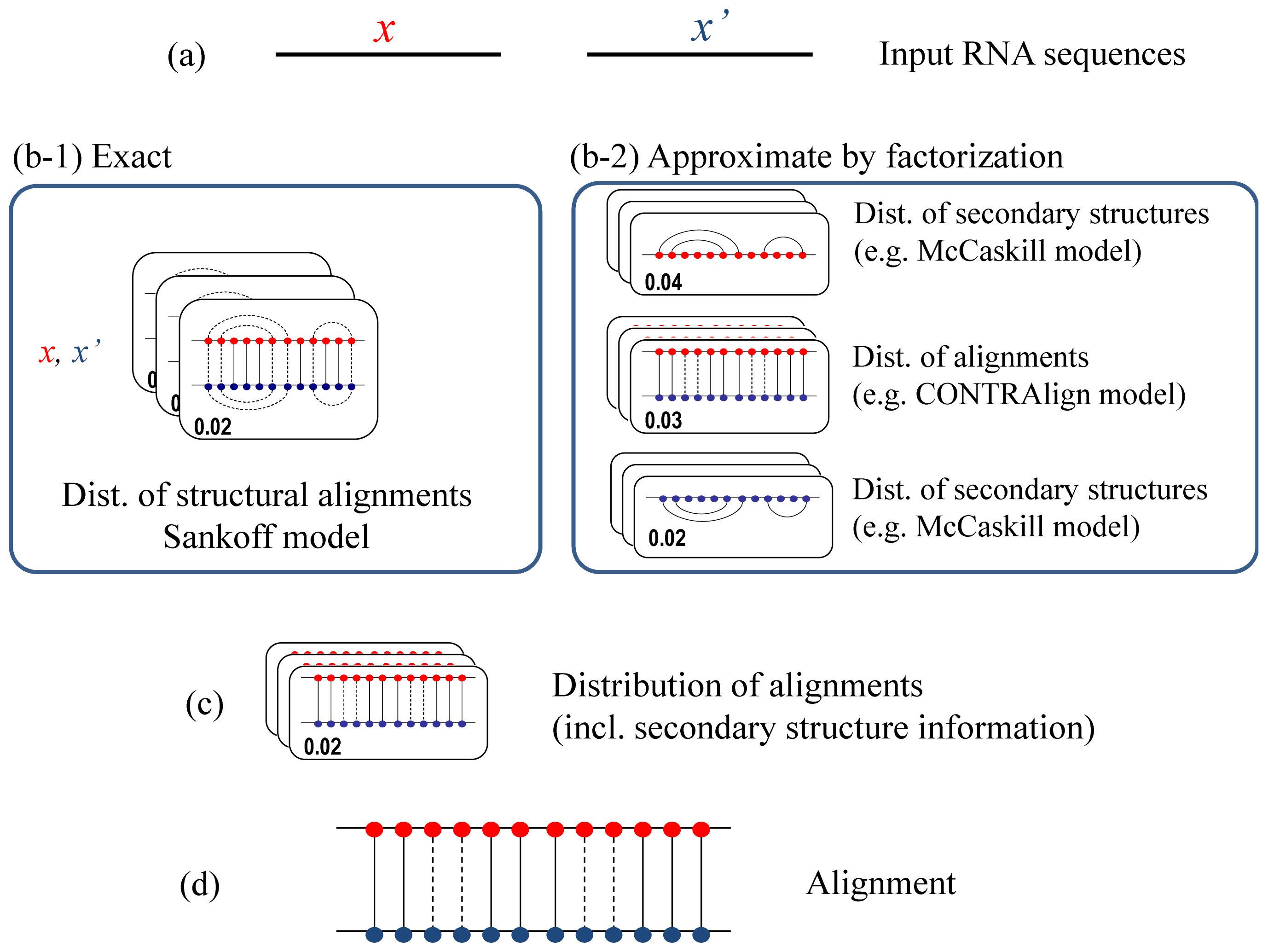
2.1. CentroidAlign
2.2. Rfold
2.3. Dataset Utilized in Computational Experiments
3. Results and Discussion
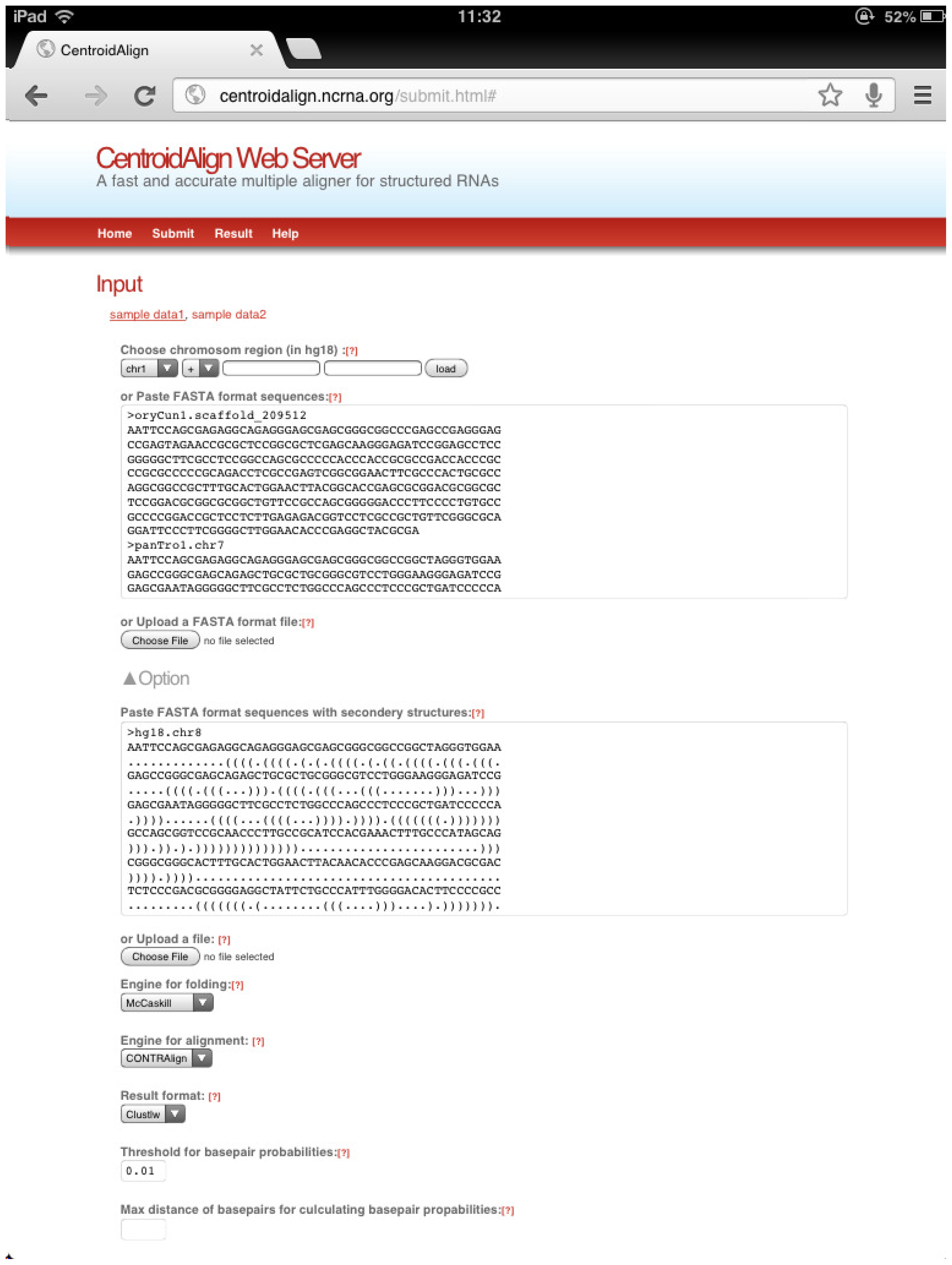
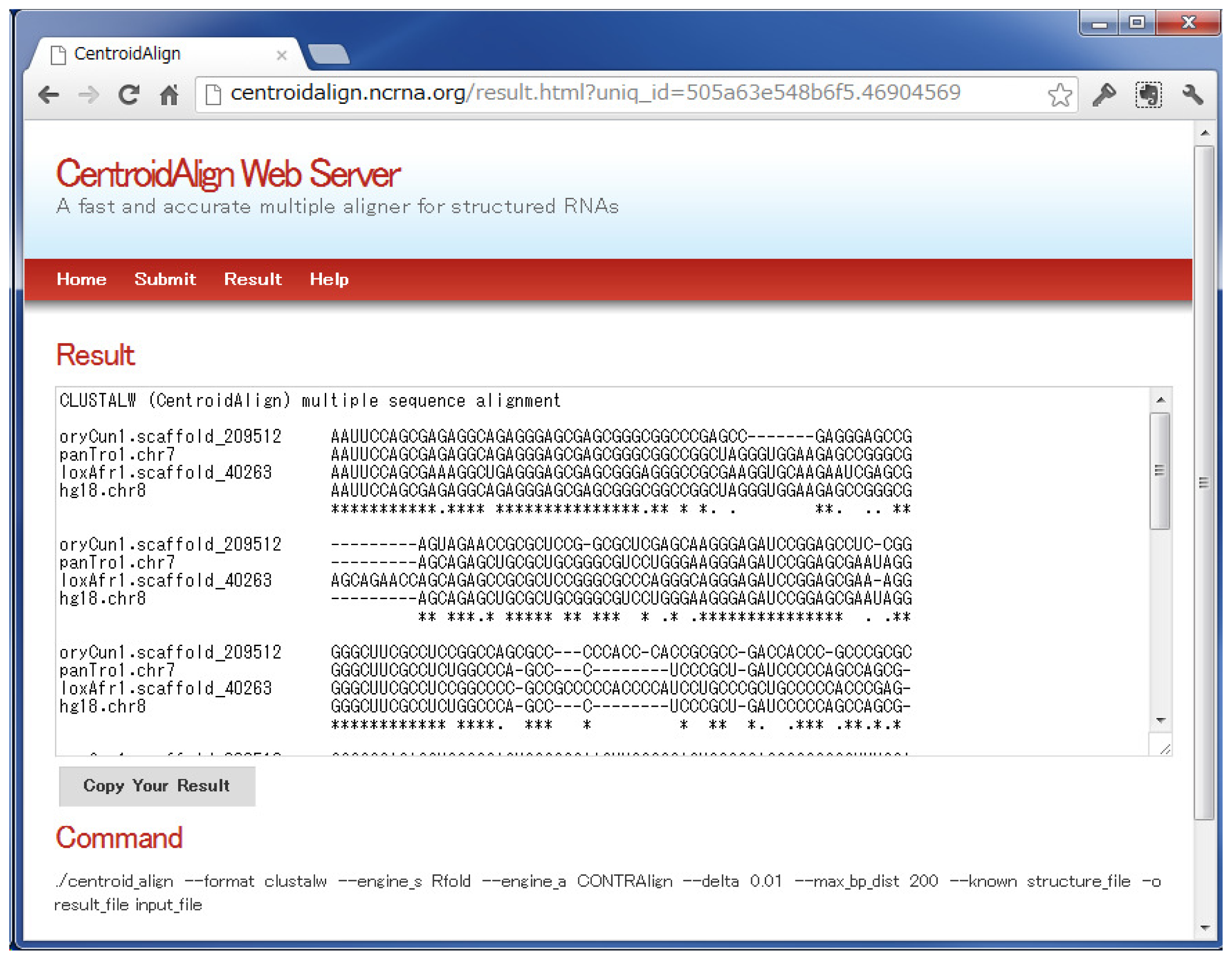
3.1. CentroidAlign Web Application (CentroidAlign-Web)
3.1.1. Incorporating the Rfold Algorithm into the Web Server
3.1.2. BPPM for an RNA Sequence with a Secondary Structure
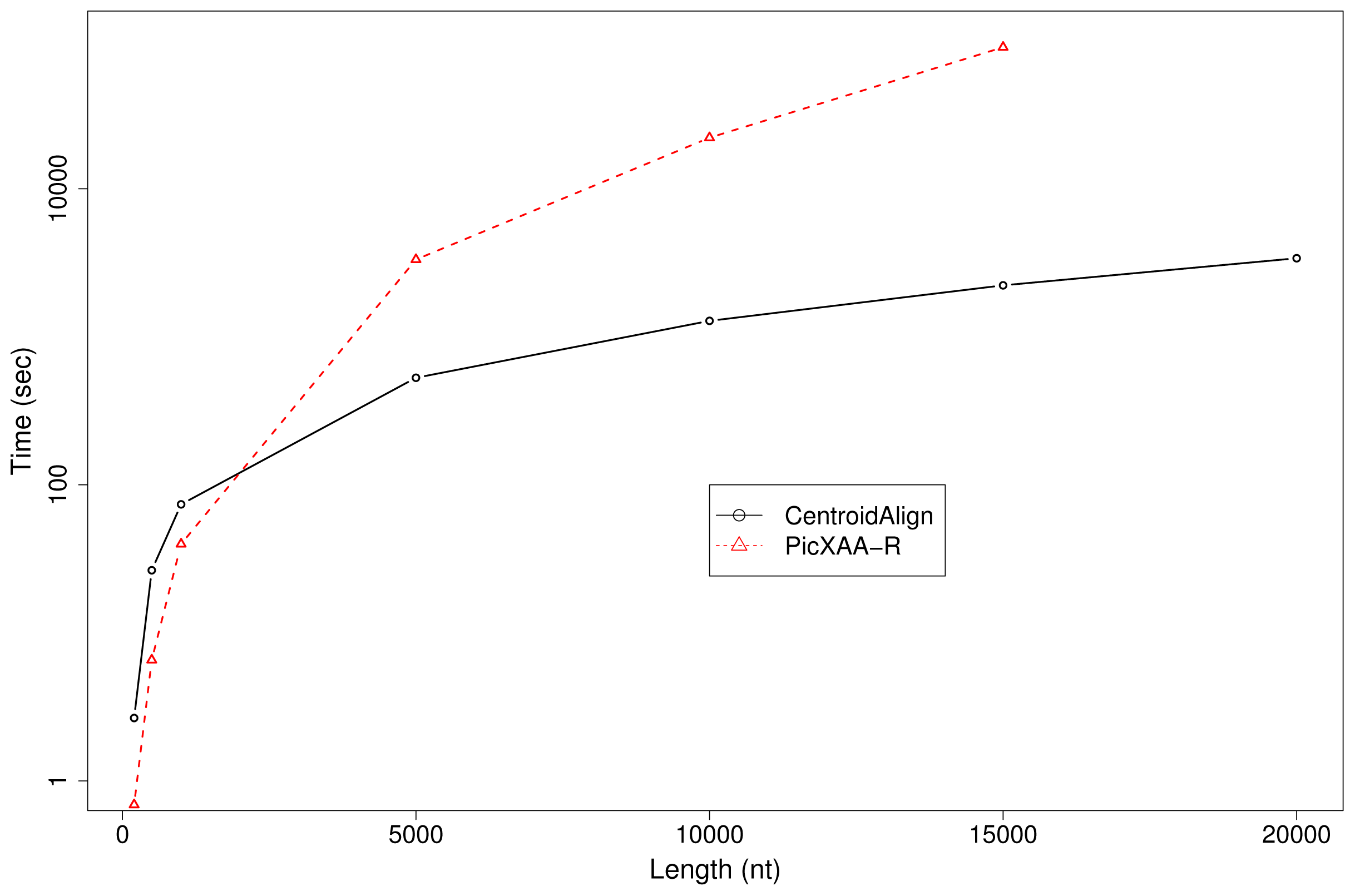
3.2. Computational Experiments
3.3. Future Work
4. Conclusions
Acknowledgments
Appendix

References
- Volders, P.J.; Helsens, K.; Wang, X.; Menten, B.; Martens, L.; Gevaert, K.; Vandesompele, J.; Mestdagh, P. LNCipedia: A database for annotated human lncRNA transcript sequences and structures. Nucl. Acids Res 2013, 41, D246–251. [Google Scholar]
- Gardner, P.P.; Daub, J.; Tate, J.; Moore, B.L.; Osuch, I.H.; Griffiths-Jones, S.; Finn, R.D.; Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R.; et al. Rfam: Wikipedia, clans and the “decimal” release. Nucl. Acids Res. 2011, 39, D141–145. [Google Scholar]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; et al. ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Sankoff, D. Simultaneous solution of the RNA folding alignment and protosequence problems. SIAM J. Appl. Math 1985, 45, 810–825. [Google Scholar]
- Sahraeian, S.M.; Yoon, B.J. PicXAA-Web: A web-based platform for non-progressive maximum expected accuracy alignment of multiple biological sequences. Nucl. Acids Res 2011, 39, 8–12. [Google Scholar]
- Moretti, S.; Wilm, A.; Higgins, D.G.; Xenarios, I.; Notredame, C. R-Coffee: A web server for accurately aligning noncoding RNA sequences. Nucl. Acids Res 2008, 36, W10–W13. [Google Scholar]
- Smith, C.; Heyne, S.; Richter, A.S.; Will, S.; Backofen, R. Freiburg RNA Tools: A web server integrating INTARNA, EXPARNA and LOCARNA. Nucl. Acids Res 2010, 38, W373–W377. [Google Scholar]
- Havgaard, J.H.; Lyngso, R.B.; Gorodkin, J. The FOLDALIGN web server for pairwise structural RNA alignment and mutual motif search. Nucl. Acids Res 2005, 33, W650–W653. [Google Scholar]
- Dalli, D.; Wilm, A.; Mainz, I.; Steger, G. STRAL: Progressive alignment of non-coding RNA using base pairing probability vectors in quadratic time. Bioinformatics 2006, 22, 1593–1599. [Google Scholar]
- Katoh, K.; Toh, H. Improved accuracy of multiple ncRNA alignment by incorporating structural information into a MAFFT-based framework. BMC Bioinforma 2008, 9, 212. [Google Scholar]
- Harmanci, A.O.; Sharma, G.; Mathews, D.H. Efficient pairwise RNA structure prediction using probabilistic alignment constraints in Dynalign. BMC Bioinform 2007, 8, 130. [Google Scholar]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; Tiedje, J.M. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucl. Acids Res 2009, 37, D141–D145. [Google Scholar]
- Bu, D.; Yu, K.; Sun, S.; Xie, C.; Skogerb, G.; Miao, R.; Xiao, H.; Liao, Q.; Luo, H.; Zhao, G.; et al. NONCODE v3.0: Integrative annotation of long noncoding RNAs. Nucl. Acids Res. 2012, 40, D210–215. [Google Scholar]
- Hamada, M.; Sato, K.; Kiryu, H.; Mituyama, T.; Asai, K. CentroidAlign: Fast and accurate aligner for structured RNAs by maximizing expected sum-of-pairs score. Bioinformatics 2009, 25, 3236–3243. [Google Scholar]
- Kiryu, H.; Kin, T.; Asai, K. Rfold: An exact algorithm for computing local base pairing probabilities. Bioinformatics 2008, 24, 367–373. [Google Scholar]
- Watts, J.M.; Dang, K.K.; Gorelick, R.J.; Leonard, C.W.; Bess, J.W.; Swanstrom, R.; Burch, C.L.; Weeks, K.M. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009, 460, 711–716. [Google Scholar]
- Pang, P.S.; Elazar, M.; Pham, E.A.; Glenn, J.S. Simplified RNA secondary structure mapping by automation of SHAPE data analysis. Nucl. Acids Res 2011, 39, e151. [Google Scholar]
- Novikova, I.V.; Hennelly, S.P.; Sanbonmatsu, K.Y. Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucl. Acids Res 2012, 40, 5034–5051. [Google Scholar]
- Will, S.; Yu, M.; Berger, B. Structure-Based Whole Genome Realignment Reveals Many Novel Non-coding RNAs. In RECOMB; Chor, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 7262, p. 341. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2002. [Google Scholar] [CrossRef]
- Do, C.B.; Mahabhashyam, M.S.; Brudno, M.; Batzoglou, S. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res 2005, 15, 330–340. [Google Scholar]
- McCaskill, J.S. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers 1990, 29, 1105–1119. [Google Scholar]
- Do, C.; Gross, S.; Batzoglou, S. CONTRAlign: Discriminative Training for Protein Sequence Alignment. In RECOMB; Apostolico, A., Guerra, C., Istrail, S., Pevzner, P.A., Waterman, M.S., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 3909, pp. 160–174. [Google Scholar]
- Hamada, M.; Asai, K. A classification of bioinformatics algorithms from the viewpoint of maximizing expected accuracy (MEA). J. Comput. Biol 2012, 19, 532–549. [Google Scholar]
- Thompson, J.D.; Plewniak, F.; Poch, O. A comprehensive comparison of multiple sequence alignment programs. Nucl. Acids Res 1999, 27, 2682–2690. [Google Scholar]
- Hamada, M.; Sato, K.; Asai, K. Improving the accuracy of predicting secondary structure for aligned RNA sequences. Nucl. Acids Res 2011, 39, 393–402. [Google Scholar]
- Do, C.B.; Woods, D.A.; Batzoglou, S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics 2006, 22, e90–e98. [Google Scholar]
- Sahraeian, S.M.; Yoon, B.J. PicXAA: Greedy probabilistic construction of maximum expected accuracy alignment of multiple sequences. Nucl. Acids Res 2010, 38, 4917–4928. [Google Scholar]
- Wan, Y.; Kertesz, M.; Spitale, R.C.; Segal, E.; Chang, H.Y. Understanding the transcriptome through RNA structure. Nat. Rev. Genet 2011, 12, 641–655. [Google Scholar]
- Hamada, M. Direct updating of an RNA base-pairing probability matrix with marginal probability constraints. J. Comput. Biol 2012, 19, 1265–1276. [Google Scholar]
- He, S.; Liu, S.; Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evol. Biol 2011, 11, 102. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset name | Accession | Num | Average length(nt) | MPI(%) | Description |
|---|---|---|---|---|---|
| SSU rRNA eukarya | RF01960 | 84 | 1791.20 | 80.00 | Eukaryotic small subunit ribosomal RNA |
| SSU rRNA bacteria | RF00177 | 93 | 1524.50 | 80.00 | Bacterial small subunit ribosomal RNA |
| SSU rRNA archaea | RF01959 | 19 | 1480.50 | 81.00 | Archaeal small subunit ribosomal RNA |
| Sacc telomerase | RF01050 | 13 | 1189.50 | 70.00 | Saccharomyces telomerase |
| snR86 | RF01272 | 5 | 998.40 | 69.00 | Small nucleolar RNA snR86 |
| RUF21 | RF01825 | 5 | 691.80 | 65.00 | RNA of unknown function 21 |
| CentroidAlign | PicXAA-R | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (a) 0% | (b) 25% | (c) 50% | (d) 75% | |||||||||
| ID | Num | Average length (nt) | SPS | Time(s) | SPS | Time(s) | SPS | Time(s) | SPS | Time(s) | SPS | Time(s) |
| RF01960 | 84 | 1791.2 | 0.9173 | 6546.08 | 0.9164 | 5924.78 | 0.9179 | 5317.99 | 0.9208 | 4687.98 | 0.9121 | 8945.82 |
| RF00177 | 93 | 1524.5 | 0.9560 | 5874.18 | 0.9576 | 5314.39 | 0.9589 | 4782.40 | 0.9572 | 4228.57 | 0.9548 | 7544.52 |
| RF01959 | 19 | 1480.5 | 0.9800 | 569.66 | 0.9816 | 474.21 | 0.9817 | 364.89 | 0.9821 | 251.17 | 0.9786 | 508.96 |
| RF01050 | 13 | 1189.5 | 0.8848 | 273.87 | 0.8840 | 219.46 | 0.8861 | 165.06 | 0.8926 | 111.81 | 0.8764 | 155.42 |
| RF01272 | 5 | 998.4 | 0.8975 | 80.06 | 0.9049 | 64.75 | 0.9152 | 48.97 | 0.9286 | 34.31 | 0.8913 | 35.58 |
| RF01825 | 5 | 691.8 | 0.8319 | 44.80 | 0.8467 | 36.38 | 0.8261 | 27.73 | 0.8572 | 18.98 | 0.8236 | 12.53 |
| Parameter name | Description | Possible | Default |
|---|---|---|---|
| Engine for folding | Probabilistic model of secondary structures | McCaskill, CONTRAfold, Rfold 1 | Rfold |
| Engine for alignment | Probabilistic model of pairwise sequence alignments | CONTRAlign, ProbCons 2 | CONTRAlign |
| Result format | Output format | ClustalW, MFA | ClustalW |
| Threshold for base-pair probabilities | Threshold for base-pairing probabilities | 0 to 1 | 0.01 |
| Max distance of base-pairs | The maximum distance of base-pairs | More than 0 | 300 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yonemoto, H.; Asai, K.; Hamada, M. CentroidAlign-Web: A Fast and Accurate Multiple Aligner for Long Non-Coding RNAs. Int. J. Mol. Sci. 2013, 14, 6144-6156. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14036144
Yonemoto H, Asai K, Hamada M. CentroidAlign-Web: A Fast and Accurate Multiple Aligner for Long Non-Coding RNAs. International Journal of Molecular Sciences. 2013; 14(3):6144-6156. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14036144
Chicago/Turabian StyleYonemoto, Haruka, Kiyoshi Asai, and Michiaki Hamada. 2013. "CentroidAlign-Web: A Fast and Accurate Multiple Aligner for Long Non-Coding RNAs" International Journal of Molecular Sciences 14, no. 3: 6144-6156. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14036144