The Role of Sulfur Dioxide in the Regulation of Mitochondrion-Related Cardiomyocyte Apoptosis in Rats with Isopropylarterenol-Induced Myocardial Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
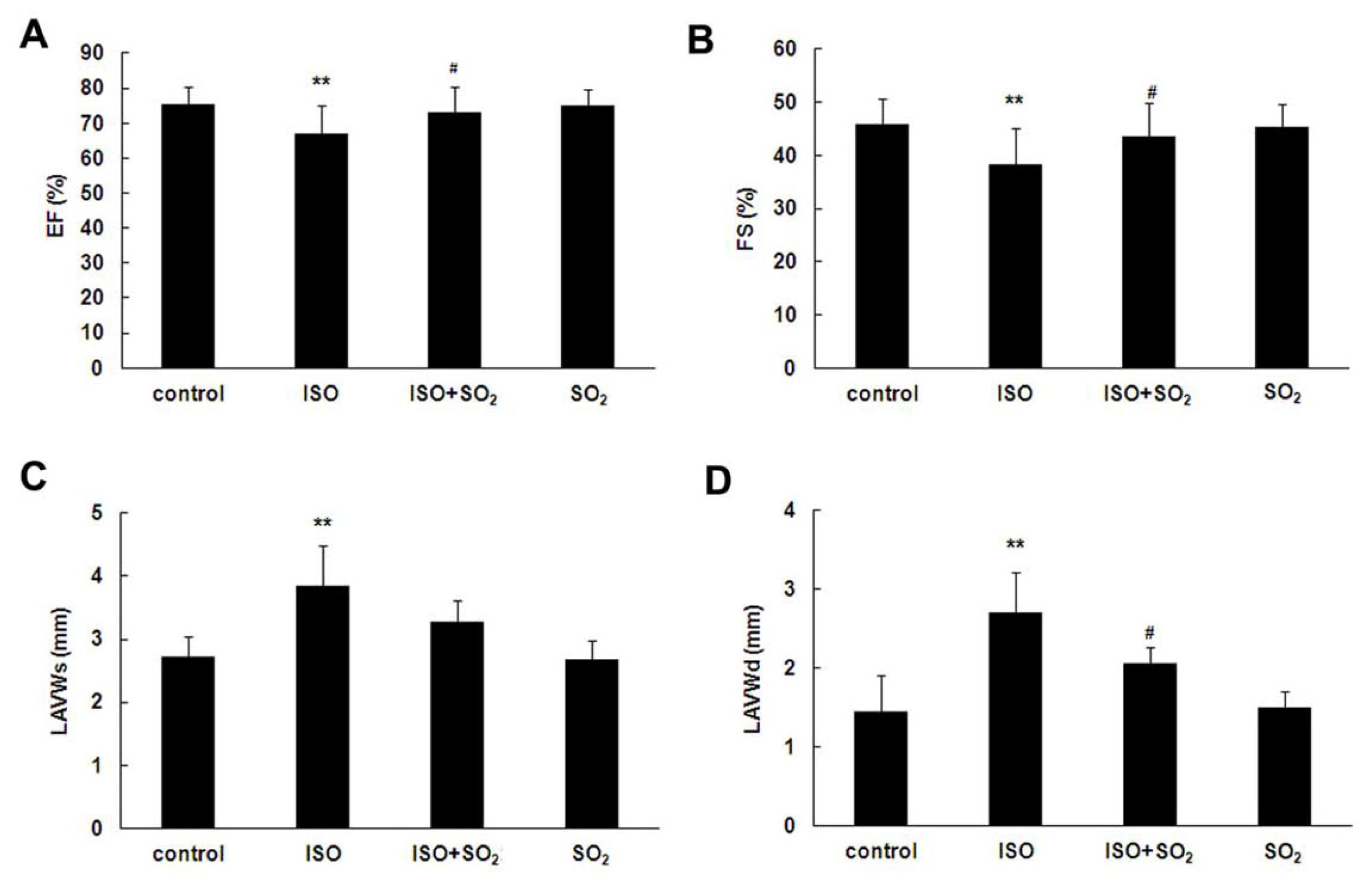
2.1. SO2 Improved Rat Heart Function in ISO-Treated Rats
2.2. SO2 Relieved Myocardial Injury in ISO-Treated Rats
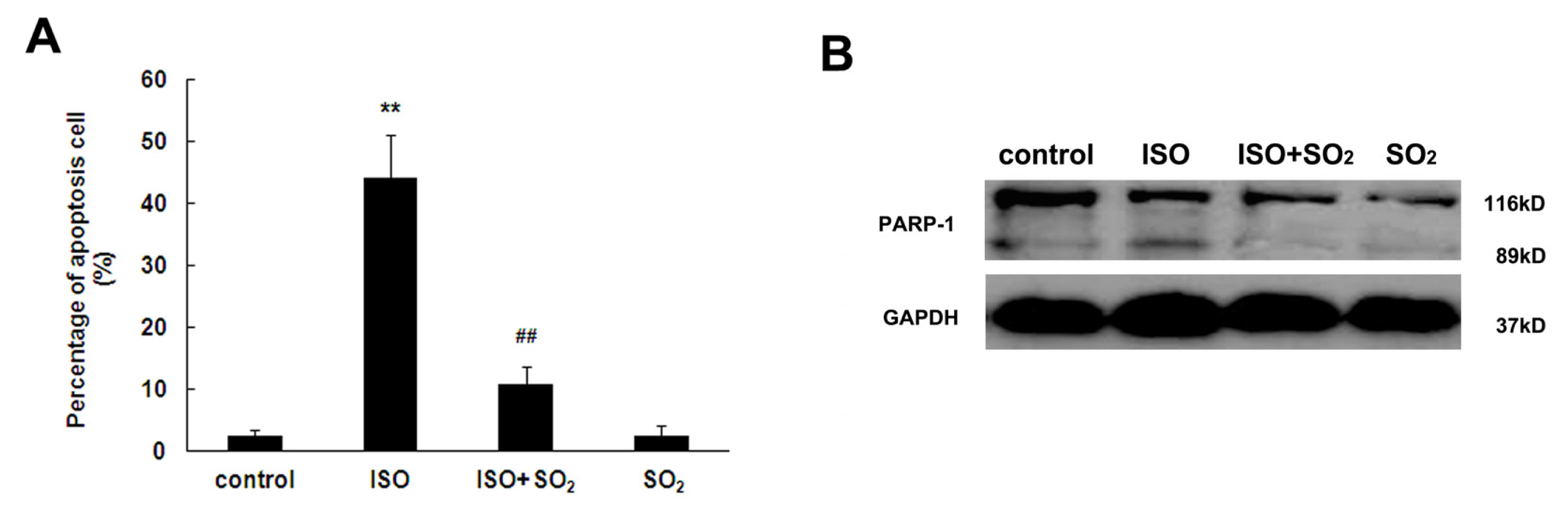
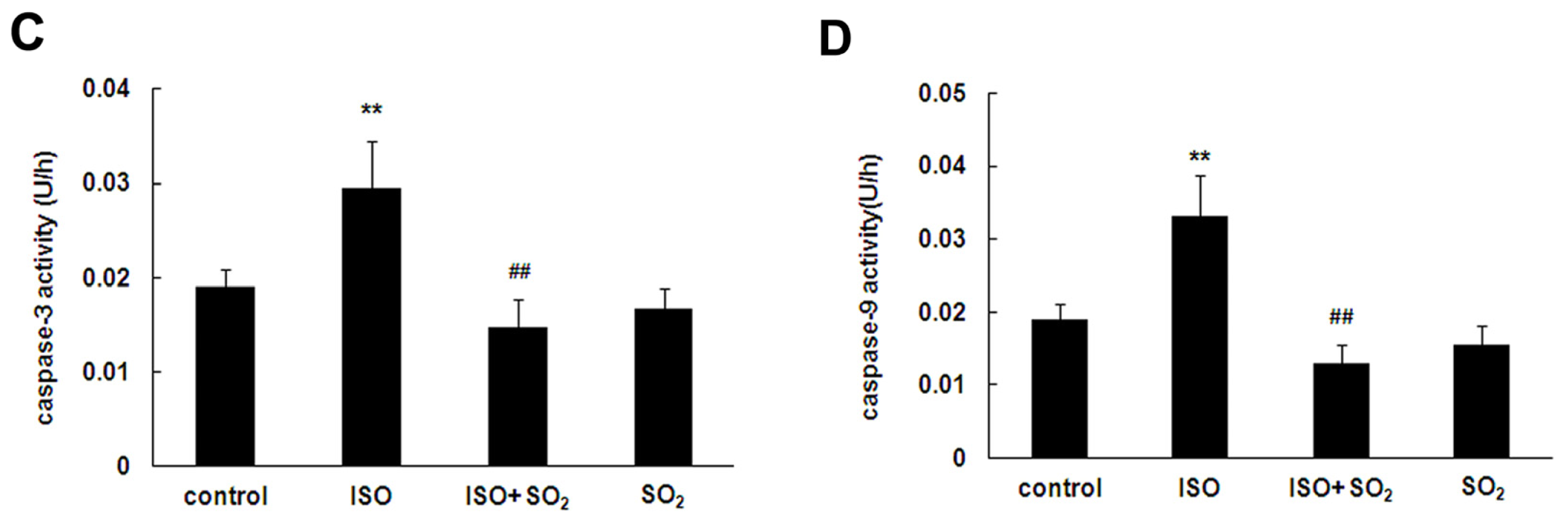
2.3. SO2 Inhibited Cell Apoptosis Induced by ISO in Rat Myocardial Tissues
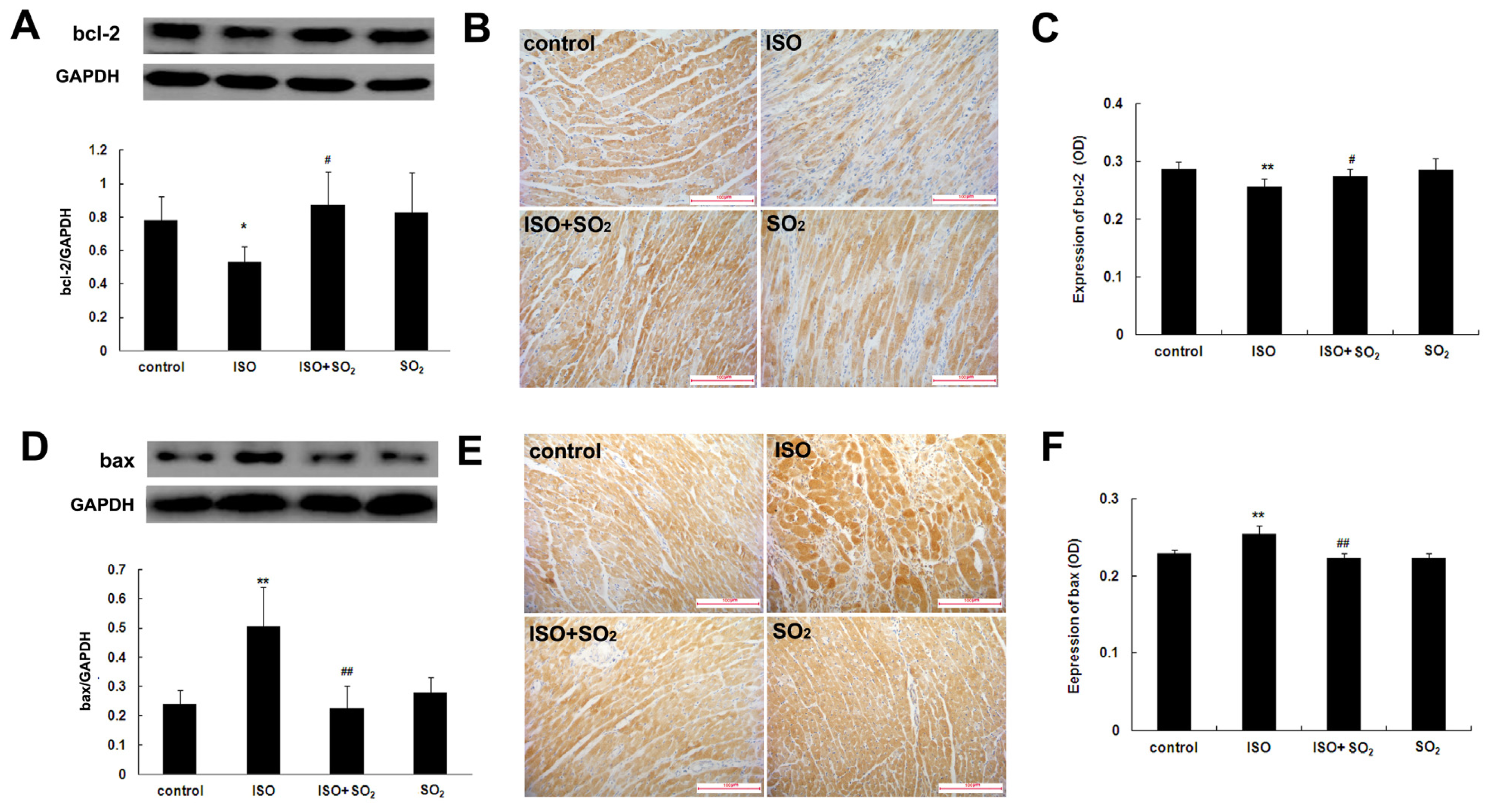
2.4. SO2 Blocked the Effect of ISO on Myocardial bcl-2 and Bax Expression
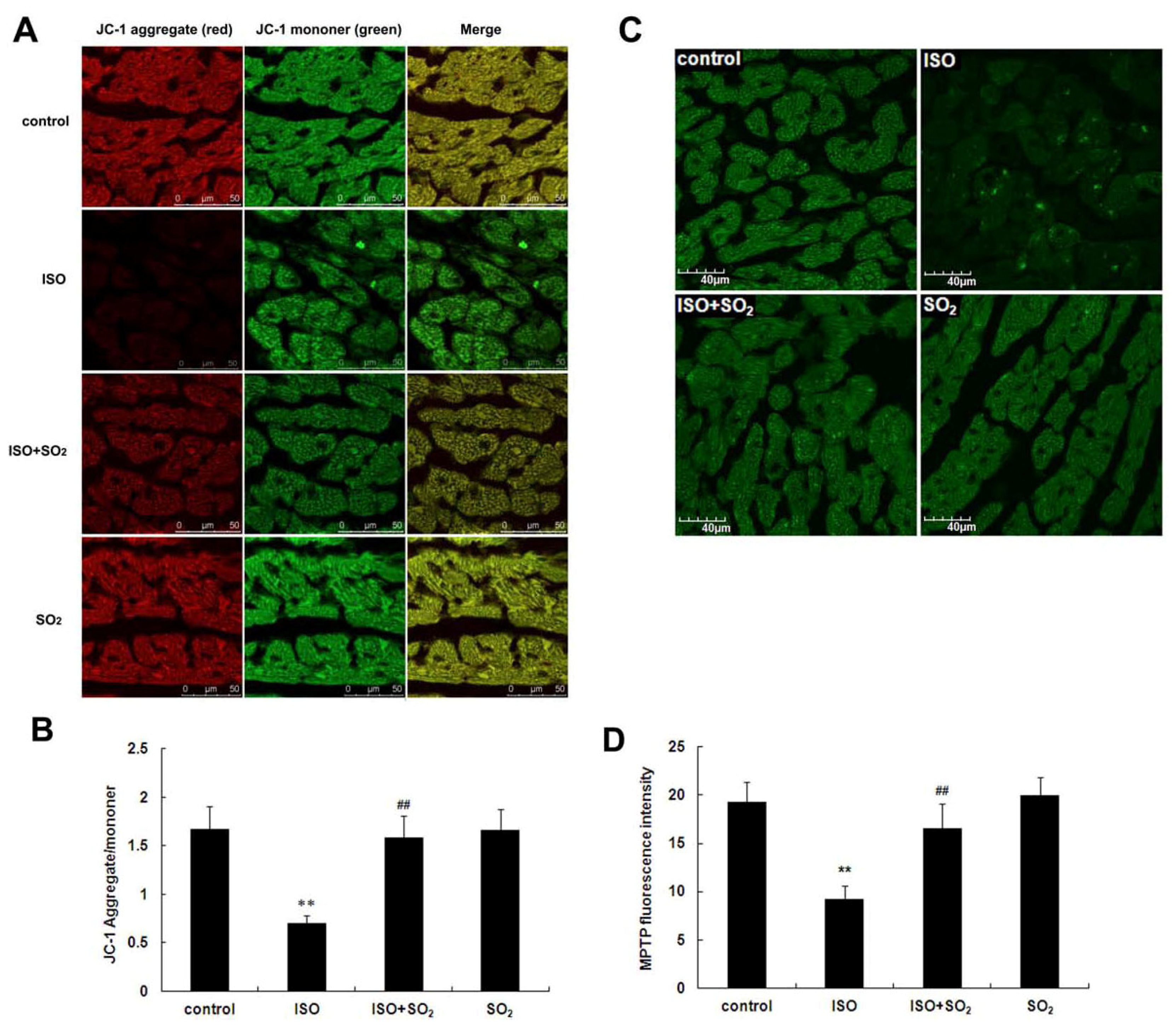
2.5. SO2 Antagonized the Inhibition of Myocardial Mitochondrial Membrane Potential by ISO in Rats
2.6. SO2 Inhibited Myocardial Mitochondrial Permeability Transition Pore (MPTP) Opening Induced by ISO
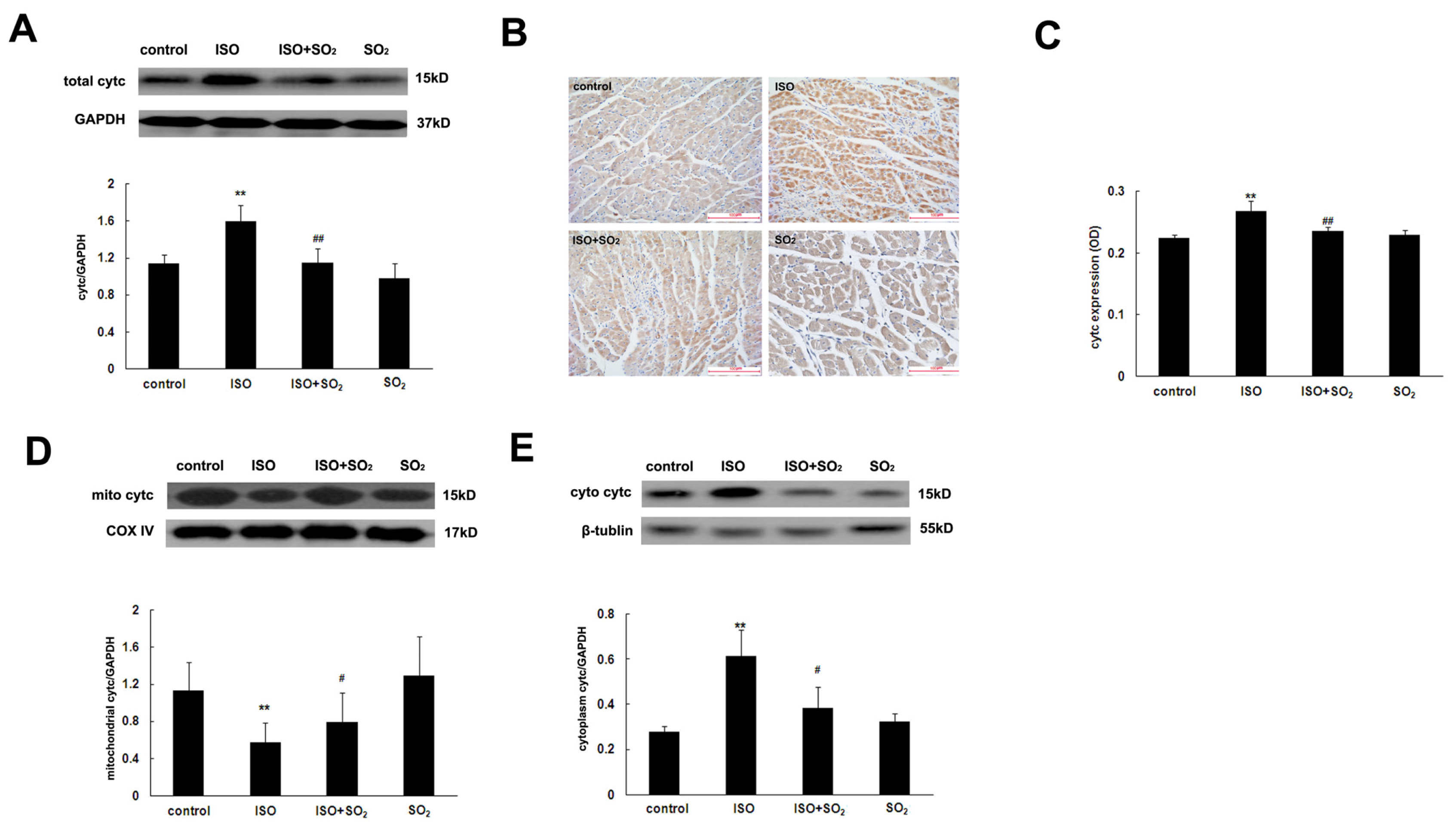
2.7. SO2 Antagonized the Increase in Myocardial Cytc Protein Expression Induced by ISO
2.8. SO2 Inhibited the ISO-Induced Release of Cytc from the Mitochondrion into the Cytoplasm of Cardiomyocytes
3. Discussion
4. Experimental Section
4.1. Animal Model
4.2. Echocardiography Analysis
4.3. Specimen Collection
4.4. Detection of Myocardial Enzymatic Activity in Plasma
4.5. TUNEL Assay
4.6. Isolation of Mitochondrial and Cytosolic Fraction
4.7. Western Blot Assay
4.8. Immunohistochemical Staining Method
4.9. Mitochondrial Membrane Potential Detection
4.10. MPTP Opening Detection
4.11. Caspase-9 Activity Assay
4.12. Caspase-3 Activity Assay
4.13. Statistical Analysis
5. Conclusion
Acknowledgments
Conflict of Interest
References
- Singh, K.P.; Jaffe, A.S.; Liang, B.T. The clinical impact of circulating caspase-3 p17 level: A potential new biomarker for myocardial injury and cardiovascular disease. Future Cardiol 2011, 7, 443–445. [Google Scholar]
- Virag, J.A.; Dries, J.L.; Easton, P.R.; Friesland, A.M.; DeAntonio, J.H.; Chintalgattu, V.; Cozzi, E.; Lehmann, B.D.; Ding, J.M.; Lust, R.M. Attenuation of myocardial injury in mice with functional deletion of the circadian rhythm gene mPer2. Am. J. Physiol. Heart Circ. Physiol 2010, 298, H1088–H1095. [Google Scholar]
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell death in the pathogenesis of heart disease: Mechanisms and significance. Annu. Rev. Physiol 2010, 72, 19–44. [Google Scholar]
- Chen, T.M.; Gokhale, J.; Shofer, S.; Kuschner, W.G. Outdoor air pollution: Nitrogen dioxide, sulfur dioxide, and carbon monoxide health effects. Am. J. Med. Sci 2007, 333, 249–256. [Google Scholar]
- Rich, D.Q.; Schwartz, J.; Mittleman, M.A.; Link, M.; Luttmann-Gibson, H.; Catalano, P.J.; Speizer, F.E.; Dockery, D.W. Association of short term ambient air pollution concentrations and ventricular arrhythmias. Am. J. Epidemiol 2005, 161, 1123–1132. [Google Scholar]
- Stipanuk, M.H. Metabolism of sulfur-containing amino acids. Annu. Rev. Nutr 1986, 6, 179–209. [Google Scholar]
- Du, S.X.; Jin, H.F.; Bu, D.F.; Zhao, X.; Geng, B.; Tang, C.S.; Du, J.B. Endogenously generated sulfur dioxide and its vasorelaxant effect in rats. Acta Pharmacol. Sin 2008, 29, 923–930. [Google Scholar]
- Luo, L.M.; Chen, S.; Jin, H.F.; Tang, C.S.; Du, J.B. Endogenous generation of sulfur dioxide in rat tissues. Biochem. Biophys. Res. Commun 2011, 415, 61–67. [Google Scholar]
- Meng, Z.; Yang, Z.; Li, J.; Zhang, Q. The vasorelaxant effect and its mechanisms of sodium bisulfite as a sulfur dioxide donor. Chemosphere 2012, 89, 579–584. [Google Scholar]
- Zhang, Q.; Meng, Z. The negative inotropic effects of gaseous sulfur dioxide and its derivatives in the isolated perfused rat heart. Environ. Toxicol 2012, 27, 175–184. [Google Scholar]
- Li, W.; Tang, C.S.; Jin, H.F.; Du, J.B. Regulatory effects of sulfur dioxide on the development of atherosclerotic lesions and vascular hydrogen sulfide in atherosclerotic rats. Atherosclerosis 2011, 215, 323–330. [Google Scholar]
- Sun, Y.; Tian, Y.; Prabha, M.; Liu, D.; Chen, S.; Zhang, R.; Liu, X.; Tang, C.; Tang, X.; Jin, H.; et al. Effects of sulfur dioxide on hypoxic pulmonary vascular structural remodeling. Lab. Invest 2010, 90, 68–82. [Google Scholar]
- Liang, Y.F.; Liu, D.; Ochs, T.; Tang, C.S.; Chen, S.; Zhang, S.Q.; Geng, B.; Jin, H.F.; Du, J.B. Endogenous sulfur dioxide protects against isoproterenol-induced myocardial injury and increases myocardial antioxidant capacity in rats. Lab. Invest 2011, 91, 12–23. [Google Scholar]
- Jin, H.F.; Wang, Y.; Wang, X.B.; Sun, Y.; Tang, C.S.; Du, J.B. Sulfur dioxide preconditioning increases antioxidative capacity in rat with myocardial ischemia reperfusion (I/R) injury. Nitric Oxide 2013. [Google Scholar] [CrossRef]
- Chen, S.S.; Du, J.B.; Liang, Y.F.; Ochs, T.; Liu, D.; Zhu, L.L.; Tang, X.Y.; Tang, C.S.; Jin, H.F. Sulfur dioxide inhibits excessively activated endoplasmic reticulum stress in rats with myocardial injury. Heart Vessels 2012, 27, 505–516. [Google Scholar]
- Chen, S.S.; Du, J.B.; Liang, Y.F.; Zhang, R.Y.; Tang, C.S.; Jin, H.F. Sulfur dioxide restores calcium homeostasis disturbance in rat with isoproterenol-induced myocardial injury. Histol. Histopathol 2012, 27, 1219–1226. [Google Scholar]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res 2012, 111, 1198–1207. [Google Scholar]
- Brunelle, J.K.; Letai, A. Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci 2009, 122, 437–441. [Google Scholar]
- Carson, P.; Giles, T.; Higginbotham, M.; Hollenberg, N.; Kannel, W.; Siragy, H.M. Angiotensin receptor blockers: Evidence for preserving target organs. Clin. Cardiol 2001, 24, 183–190. [Google Scholar]
- Hassan, M.A.; Ketat, A.F. Sildenafil citrate increases myocardial cGMP content in rat heart, decreases its hypertrophic response to isoproterenol and decreases myocardial leak of creatine kinase and troponin T. BMC Pharmacol. 2005, 5. [Google Scholar] [CrossRef] [Green Version]
- Geng, B.; Chang, L.; Pan, C.S.; Qi, Y.F.; Zhao, J.; Pang, Y.Z.; Du, J.B.; Tang, C.S. Endogenous hydrogen sulfide regulation of myocardial injury induced by isoproterenol. Biochem. Biophys. Res. Commun 2004, 318, 756–763. [Google Scholar]
- Yang, J.; Maity, B.; Huang, J.; Gao, Z.; Stewart, A.; Weiss, R.M.; Anderson, M.E.; Fisher, R.A. G-protein inactivator RGS6 mediates myocardial cell apoptosis and cardiomyopathy caused by doxorubicin. Cancer Res 2013, 73, 1662–1667. [Google Scholar]
- Murriel, C.L.; Churchill, E.; Inagaki, K.; Szweda, L.I.; Mochly-Rosen, D. Protein kinase activation induces apoptosis in response to cardiac ischemia and reperfusion damage: A mechanism involving BAD and the mitochondria. J. Biol. Chem 2004, 279, 47985–47991. [Google Scholar]
- Yang, B.; Ye, D.; Wang, Y. Caspase-3 as a therapeutic target for heart failure. Expert Opin. Ther. Targets 2013, 17, 255–263. [Google Scholar]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem 1999, 274, 22932–22940. [Google Scholar]
- Kuida, K.; Haydar, T.F.; Kuan, C.Y.; Gu, Y.; Taya, C.; Karasuyama, H.; Su, M.S.; Rakic, P.; Flavell, R.A. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 1998, 94, 325–337. [Google Scholar]
- Lin, J.W.; Chen, J.T.; Hong, C.Y.; Lin, Y.L.; Wang, K.T.; Yao, C.J.; Lai, G.M.; Chen, R.M. Honokiol traverses the blood-brain barrier and induces apoptosis of neuroblastoma cells via an intrinsic bax-mitochondrion-cytochrome c-caspase protease pathway. Neuro Oncol 2012, 14, 302–314. [Google Scholar]
- Adams, J.M.; Cory, S. Bcl-2-regulated apoptosis: Mechanism and therapeutic potential. Curr. Opin. Immunol 2007, 19, 488–496. [Google Scholar]
- Kirkland, R.A.; Franklin, J.L. Bax, reactive oxygen, and cytochrome c release in neuronal apoptosis. Antioxid. Redox Signal 2003, 5, 589–596. [Google Scholar]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar]
- Wang, J.; Yuan, L.; Xiao, H.; Xiao, C.; Wang, Y.; Liu, X. Momordin Ic induces HepG2 cell apoptosis through MAPK and PI3K/Akt-mediated mitochondrial pathways. Apoptosis 2013, 18, 751–765. [Google Scholar]
- Zhao, M.M.; Yang, J.Y.; Wang, X.B.; Tang, C.S.; Du, J.B.; Jin, H.F. The PI3K/Akt pathway mediates the protection of SO2 preconditioning against myocardial ischemia/reperfusion injury in rats. Acta Pharmacol. Sin 2013, 34, 501–506. [Google Scholar]
- Caroppi, P.; Sinibaldi, F.; Fiorucci, L.; Santucci, R. Apoptosis and human diseases: Mitochondrion damage and lethal role of released cytochrome C as proapoptotic protein. Curr. Med. Chem 2009, 16, 4058–4065. [Google Scholar]
- Loor, G.; Kondapalli, J.; Iwase, H.; Chandel, N.S.; Waypa, G.B.; Guzy, R.D.; Vanden Hoek, T.L.; Schumacker, P.T. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim. Biophys. Acta 2011, 1813, 1382–1394. [Google Scholar]
- Cunha, D.A.; Igoillo-Esteve, M.; Gurzov, E.N.; Germano, C.M.; Naamane, N.; Marhfour, I.; Fukaya, M.; Vanderwinden, J.M.; Gysemans, C.; Mathieu, C.; et al. Death protein 5 and p53-upregulated modulator of apoptosis mediate the endoplasmic reticulum stress-mitochondrial dialog triggering lipotoxic rodent and human β-cell apoptosis. Diabetes 2012, 61, 2763–2775. [Google Scholar]
- Shi, Y.X.; Chen, Y.; Zhu, Y.Z.; Huang, G.Y.; Moore, P.K.; Huang, S.H.; Yao, T.; Zhu, Y.C. Chronic sodium hydrosulfide treatment decreases medial thickening of intramyocardial coronary arterioles, interstitial fibrosis, and ROS production in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol 2007, 293, H2093–H2100. [Google Scholar]
- Gavrieli, Y.; Sherman, Y.; Ben-Sasson, S.A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol 1992, 119, 493–501. [Google Scholar]
- Reers, M.; Smiley, S.T.; Mottola-Hartshorn, C.; Chen, A.; Lin, M.; Chen, L.B. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol 1995, 260, 406–417. [Google Scholar]
- Zhao, C.Q.; Zhang, Y.H.; Jiang, S.D.; Jiang, L.S.; Dai, L.Y. Both endoplasmic reticulum and mitochondria are involved in disc cell apoptosis and intervertebral disc degeneration in rats. Age (Dordr) 2010, 32, 161–171. [Google Scholar]
- Angeloni, C.; Hrelia, S. Quercetin reduces inflammatory responses in LPS-stimulated cardiomyoblasts. Oxid. Med. Cell. Longev. 2012, 2012. [Google Scholar] [CrossRef]







© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jin, H.; Liu, A.D.; Holmberg, L.; Zhao, M.; Chen, S.; Yang, J.; Sun, Y.; Chen, S.; Tang, C.; Du, J. The Role of Sulfur Dioxide in the Regulation of Mitochondrion-Related Cardiomyocyte Apoptosis in Rats with Isopropylarterenol-Induced Myocardial Injury. Int. J. Mol. Sci. 2013, 14, 10465-10482. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms140510465
Jin H, Liu AD, Holmberg L, Zhao M, Chen S, Yang J, Sun Y, Chen S, Tang C, Du J. The Role of Sulfur Dioxide in the Regulation of Mitochondrion-Related Cardiomyocyte Apoptosis in Rats with Isopropylarterenol-Induced Myocardial Injury. International Journal of Molecular Sciences. 2013; 14(5):10465-10482. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms140510465
Chicago/Turabian StyleJin, Hongfang, Angie Dong Liu, Lukas Holmberg, Manman Zhao, Siyao Chen, Jinyan Yang, Yan Sun, Shanshan Chen, Chaoshu Tang, and Junbao Du. 2013. "The Role of Sulfur Dioxide in the Regulation of Mitochondrion-Related Cardiomyocyte Apoptosis in Rats with Isopropylarterenol-Induced Myocardial Injury" International Journal of Molecular Sciences 14, no. 5: 10465-10482. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms140510465