Molecular Motors and Apical CFTR Traffic in Epithelia

Abstract
:1. Introduction
1.1. CFTR in Organs and Tissues
1.2. Regulation of CFTR by Trafficking
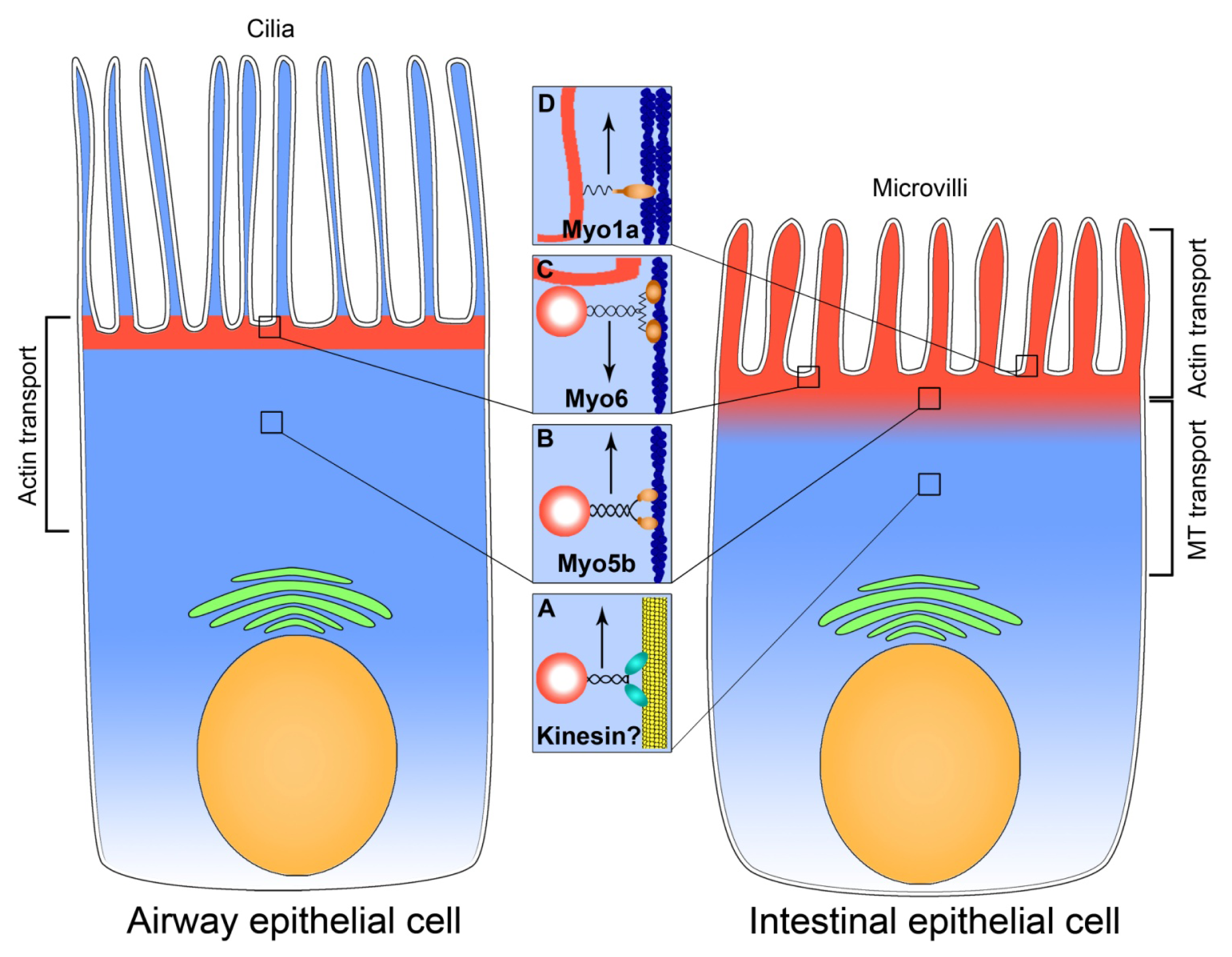
2. Microtubule-Dependent Transport in CFTR Trafficking
2.1. Intestinal Epithelium
2.2. Airway Epithelium
2.3. Renal Epithelium
2.4. Summary: Microtubule-Dependent Delivery
3. Actin-Based Transport in CFTR Trafficking
3.1. Intestinal Epithelium
3.1.1. Myosin Vb and Exocytosis of CFTR
3.1.2. Myosin VI Regulates Endocytosis of CFTR in the Intestine
3.1.3. Myosin Ia is a Brush Border-Specific Regulator of CFTR
3.2. Airway Epithelium
3.2.1. Myosin VI Facilitates Endocytosis of CFTR
3.2.2. Myosin Vb Regulates Exocytosis of CFTR in the Airway Epithelial Cells
3.3. Summary on Myosins
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Gadsby, D.C.; Vergani, P.; Csanady, L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 2006, 440, 477–483. [Google Scholar]
- Harris, A.; Charkley, G.; Goodman, S.; Coleman, L. Expression of the cystic fibrosis gene in human development. Development 1991, 113, 305–310. [Google Scholar]
- O’Loughlin, E.V.; Hunt, D.M.; Gaskin, K.J.; Steil, D.; Bruzuszcak, I.M.; Martin, H.C.O.; Bambach, C.; Smith, R. X-ray microanalysis of cell elements in normal and cystic fibrosis Jejunum: Evidence for chloride secretion in villi. Gastroenterology 1996, 110, 411–418. [Google Scholar]
- Pratha, V.S.; Hogan, D.L.; Martensson, B.A.; Bernard, J.; Zhou, R.; Isenberg, J.I. Identification of transport abnormalities in duodenal mucosa and duodenal enterocytes from patients with cystic fibrosis. Gastroenterology 2000, 118, 1051–1060. [Google Scholar]
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic fibrosis. N. Engl. J. Med 2005, 352, 1992–2001. [Google Scholar]
- Pilewski, J.M.; Frizzell, R.A. Role of CFTR in airway disease. Physiol. Rev 1999, 79, S215–S255. [Google Scholar]
- Terheggen-Lagro, S.W.; Rijkers, G.T.; van der Ent, C.K. The role of airway epithelium and blood neutrophils in the inflammatory response in cystic fibrosis. J. Cyst. Fibros 2005, 4, 15–23. [Google Scholar]
- Machen, T.E. Innate immune response in CF airway epithelia: Hyperinflammatory? Am. J. Physiol. Cell Physiol 2006, 291, C218–C230. [Google Scholar]
- Bridges, R.J. Mechanisms of bicarbonate secretion: Lessons from the airways. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Jakab, R.L.; Collaco, A.M.; Ameen, N.A. Physiological relevance of cell-specific distribution patterns of CFTR, NKCC1, NBCe1, and NHE3 along the crypt-villus axis in the intestine. Am. J. Physiol. Gastrointest. Liver Physiol 2011, 300, G82–G98. [Google Scholar]
- Ameen, N.; Silvis, M.; Bradbury, N.A. Endocytic trafficking of CFTR in health and disease. J. Cyst. Fibros 2007, 6, 1–14. [Google Scholar]
- Lukacs, G.L.; Chang, X.B.; Bear, C.; Kartner, N.; Mohamed, A.; Riordan, J.R.; Grinstein, S. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J. Biol. Chem 1993, 268, 21592–21598. [Google Scholar]
- Thiagarajah, J.R.; Verkman, A.S. CFTR pharmacology and its role in intestinal fluid secretion. Curr. Opin. Pharmacol 2003, 3, 594–599. [Google Scholar]
- Ameen, N.; Alexis, J.; Salas, P. Cellular localization of the cystic fibrosis transmembrane conductance regulator in mouse intestinal tract. Histochem. Cell Biol 2000, 114, 69–75. [Google Scholar]
- Ameen, N.A.; van Donselaar, E.; Posthuma, G.; de Jonge, H.; McLaughlin, G.; Geuze, H.J.; Marino, C.; Peters, P.J. Subcellular distribution of CFTR in rat intestine supports a physiologic role for CFTR regulation by vesicle traffic. Histochem. Cell Biol 2000, 114, 219–228. [Google Scholar]
- Guggino, W.B.; Stanton, B.A. New insights into cystic fibrosis: Molecular switches that regulate CFTR. Nat. Rev. Mol. Cell Biol 2006, 7, 426–436. [Google Scholar]
- Collaco, A.; Marathe, J.; Kohnke, H.; Kravstov, D.; Ameen, N. Syntaxin 3 is necessary for cAMP- and cGMP-regulated exocytosis of CFTR: Implications for enterotoxigenic diarrhea. Am. J. Physiol. Cell Physiol 2010, 299, C1450–C1460. [Google Scholar]
- Kravtsov, D.V.; Caputo, C.; Collaco, A.; Hoekstra, N.; Egan, M.E.; Mooseker, M.S.; Ameen, N.A. Myosin Ia is required for CFTR brush border membrane trafficking and ion transport in the mouse small intestine. Traffic 2012, 13, 1072–1082. [Google Scholar]
- Gentzsch, M.; Chang, X.B.; Cui, L.; Wu, Y.; Ozols, V.V.; Choudhury, A.; Pagano, R.E.; Riordan, J.R. Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2004, 15, 2684–2696. [Google Scholar]
- Swiatecka-Urban, A.; Talebian, L.; Kanno, E.; Moreau-Marquis, S.; Coutermarsh, B.; Hansen, K.; Karlson, K.H.; Barnaby, R.; Cheney, R.E.; Langford, G.M.; et al. Myosin Vb is required for trafficking of the cystic fibrosis transmembrane conductance regulator in Rab11a-specific apical recycling endosomes in polarized human airway epithelial cells. J. Biol. Chem 2007, 282, 23725–23736. [Google Scholar]
- Silvis, M.R.; Bertrand, C.A.; Ameen, N.; Golin-Bisello, F.; Butterworth, M.B.; Frizzell, R.A.; Bradbury, N.A. Rab11b regulates the apical recycling of the cystic fibrosis transmembrane conductance regulator in polarized intestinal epithelial cells. Mol. Biol. Cell 2009, 20, 2337–2350. [Google Scholar]
- Cheng, S.H.; Rich, D.P.; Marshall, J.; Gregory, R.J.; Welsh, M.J.; Smith, A.E. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell 1991, 66, 1027–1036. [Google Scholar]
- Tien, X.-Y.; Brasitus, T.A.; Kaetzel, M.A.; Dedman, J.R.; Nelson, D.J. Activation of the cystic fibrosis transmembrane conductance regulator by cGMP in the human colonic cancer cell line, Caco-2. J. Biol. Chem 1994, 269, 51–54. [Google Scholar]
- Schwiebert, E.M.; Gesek, F.; Ercolani, L.; Wjasow, C.; Gruenert, D.C.; Karlson, K.; Stanton, B.A. Heterotrimeric G proteins, vesicle trafficking, and CFTR Cl− channels. Am. J. Physiol 1994, 267, C272–C281. [Google Scholar]
- Ameen, N.A.; Marino, C.; Salas, P.J. cAMP-dependent exocytosis and vesicle traffic regulate CFTR and fluid transport in rat jejunum in vivo. Am. J. Physiol. Cell Physiol 2003, 284, C429–C438. [Google Scholar]
- Rogan, M.P.; Stoltz, D.A.; Hornick, D.B. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest 2011, 139, 1480–1490. [Google Scholar]
- Vale, R.D. The molecular motor toolbox for intracellular transport. Cell 2003, 112, 467–480. [Google Scholar]
- Grotmol, T.; Van Dyke, R.W. Prostaglandin- and theophylline-induced C1 secretion in rat distal colon is inhibited by microtubule inhibitors. Dig. Dis. Sci 1992, 37, 1709–1717. [Google Scholar]
- Morris, A.P.; Cunningham, S.A.; Tousson, A.; Benos, D.J.; Frizzell, R.A. Polarization-dependent apical membrane CFTR targeting underlies cAMP-stimulated Cl− secretion in epithelial cells. Am. J. Physiol 1994, 266, C254–C268. [Google Scholar]
- Tousson, A.; Fuller, C.M.; Benos, D.J. Apical recruitment of CFTR in T84 cells is dependent on cAMP and microtubules but not calcium or microfilaments. J. Cell Sci 1996, 109, 1325–1334. [Google Scholar]
- Hiebsch, R.R.; Raub, T.J.; Wattenberg, B.W. Primaquine blocks transport by inhibiting the formation of functional transport vesicles. Studies in a cell-free assay of protein transport through the Golgi apparatus. J. Biol. Chem 1991, 266, 20323–20328. [Google Scholar]
- Loffing, J.; Moyer, B.D.; McCoy, D.; Stanton, B.A. Exocytosis is not involved in activation of Cl− secretion via CFTR in Calu-3 airway epithelial cells. Am. J. Physiol 1998, 275, C913–C920. [Google Scholar]
- Jin, M.; Snider, M.D. Role of microtubules in transferrin receptor transport from the cell surface to endosomes and the Golgi complex. J. Biol. Chem 1993, 268, 18390–18397. [Google Scholar]
- Morris, R.G.; Tousson, A.; Benos, D.J.; Schafer, J.A. Microtubule disruption inhibits AVT-stimulated Cl− secretion but not Na+ reabsorption in A6 cells. Am. J. Physiol 1998, 274, F300–F314. [Google Scholar]
- Moyer, B.D.; Loffing, J.; Schwiebert, E.M.; Loffing-Cueni, D.; Halpin, P.A.; Karlson, K.H.; Ismailov, I.L.; Guggino, W.B.; Langford, G.M.; Stanton, B.A. Membrane trafficking of the cystic fibrosis gene product,cystic fibrosis transmembrane conductance regulator,tagged with green fluorescent protein in madin-darby kidney cells. J. Biol. Chem 1998, 21, 21759–21768. [Google Scholar]
- Johnston, J.A.; Illing, M.E.; Kopito, R.R. Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil. Cytoskeleton 2002, 53, 26–38. [Google Scholar]
- Bannykh, S.I.; Bannykh, G.I.; Fish, K.N.; Moyer, B.D.; Riordan, J.R.; Balch, W.E. Traffic pattern of cystic fibrosis transmembrane regulator through the early exocytic pathway. Traffic 2000, 1, 852–870. [Google Scholar]
- Burnett, B.G.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin 3 regulates aggresome formation. Proc. Natl. Acad. Sci. USA 2005, 102, 4330–4335. [Google Scholar]
- Prat, A.G.; Xiao, Y.F.; Ausiello, D.A.; Cantiello, H.F. cAMP-independent regulation of CFTR by the actin cytoskeleton. Am. J. Physiol 1995, 268, C1552–C1561. [Google Scholar]
- Hug, T.; Koslowsky, T.; Ecke, D.; Greger, R.; Kunzelmann, K. Actin-dependent activation of ion conductances in bronchial epithelial cells. Pflugers Arch 1995, 429, 682–690. [Google Scholar]
- Halm, D.R.; Halm, S.T.; DiBona, D.R.; Frizzell, R.A.; Johnson, R.D. Selective stimulation of epithelial cells in colonic crypts: Relation to active chloride secretion. Am. J. Physiol 1995, 269, C929–C942. [Google Scholar]
- Krendel, M.; Mooseker, M.S. Myosins: Tails (and heads) of functional diversity. Physiology 2005, 20, 239–251. [Google Scholar]
- Heintzelman, M.B.; Hasson, T.; Mooseker, M.S. Multiple unconventional myosin domains of the intestinal brush border cytoskeleton. J. Cell Sci 1994, 107, 3535–3543. [Google Scholar]
- Henn, A.; De La Cruz, E.M. Vertebrate myosin VIIb is a high duty ratio motor adapted for generating and maintaining tension. J. Biol. Chem 2005, 280, 39665–39676. [Google Scholar]
- Mermall, V.; Post, P.L.; Mooseker, M.S. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science 1998, 279, 527–533. [Google Scholar]
- Ameen, N.A.; Salas, P.J. Microvillus inclusion disease: A genetic defect affecting apical membrane protein traffic in intestinal epithelium. Traffic 2000, 1, 76–83. [Google Scholar]
- Ukarapol, N.; Chotinaruemol, S.; Lertprasertsuk, N.; Wongsawasdi, L. Microvillus inclusion disease as a cause of severe protracted diarrhea in infants. J Med. Assoc. Thail 2001, 84, 1356–1360. [Google Scholar]
- Cutz, E.; Rhoads, J.M.; Drumm, B.; Sherman, P.M.; Durie, P.R.; Forstner, G.G. Microvillus inclusion disease: An inherited defect of brush-border assembly and differentiation. N. Engl. J. Med 1989, 320, 646–651. [Google Scholar]
- Muller, T.; Hess, M.W.; Schiefermeier, N.; Pfaller, K.; Ebner, H.L.; Heinz-Erian, P.; Ponstingl, H.; Partsch, J.; Rollinghoff, B.; Kohler, H.; et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat. Genet 2008, 40, 1163–1165. [Google Scholar]
- Ruemmele, F.M.; Muller, T.; Schiefermeier, N.; Ebner, H.L.; Lechner, S.; Pfaller, K.; Thoni, C.E.; Goulet, O.; Lacaille, F.; Schmitz, J.; et al. Loss-of-function of MYO5B is the main cause of microvillus inclusion disease: 15 novel mutations and a CaCo-2 RNAi cell model. Hum. Mutat 2010, 31, 544–551. [Google Scholar]
- Ameen, N.; Apodaca, G. Defective CFTR apical endocytosis and enterocyte brush border in myosin VI-deficient mice. Traffic 2007, 8, 998–1006. [Google Scholar]
- Tyska, M.J.; Mackey, A.T.; Huang, J.D.; Copeland, N.G.; Jenkins, N.A.; Mooseker, M.S. Myosin-1a is critical for normal brush border structure and composition. Mol. Biol. Cell 2005, 16, 2443–2457. [Google Scholar]
- Tyska, M.J.; Mooseker, M.S. A role for myosin-1A in the localization of a brush border disaccharidase. J. Cell Biol 2004, 165, 395–405. [Google Scholar]
- Buss, F.; Arden, S.D.; Lindsay, M.; Luzio, J.P.; Kendrick-Jones, J. Myosin VI isoform localized to clathrin-coated vesicles with a role in clathrin-mediated endocytosis. EMBO J 2001, 20, 3676–3684. [Google Scholar]
- Buss, F.; Luzio, J.P.; Kendrick-Jones, J. Myosin VI, a new force in clathrin mediated endocytosis. FEBS Lett 2001, 508, 295–299. [Google Scholar]
- Swiatecka-Urban, A.; Boyd, C.; Coutermarsh, B.; Karlson, K.H.; Barnaby, R.; Aschenbrenner, L.; Langford, G.M.; Hasson, T.; Stanton, B.A. Myosin VI regulates endocytosis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem 2004, 279, 38025–38031. [Google Scholar]
- Coluccio, L.M.; Myosin, I. Myosins A Superfamily of Molecular Motors; Coluccio, L.M., Ed.; Springer: Dordrecht, The Netherlands, 1998; pp. 95–124. [Google Scholar]
- Madden, D.R.; Swiatecka-Urban, A. Tissue-specific control of CFTR endocytosis by Dab2: Cargo recruitment as a therapeutic target. Commun. Integr. Biol 2012, 5, 473–476. [Google Scholar]
- Swiatecka-Urban, A.; Brown, A.; Moreau-Marquis, S.; Renuka, J.; Coutermarsh, B.; Barnaby, R.; Karlson, K.H.; Flotte, T.R.; Fukuda, M.; Langford, G.M.; et al. The short apical membrane half-life of rescued {Delta}F508-cystic fibrosis transmembrane conductance regulator (CFTR) results from accelerated endocytosis of {Delta}F508-CFTR in polarized human airway epithelial cells. J. Biol. Chem 2005, 280, 36762–36772. [Google Scholar]
- Tang, B.L.; Gee, H.Y.; Lee, M.G. The cystic fibrosis transmembrane conductance regulator’s expanding SNARE interactome. Traffic 2011, 12, 364–371. [Google Scholar]
- Kreda, S.M.; Mall, M.; Mengos, A.; Rochelle, L.; Yankaskas, J.; Riordan, J.R.; Boucher, R.C. Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol. Biol. Cell 2005, 16, 2154–2167. [Google Scholar]
- Chen, P.; Hwang, T.C.; Gillis, K.D. The relationship between cAMP, Ca2+, and transport of CFTR to the plasma membrane. J. Gen. Physiol 2001, 118, 135–144. [Google Scholar]
- Chang, S.Y.; Di, A.; Naren, A.P.; Palfrey, H.C.; Kirk, K.L.; Nelson, D.J. Mechanisms of CFTR regulation by syntaxin 1A and PKA. J. Cell Sci 2002, 115, 783–791. [Google Scholar]


{kind=link}
| N 30–33 μM | Col 100 μM | Col 33 μM | T, 20 μM | T, 10 μM | BFA, 5 μg/mL | Control | ||
|---|---|---|---|---|---|---|---|---|
| Grotmol, 1992 Rat colon 10 μM PGE2 + 10 μM Theophylline | Basal | 35 (24–46) | 20 (2–37) | 36 (21–40) | 30 (21–48) | |||
| Stimulated | 34 (24–45) | 37 (29–49) | 33 (20–60) | 56 (46–93) | ||||
| Morris, 1994 HT-29 intestinal cells 10 μM FSK | Stimulated | 6.3 ± 1.0 | 20 ± 1.9 | |||||
| Morris, 1998 A6 kidney cells 0.1 μM AVT | Basal | 5.1 ± 0.4 | 3.2 ± 0.8 | 15.3 ± 2.7 | ||||
| Stimulated | 19.7 ± 1.9 | 6.3 ± 1.7 | 38.2 ± 4.1 | |||||
| Moyer, 1998 MDCK kidney cells cAMP mix | Stimulated | 11.9 ± 0.4 | 10.2 ± 0.7 | |||||
| Loffing, 1998 Calu3 airway cells | Stimulated | 61.9 ± 3.1 | 44.8 ± 2.0 | 60.5 ± 1.8 | ||||
| Group | Cell type | Agent used |
|---|---|---|
| Grotmol, 1992 | Rat colon | 10 μM PGE2 + 10 mM theophylline |
| Morris, 1994 | HT-29 | 10 μM FSK |
| Tousson, 1996 | T-84 | 10 μM FSK |
| Loffing, 1998 | Calu3 | 100 μM CPT-cAMP |
| Morris, 1998 | A6 | 0.1 μM AVT |
| Moyer, 1998 | MDCK | 100 μM CPT-cAMP + 100 μM sobutylmethylxanthine + 20 μM FSK |
| Ameen, 2003 | Rat jejunum | 1 mM 8-BrcAMP |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kravtsov, D.V.; Ameen, N.A. Molecular Motors and Apical CFTR Traffic in Epithelia. Int. J. Mol. Sci. 2013, 14, 9628-9642. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14059628
Kravtsov DV, Ameen NA. Molecular Motors and Apical CFTR Traffic in Epithelia. International Journal of Molecular Sciences. 2013; 14(5):9628-9642. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14059628
Chicago/Turabian StyleKravtsov, Dmitri V., and Nadia A. Ameen. 2013. "Molecular Motors and Apical CFTR Traffic in Epithelia" International Journal of Molecular Sciences 14, no. 5: 9628-9642. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14059628