Design, Synthesis and DNA Interaction Study of New Potential DNA Bis-Intercalators Based on Glucuronic Acid

Abstract
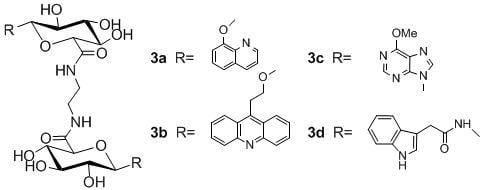
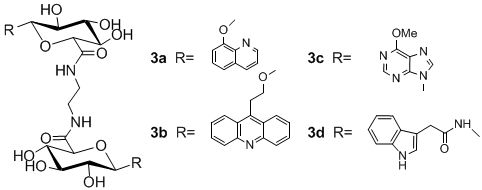
:
1. Introduction
2. Results and Discussion
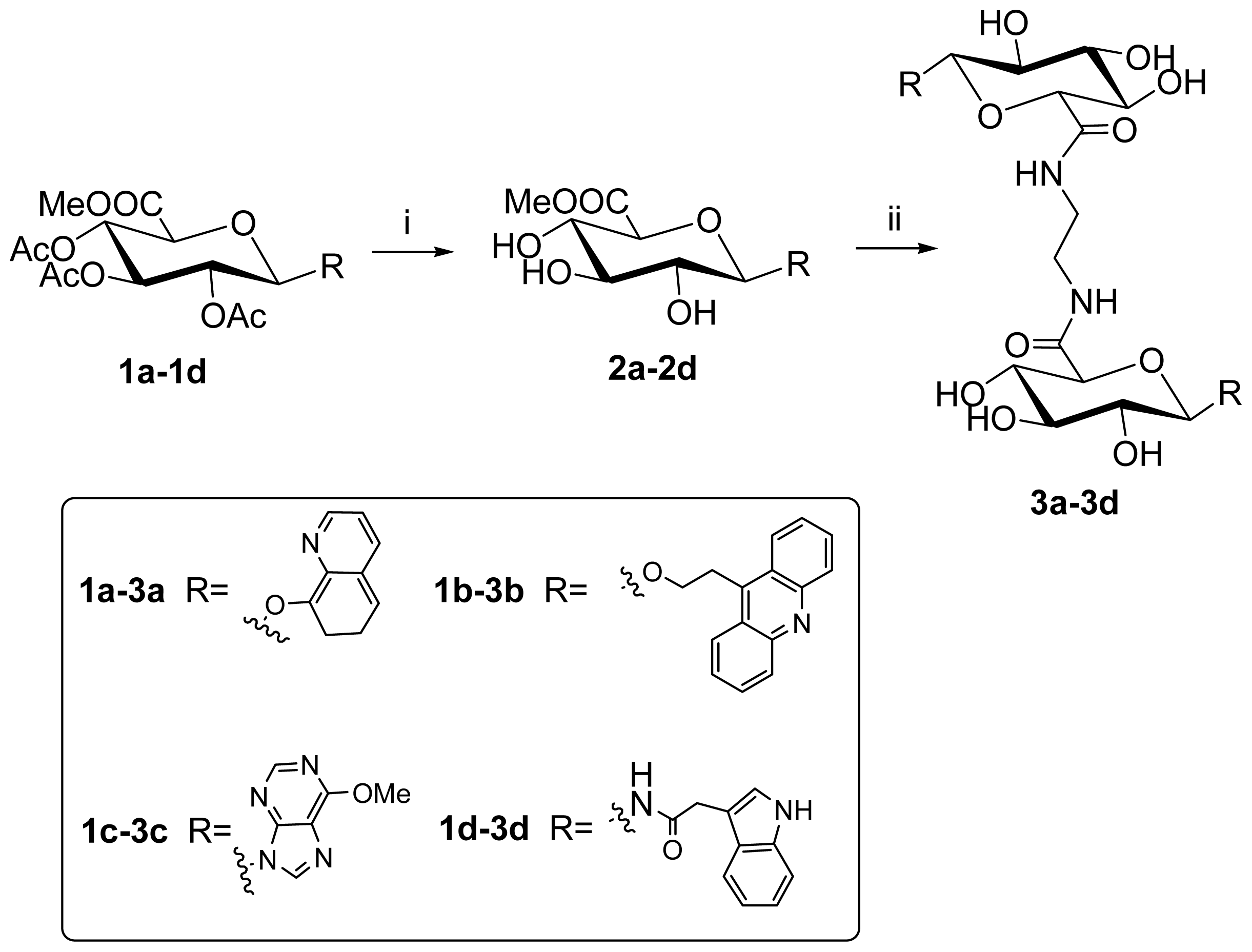
2.1. Synthesis
2.2. DNA Binding Properties
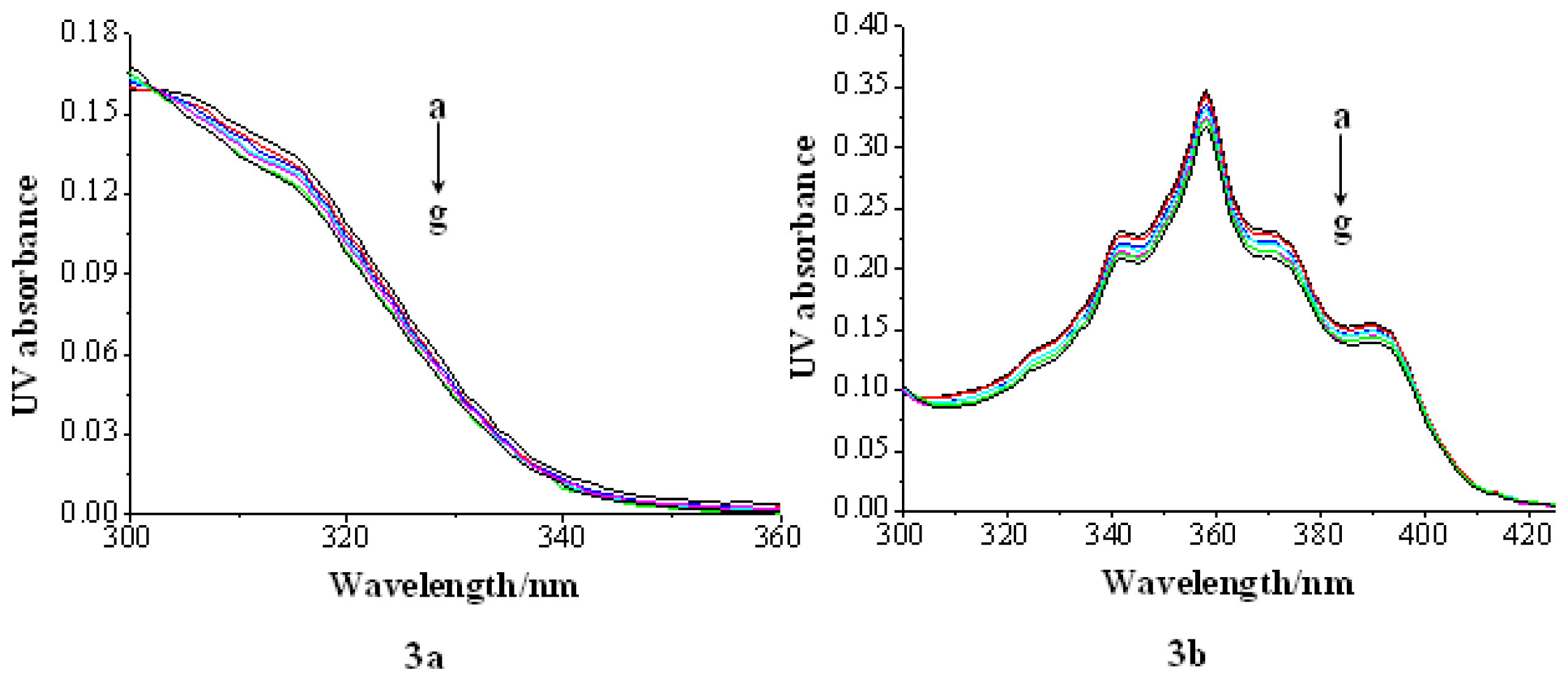
2.2.1. UV Absorption
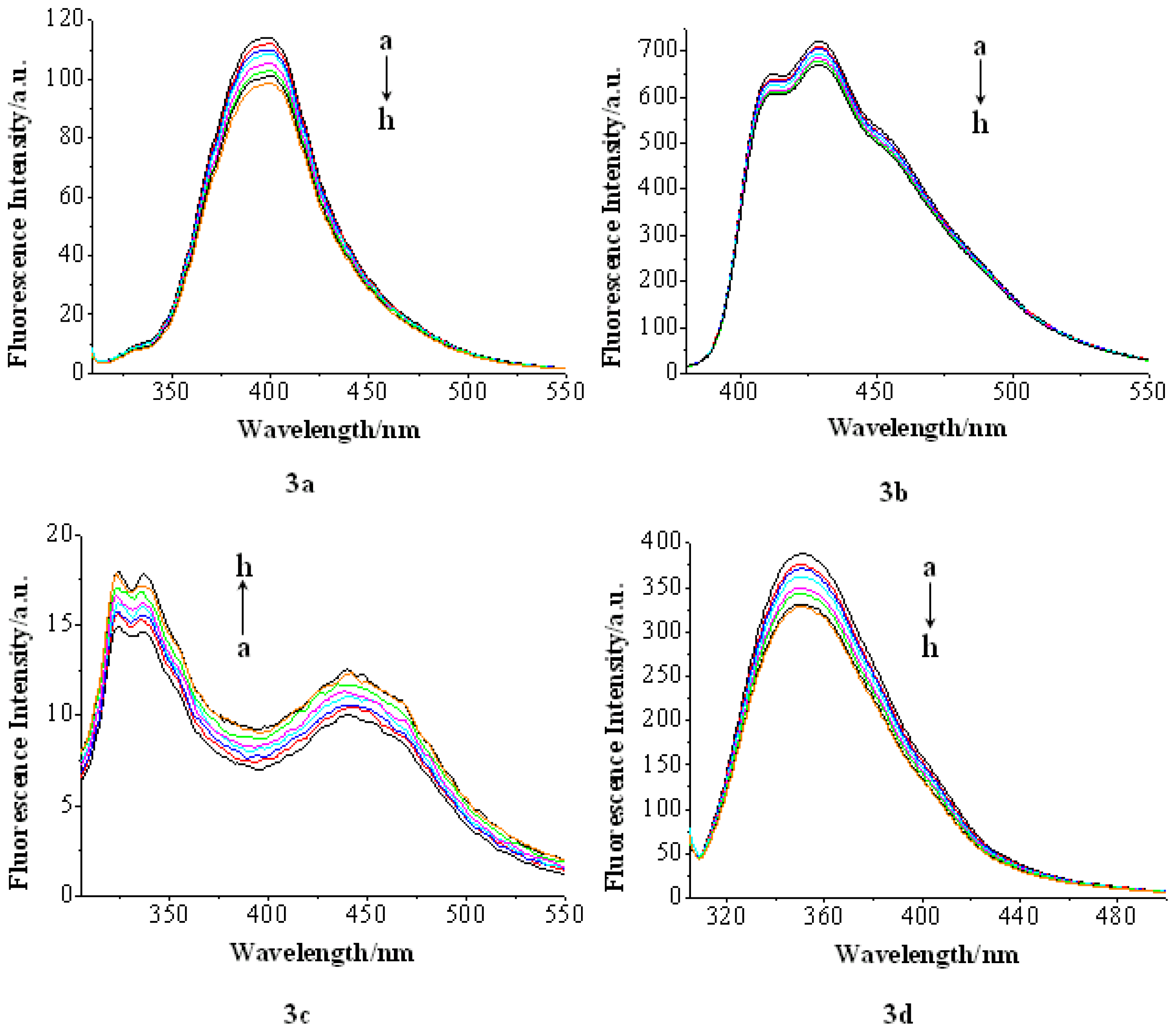
2.2.2. Fluorescence Emission
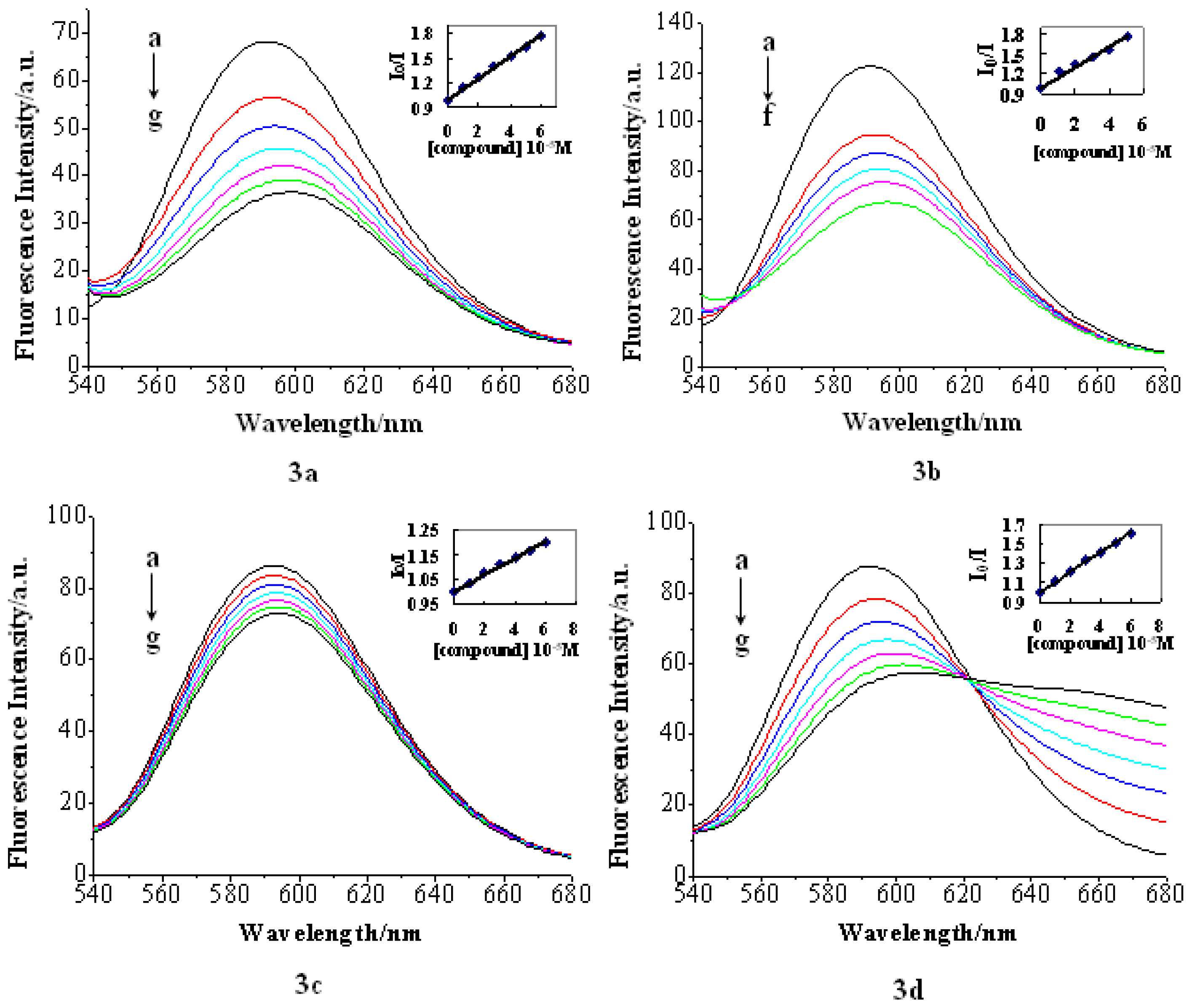
2.2.3. EB Competition Assay
3. Experimental Section
3.1. General Methods
3.2. DNA-Binding Studies
3.3. Synthesis
8-O-(methyl 2,3,4-tri-O-acetyl-β-d-glucuronosyluronate)quinolin (1a)
8-O-(methyl β-d-glucuronosyluronate)quinolin (2a)
N,N-1,2-di-(1-O-(quinolin-8-yl)-β-d-glucuronamide)ethane (3a)
9-O-(methyl 2,3,4-tri-O-acetyl-β-d-glucuronosyluronate)ethyl acridine (1b)
9-O-(methyl β-d-glucuronosyluronate)ethyl acridine (2b)
N,N-1,2-di-(1-O-(2-(acridine-9-yl)ethyl)-β-d-glucuronamide) ethane (3b)
6-Cloro-9-(methyl 2,3,4-tri-O-acetyl-1-deoxy-β-d-glucuronosyluronate)purine (1c)
6-methoxyl-9-(methyl β-d-glucuronosyluronate)purine (2c)
N,N-1,2-di-(1-deoxy-1-(6-chloropurine-9-yl)-β-d-glucuronamide)ethane (3c)
N-(methyl-2,3,4-tri-O-acetyl-1-deoxy-β-d-glucuronosyluronate)-2-(indol-3-yl) acetamide (1d)
N-(methyl β-d-glucuronosyluronate)-2-(indol-3-yl)acetamide (2d)
N,N-1,2-di-(1-deoxy-1-N-(2-(indol-3-yl)acetamide)-β-d-glucuronamide)ethane (3d)
4. Conclusions
Supporting Information Available
Acknowledgements
Conflicts of Interest
References
- Hartley, J.A.; Hochhauser, D. Small molecule drugs-optimizing DNA damaging agent-based therapeutics. Curr. Opin. Pharmacol 2012, 12, 398–402. [Google Scholar]
- Srinivasan, A.; Gold, B. Small-molecule inhibitors of DNA damage-repair pathways: An approach to overcome tumor resistance to alkylating anticancer drugs. Future Med. Chem 2012, 4, 1093–1111. [Google Scholar]
- Li, V.S.; Choi, D.; Wang, Z.; Jimenez, L.S.; Tang, M.S.; Kohn, H. Role of the C-10 substituent in mitomycin C-1-DNA bonding. J. Am. Chem. Soc 1996, 118, 2326–2331. [Google Scholar]
- Zuber, G.; Quada, J.C.; Hecht, S.M. Sequence selective cleavage of a DNA octanucleotide by chlorinated bithiazoles and bleomycins. J. Am. Chem. Soc 1998, 120, 9368–9369. [Google Scholar]
- Hecht, S.M. Bleomycin: New perspectives on the mechanism of action. J. Nat. Prod 2000, 63, 158–168. [Google Scholar]
- Dervan, P.B. Molecular recognition of DNA by small molecules. Bioorg. Med. Chem 2001, 9, 2215–2235. [Google Scholar]
- Neidle, S. DNA minor-groove recognition by small molecules. Nat. Prod. Rep 2001, 18, 291–309. [Google Scholar]
- Xi, H.; Davis, E.; Ranjan, N.; Xue, L.; Hyde-Volpe, D.; Arya, D.P. Thermodynamics of nucleic acid “shape readout” by an aminosugar. Biochemistry 2011, 50, 9088–9113. [Google Scholar]
- Kumar, S.; Xue, L.; Arya, D.P. Neomycin-neomycin dimer: An all-carbohydrate scaffold with high affinity for AT-rich DNA duplexes. J. Am. Chem. Soc 2011, 133, 7361–7375. [Google Scholar]
- Hamilton, P.L.; Arya, D.P. Natural product DNA major groove binders. Nat. Prod. Rep 2012, 29, 134–143. [Google Scholar]
- Greguric, I.; Aldrich-Wright, J.R.; Collins, J.G. A1H NMR study of the binding of Δ-[Ru(phen)2DPQ]2+ to the hexanucleotide d(GTCGAC)2. evidence for intercalation from the minor groove. J. Am. Chem. Soc 1997, 119, 3621–3622. [Google Scholar]
- Filichev, V.V.; Pedersen, E.B. DNA-Conjugated Organic Chromophores in DNA Stacking Interactions. Wiley Encycl. Chem. Biol. 2008. [Google Scholar] [CrossRef]
- Brana, M.F.; Cacho, M.; Gradillas, A.; de Pascual-Teresa, B.; Ramos, A. Intercalators as anticancer drugs. Curr. Pharm. Des 2001, 7, 1745–1780. [Google Scholar]
- Ross, W.E.; Bradley, M.O. DNA double-strand breaks in mammalian-cells after exposure to intercalating agents. Biochim. Biophys. Acta 1981, 654, 129–134. [Google Scholar]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar]
- Chaires, J.B.; Leng, F.; Przewloka, T.; Fokt, I.; Ling, Y.H.; Perez-Soler, R.; Priebe, W. Structure-based design of a new bisintercalating anthracycline antibiotic. J. Med. Chem 1997, 40, 261–266. [Google Scholar]
- Kuruvilla, E.; Joseph, J.; Ramaiah, D. Novel bifunctional acridine-acridinium conjugates: Synthesis and study of their chromophore-selective electron-transfer and DNA-binding properties. J. Phys. Chem. B 2005, 109, 21997–22002. [Google Scholar]
- Lorente, A.; Vazquez, Y.G.; Fernandez, M.J.; Ferrandez, A. Bisacridines with aromatic linking chains. Synthesis, DNA interaction, and antitumor activity. Bioorg. Med. Chem 2004, 12, 4307–4312. [Google Scholar]
- Xue, L.; Xi, H.; Kumar, S.; Gray, D.; Davis, E.; Hamilton, P.; Skriba, M.; Arya, D.P. Probing the recognition surface of a DNA triplex: Binding studies with intercalator-neomycin conjugates. Biochemistry 2010, 49, 5540–5552. [Google Scholar]
- Willis, B.; Arya, D.P. Triple recognition of B-DNA by a neomycin-Hoechst 33258-pyrene conjugate. Biochemistry 2010, 49, 452–469. [Google Scholar]
- Willis, B.; Arya, D.P. Triple recognition of B-DNA. Bioorg. Med. Chem. Lett 2009, 19, 4974–4979. [Google Scholar]
- Shaw, N.N.; Xi, H.; Arya, D.P. Molecular recognition of a DNA:RNA hybrid: Sub-nanomolar binding by a neomycin-methidium conjugate. Bioorg. Med. Chem. Lett 2008, 18, 4142–4145. [Google Scholar]
- Arya, D.P.; Xue, L.; Tennant, P. Combining the best in triplex recognition: Synthesis and nucleic acid binding of a BQQ-neomycin conjugate. J. Am. Chem. Soc 2003, 125, 8070–8071. [Google Scholar]
- Xue, L.; Charles, I.; Arya, D.P. Pyrene-neomycin conjugate: Dual recognition of a DNA triple helix. Chem. Commun. 2002, 70–71. [Google Scholar]
- Wunberg, T.; Kallus, C.; Opatz, T.; Henke, S.; Schmidt, W.; Kunz, H. Carbohydrates as multifunctional chiral scaffolds in combinatorial synthesis. Angew. Chem. Int. Edit 1998, 37, 2503–2505. [Google Scholar]
- Velasco-Torrijos, T.; Murphy, P.V. Synthesis and conformational analysis of novel water soluble macrocycles incorporating carbohydrates, including a β-cyclodextrin mimic. Tetrahedron 2005, 16, 261–272. [Google Scholar]
- Liu, C.H.; Liu, H.; Han, X.Y.; Wu, B.; Zhong, B.H.; Gong, Z.H. Synthesis and characterization of thienorphine and its glucuronide conjugate. Synthetic. Commun 2005, 35, 701–710. [Google Scholar]
- Chiba, T.; Sinay, P. Application of a radical reaction to the synthesis of l-iduronic acid-derivatives from d-glucuronic acid analogs. Carbohyd. Res 1986, 151, 379–389. [Google Scholar]
- Florio, P.; Thomson, R.J.; von Itzstein, M. Rapid access to uronic acid-based mimetics of Kdn2en from d-glucurono-6,3-lactone. Carbohydr. Res 2000, 328, 445–448. [Google Scholar]
- Pitt, N.; Duane, R.M.; O’Brien, A.; Bradley, H.; Wilson, S.J.; O’Boyle, K.M.; Murphy, P.V. Synthesis of a glucuronic acid and glucose conjugate library and evaluation of effects on endothelial cell growth. Carbohydr. Res 2004, 339, 1873–1887. [Google Scholar]
- Wolfrom, M.L.; McWain, P. Nucleosides of d-glucuronic acid and of d-glucofuranose and d-galactofuranose. J. Org. Chem 1965, 30, 1099–1101. [Google Scholar]
- Liu, Z.Q.; Li, Y.T.; Wu, Z.Y.; Song, Y.L. A two-dimensional copper(II) polymer with bridging μ-trans-oxamidate and μ2-picrate ligands: Synthesis, crystal structure and DNA binding studies. Inorg. Chim. Acta 2008, 361, 226–232. [Google Scholar]
- Zhou, J.H.; Xia, S.Q.; Chen, J.R.; Wang, X.S.; Zhang, B.W.; Zhang, H.J.; Zou, P.; Ai, X.C.; Zhang, J.P. Surface binding and improved photodamage of the lanthanum ion complex of hypocrellin A to calf thymus DNA. J. Photoch. Photobio. A 2004, 165, 143–147. [Google Scholar]
- Zhang, Q.Q.; Zhang, F.; Wang, W.G.; Wang, X.L. Synthesis, crystal structure and DNA binding studies of a binuclear copper(II) complex with phenanthroline. J. Inorg. Biochem 2006, 100, 1344–1352. [Google Scholar]
- Song, Y.F.; Yang, P. Mononuclear tetrapyrido[3,2-a:2′,3′-c:3″,2″-h:2‴,3‴-j]phenazine (tpphz) cobalt complex. Polyhedron 2001, 20, 501–506. [Google Scholar]
- Han, M.J.; Duan, Z.M.; Hao, Q.; Zheng, S.Z.; Wang, K.Z. Molecular light switches for calf thymus DNA based on three Ru(II) bipyridyl complexes with variations of heteroatoms. J. Phys. Chem. C 2007, 111, 16577–16585. [Google Scholar]
- Patel, D.J. Nuclear magnetic-resonance studies of drug-nucleic acid interactions at the synthetic DNA level in solution. Accounts Chem. Res 1979, 12, 118–125. [Google Scholar]
- Kumar, C.V.; Barton, J.K.; Turro, N.J. Photophysics of ruthenium complexes bound to double helical DNA. J. Am. Chem. Soc 1985, 107, 5518–5523. [Google Scholar]
- Chen, Y.D.; Cai, M.Y.; Zhang, Y.; Zheng, W.L.; Ma, W.L. Study on the mechanism of chitosan complex formation with PEGF-PC3DNA (in Chinese). Pharm. Biotechnol 2005, 12, 291–293. [Google Scholar]
- Stern, O.; Volmer, M. Uber die abklingungszeit der fluoreszenz. Phys. Z 1919, 20, 183–188. [Google Scholar]
- Marmur, J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol 1961, 3, 208–218. [Google Scholar]
- Reichmann, M.E.; Rice, S.A.; Thomas, C.A.; Doty, P. A further examination of the molecular weight and size of desoxypentose nucleic acid. J. Am. Chem. Soc 1954, 76, 3047–3053. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound 3a | Compound 3b | Compound 3c | Compound 3d |
|---|---|---|---|---|
| ksv | 1.30 × 104 | 1.54 × 104 | 3.4 × 103 | 1.04 × 104 |
| R (for 5 points) | 0.998 | 0.982 | 0.995 | 0.998 |
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhao, J.; Li, W.; Ma, R.; Chen, S.; Ren, S.; Jiang, T. Design, Synthesis and DNA Interaction Study of New Potential DNA Bis-Intercalators Based on Glucuronic Acid. Int. J. Mol. Sci. 2013, 14, 16851-16865. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms140816851
Zhao J, Li W, Ma R, Chen S, Ren S, Jiang T. Design, Synthesis and DNA Interaction Study of New Potential DNA Bis-Intercalators Based on Glucuronic Acid. International Journal of Molecular Sciences. 2013; 14(8):16851-16865. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms140816851
Chicago/Turabian StyleZhao, Jiuyang, Wei Li, Rui Ma, Shaopeng Chen, Sumei Ren, and Tao Jiang. 2013. "Design, Synthesis and DNA Interaction Study of New Potential DNA Bis-Intercalators Based on Glucuronic Acid" International Journal of Molecular Sciences 14, no. 8: 16851-16865. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms140816851