Assessing the Effect of Loop Mutations in the Folding Space of β2-Microglobulin with Molecular Dynamics Simulations

Abstract
:
1. Introduction
2. Results and Discussion
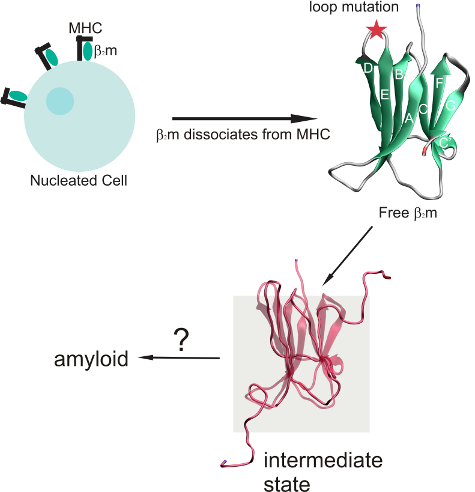
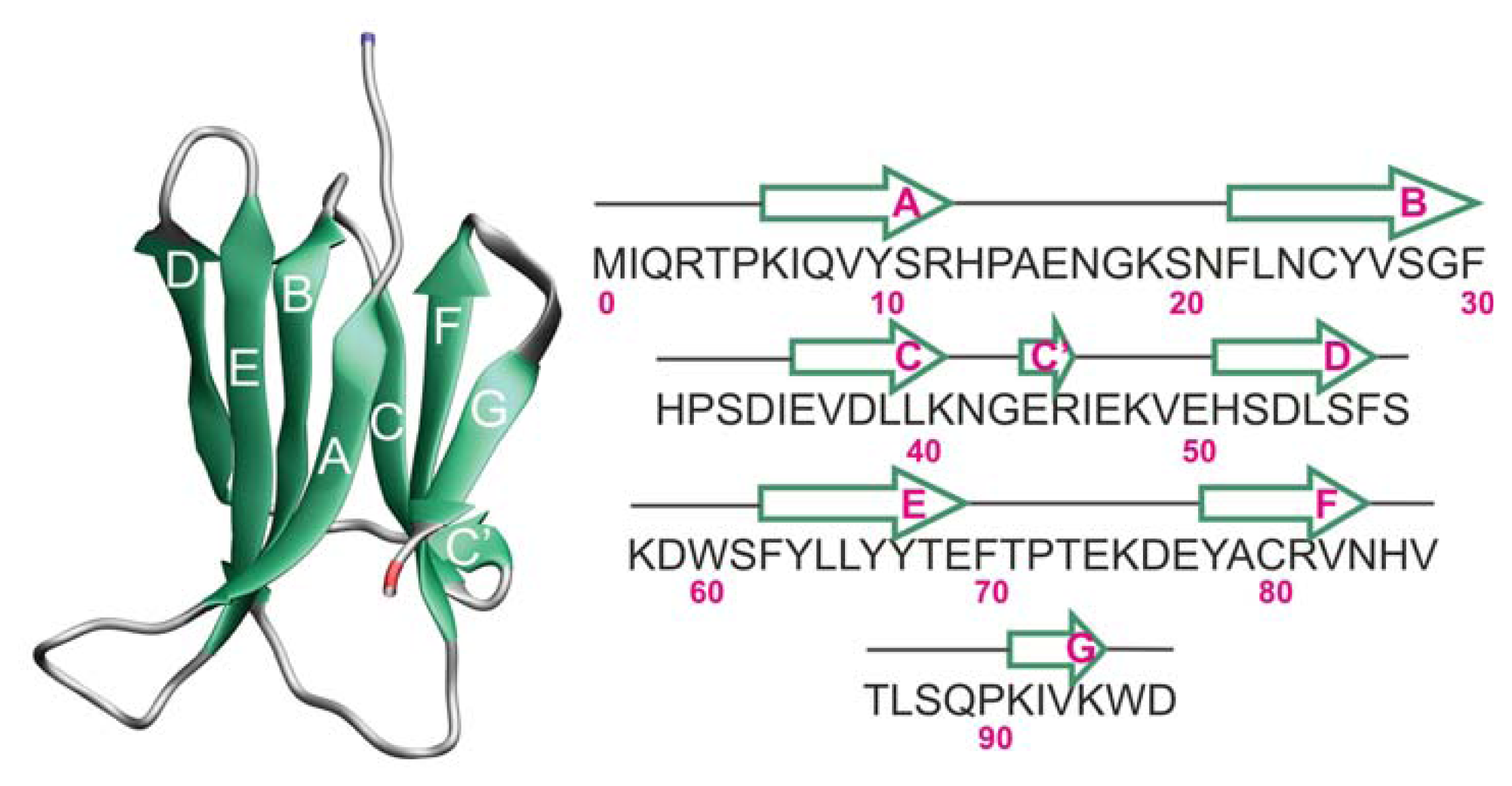
2.1. Proteins Studied
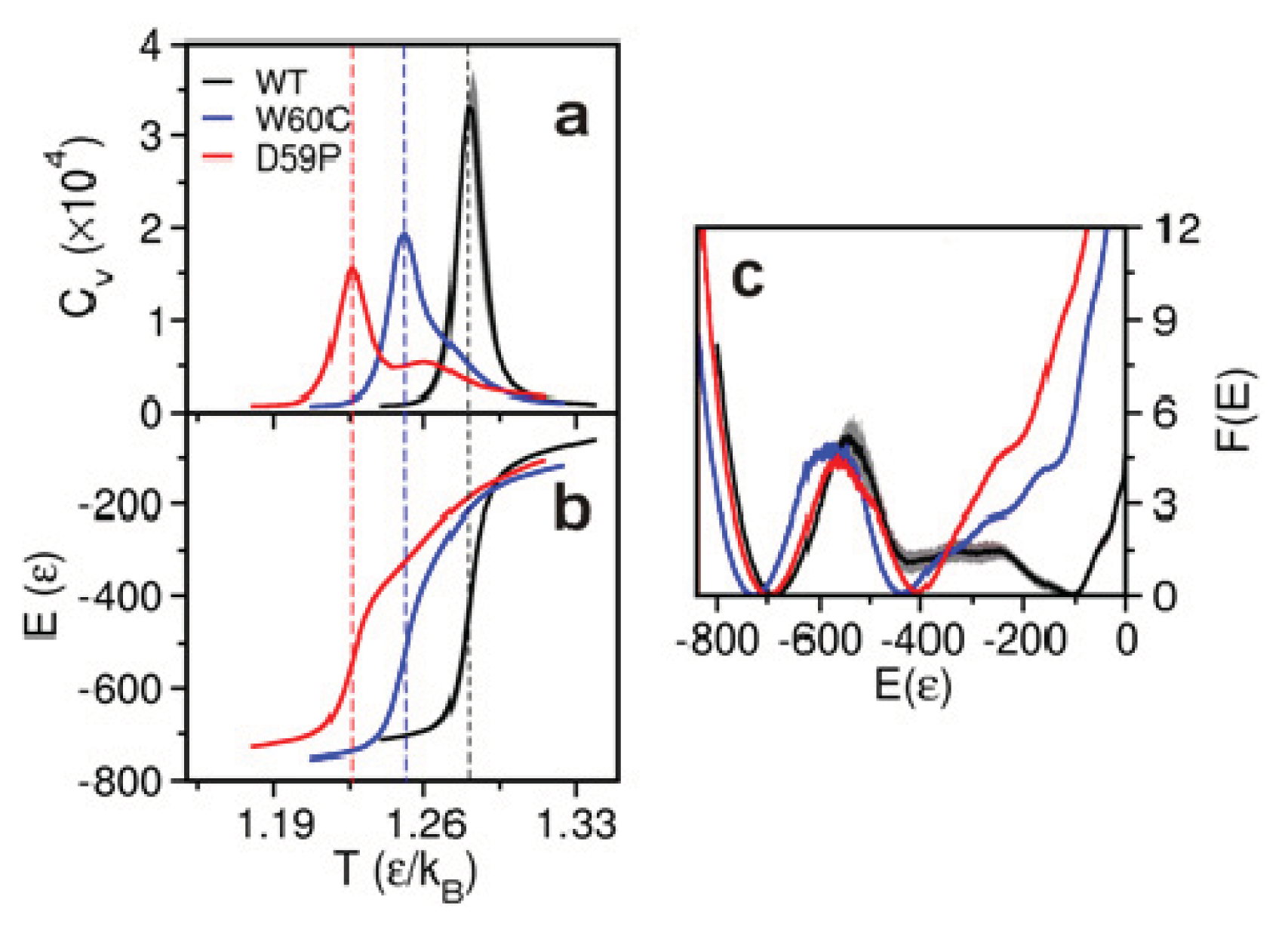
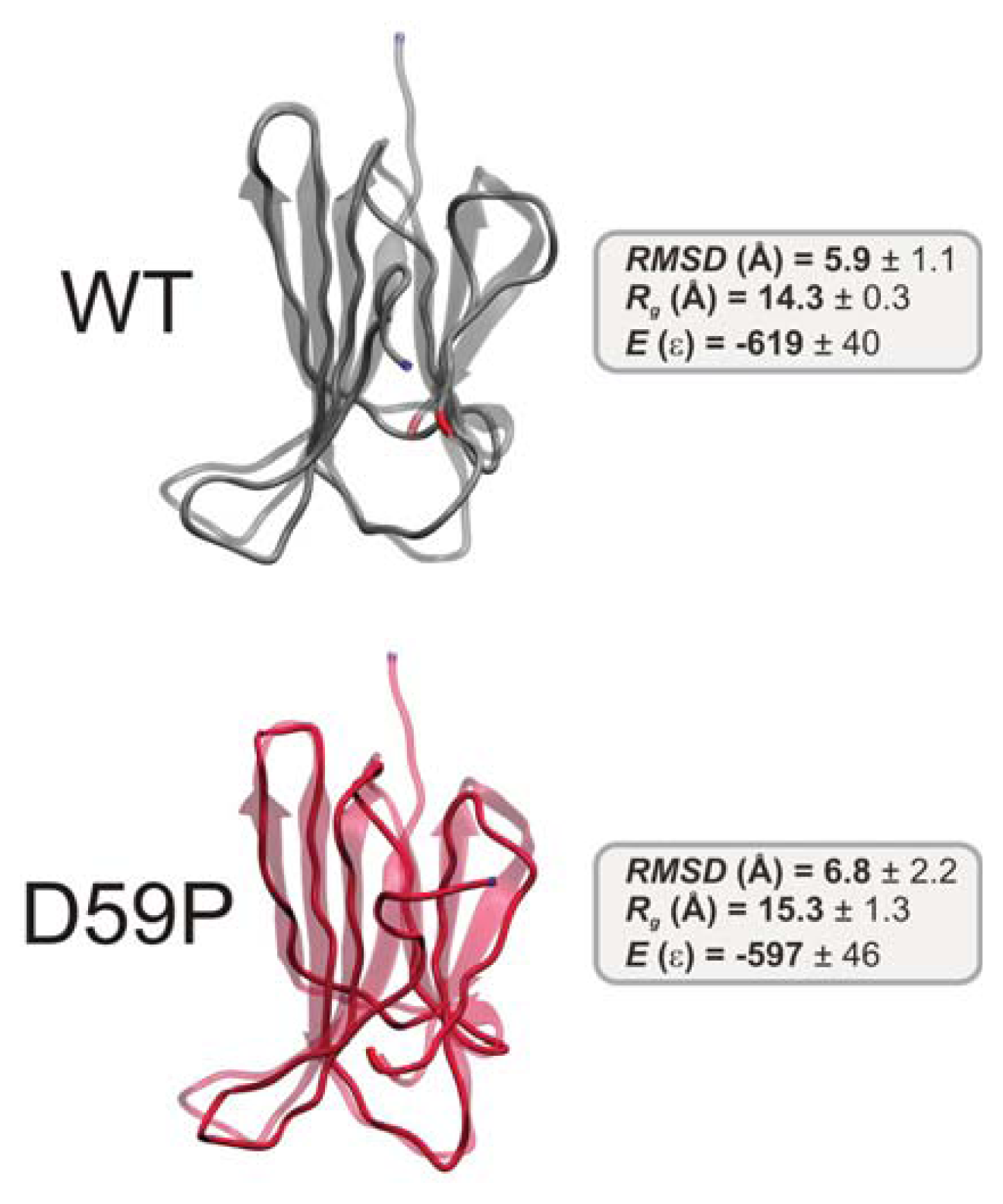
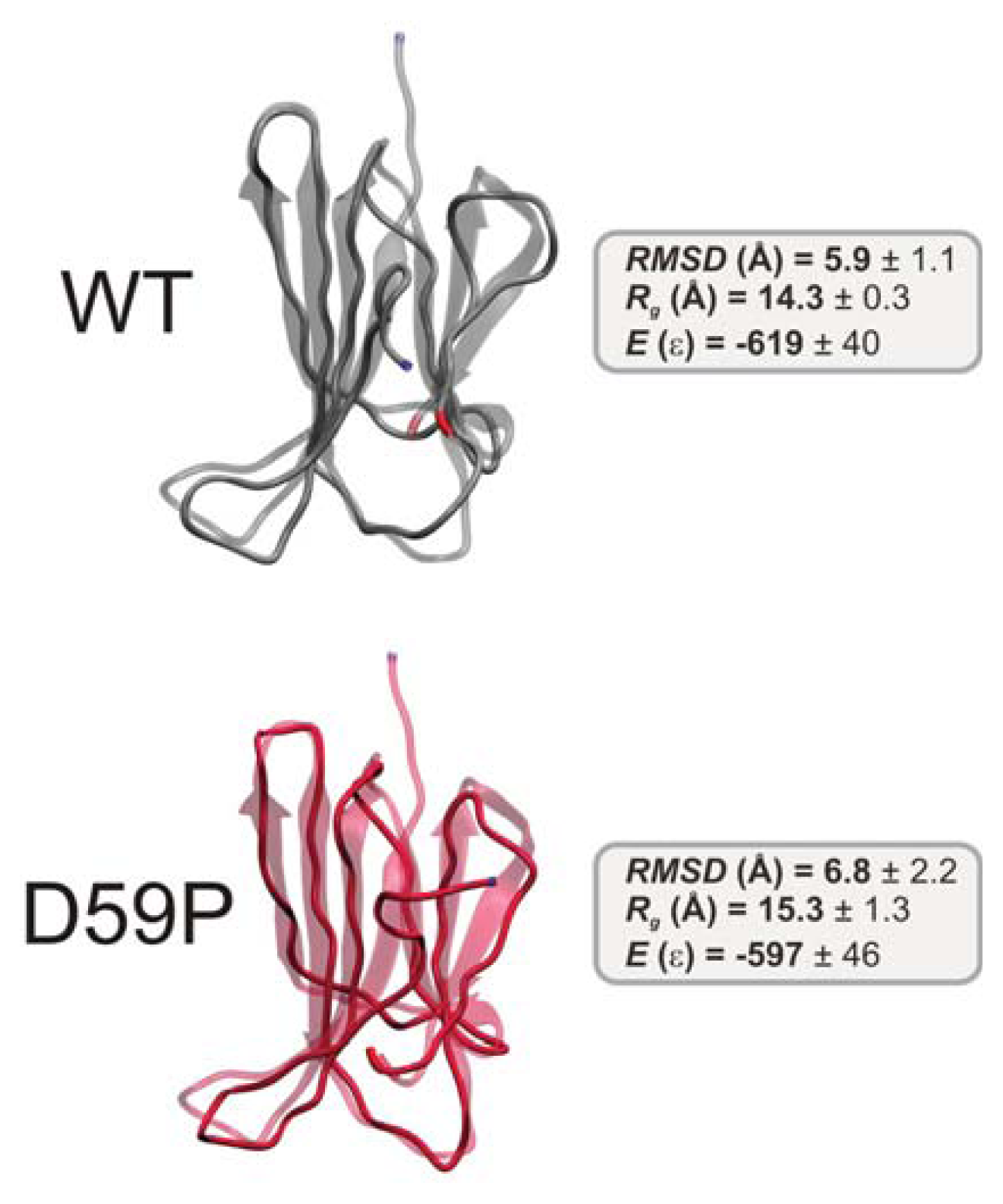
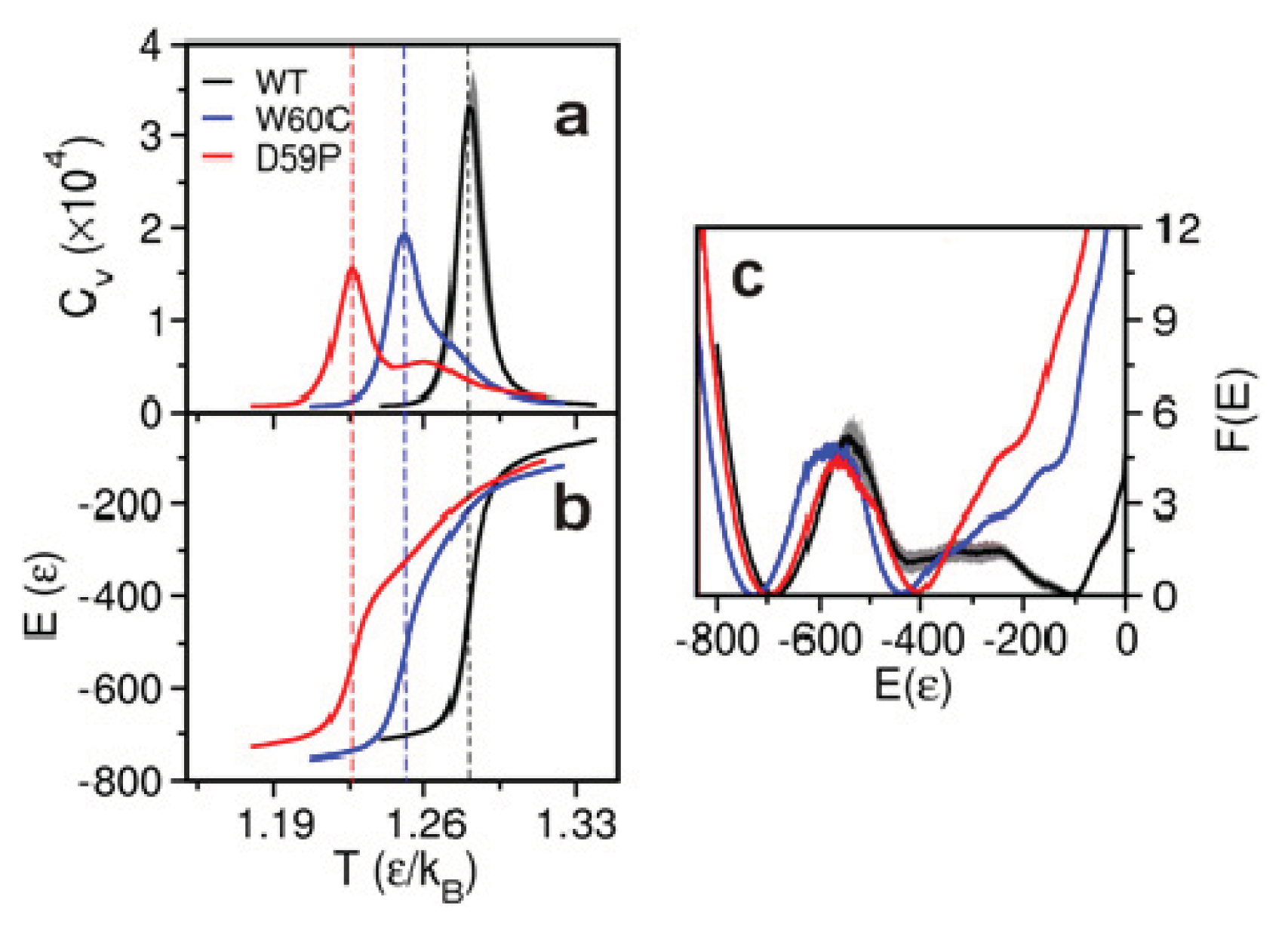
2.2. The Loop Mutations Decrease Thermal Stability
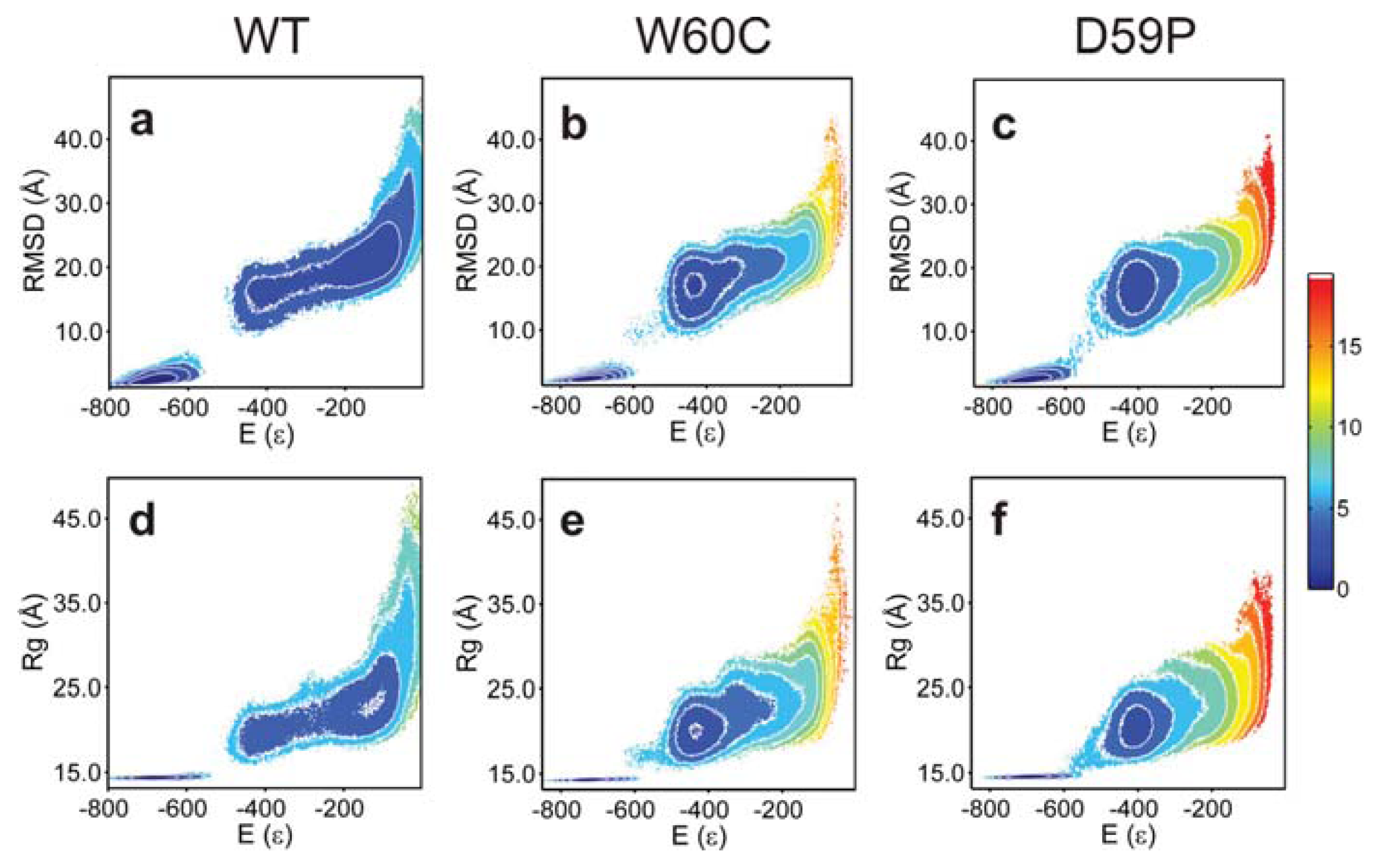
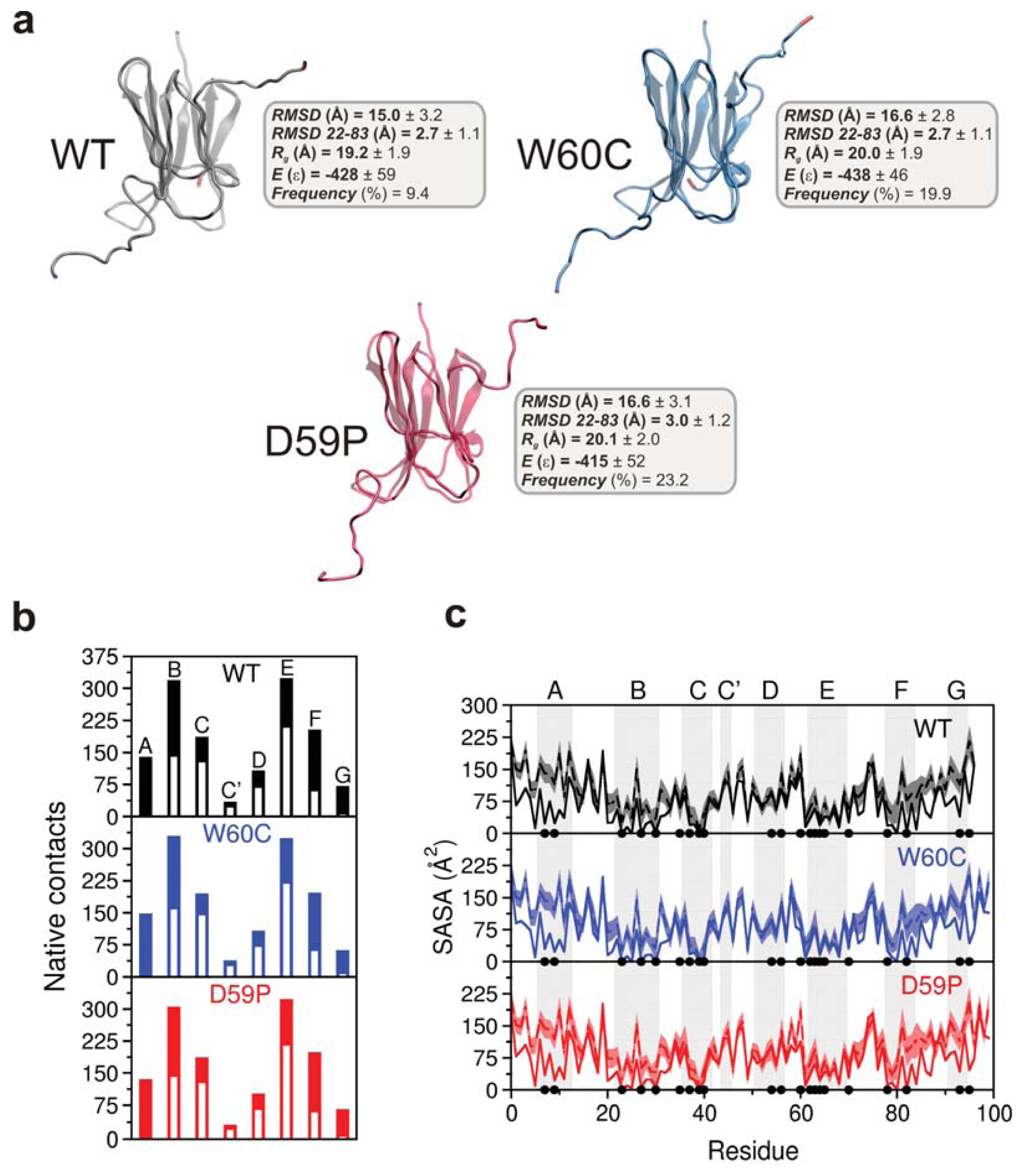
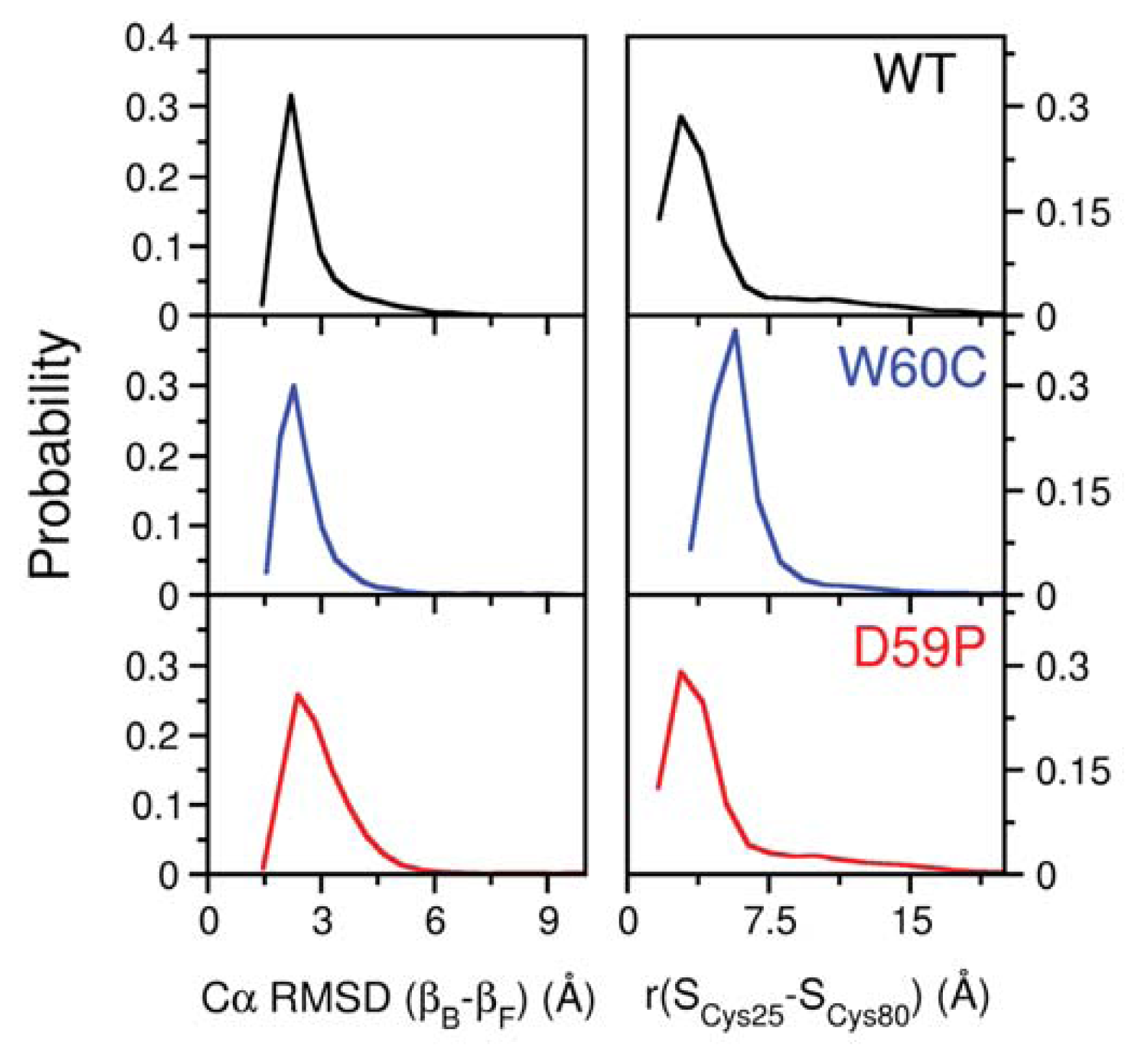
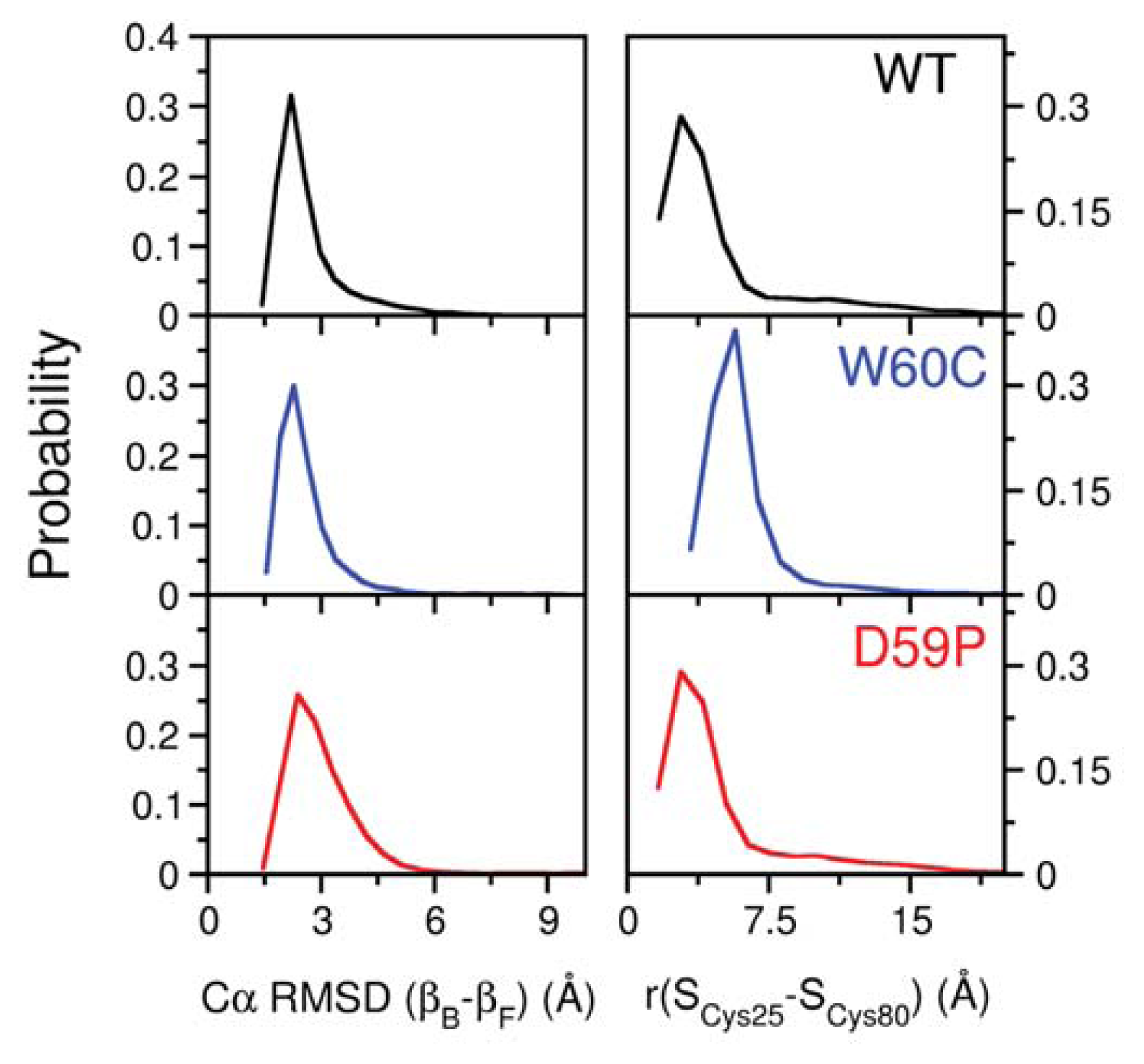
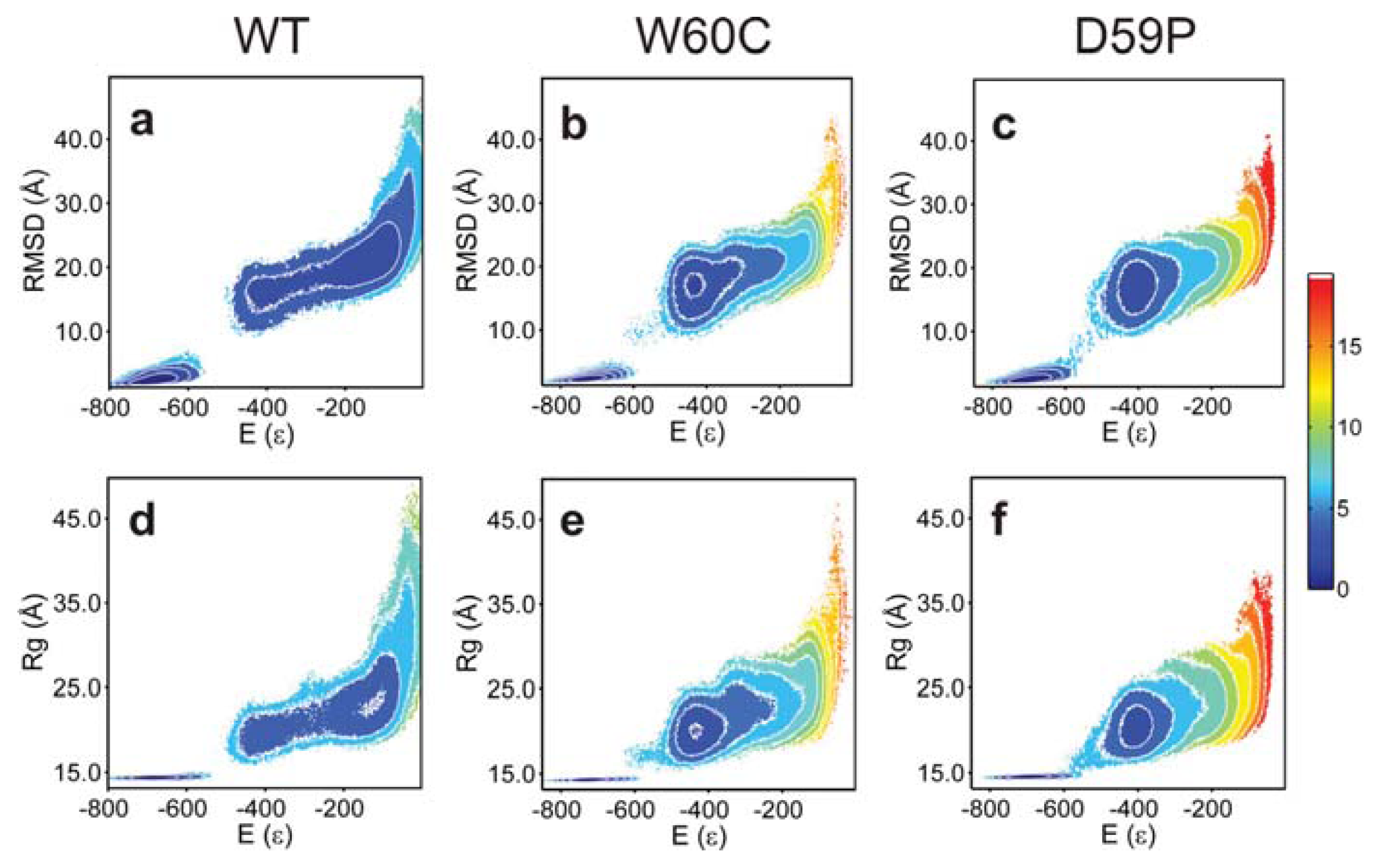
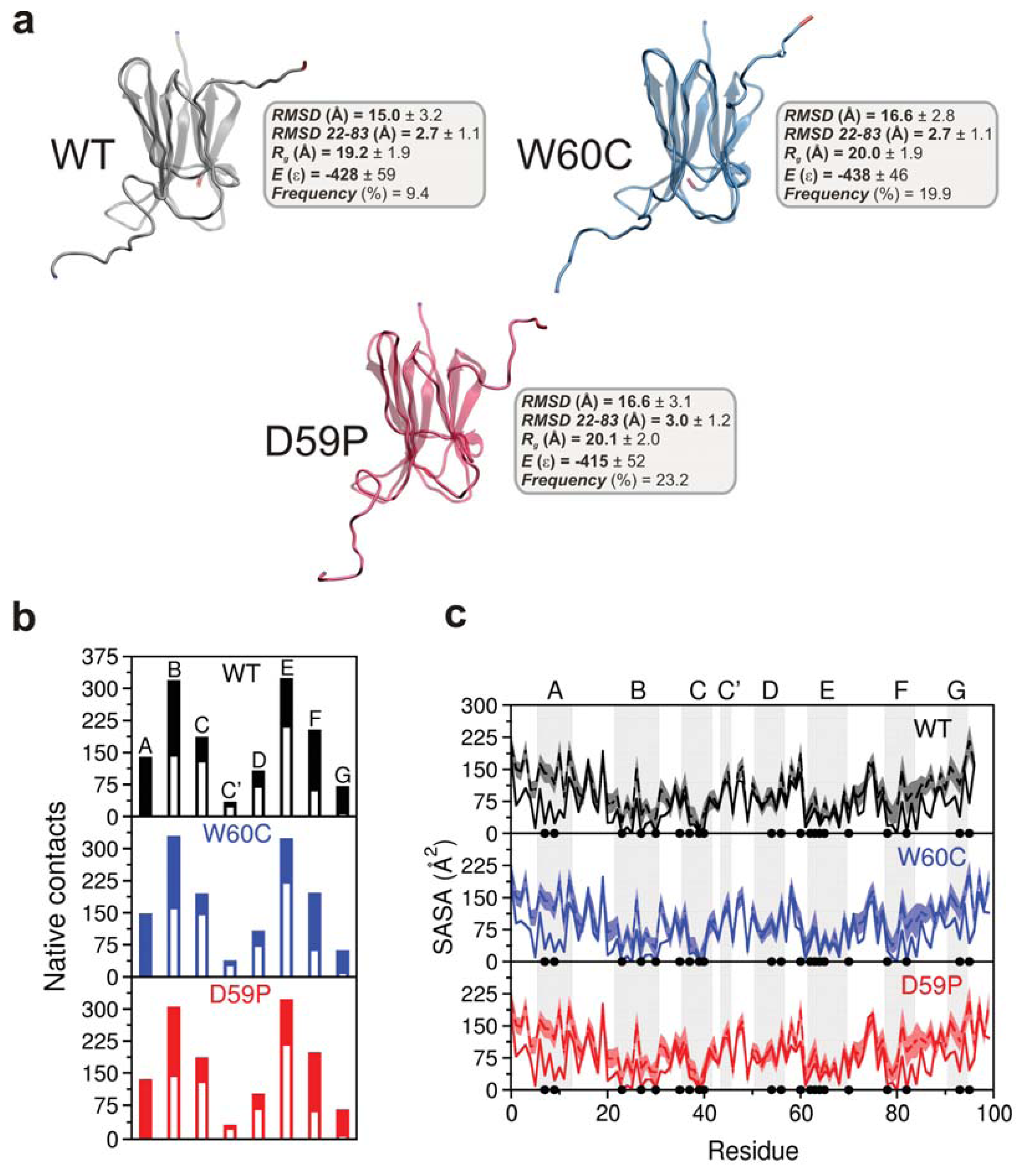
2.3. The Loop Mutations Trigger the Population of a Partially Folded Intermediate State
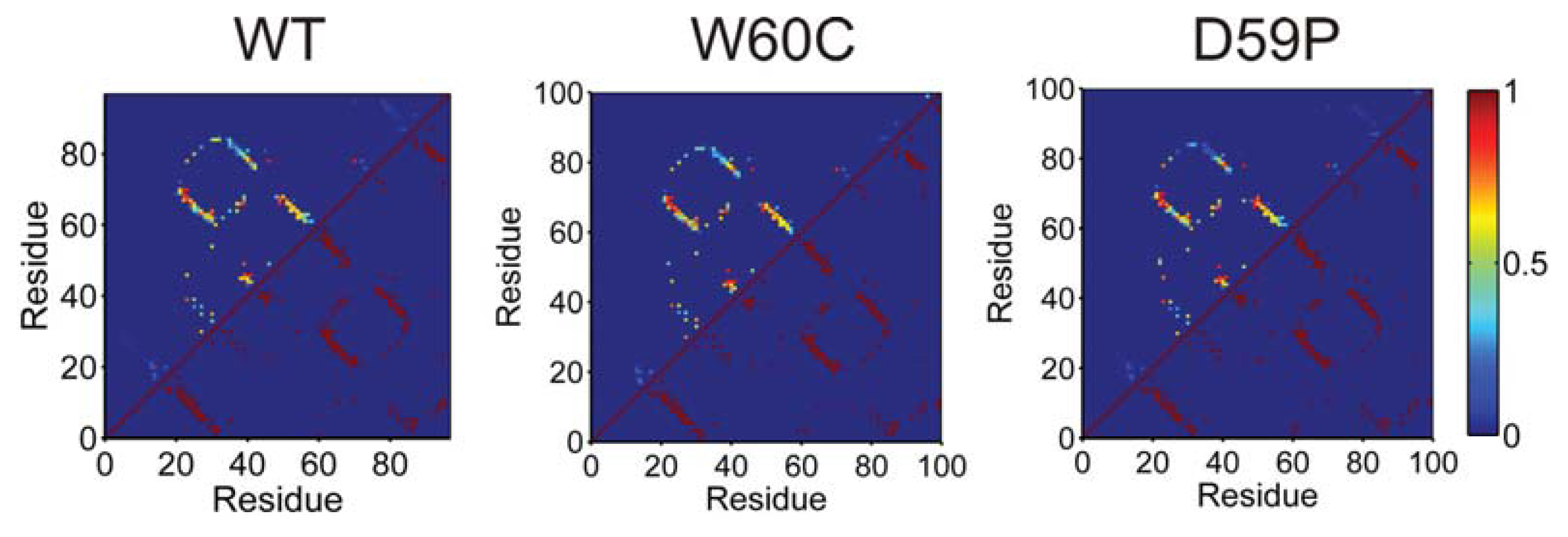
2.4. The Intermediate State Has a Molten Globule-Like Nature
2.5. The Intermediate State Exhibits Aggregation-Prone Traits
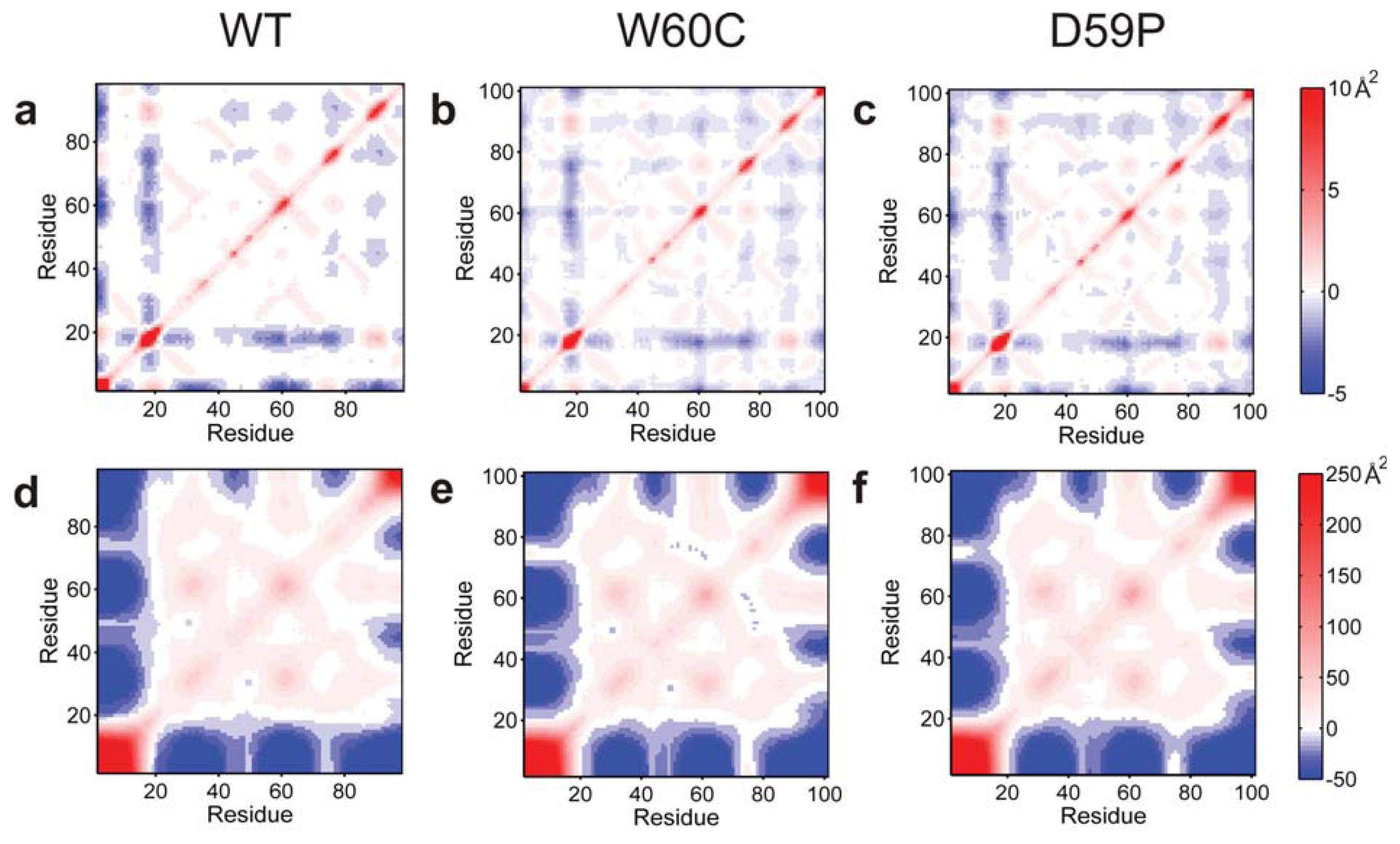
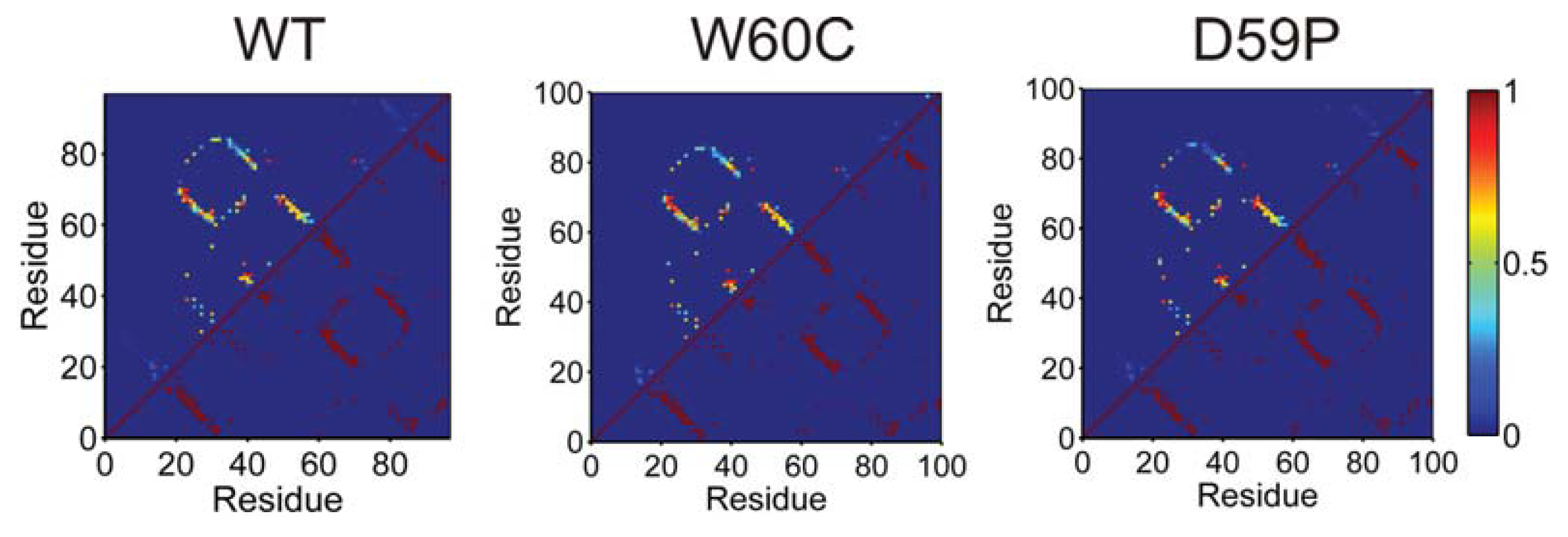
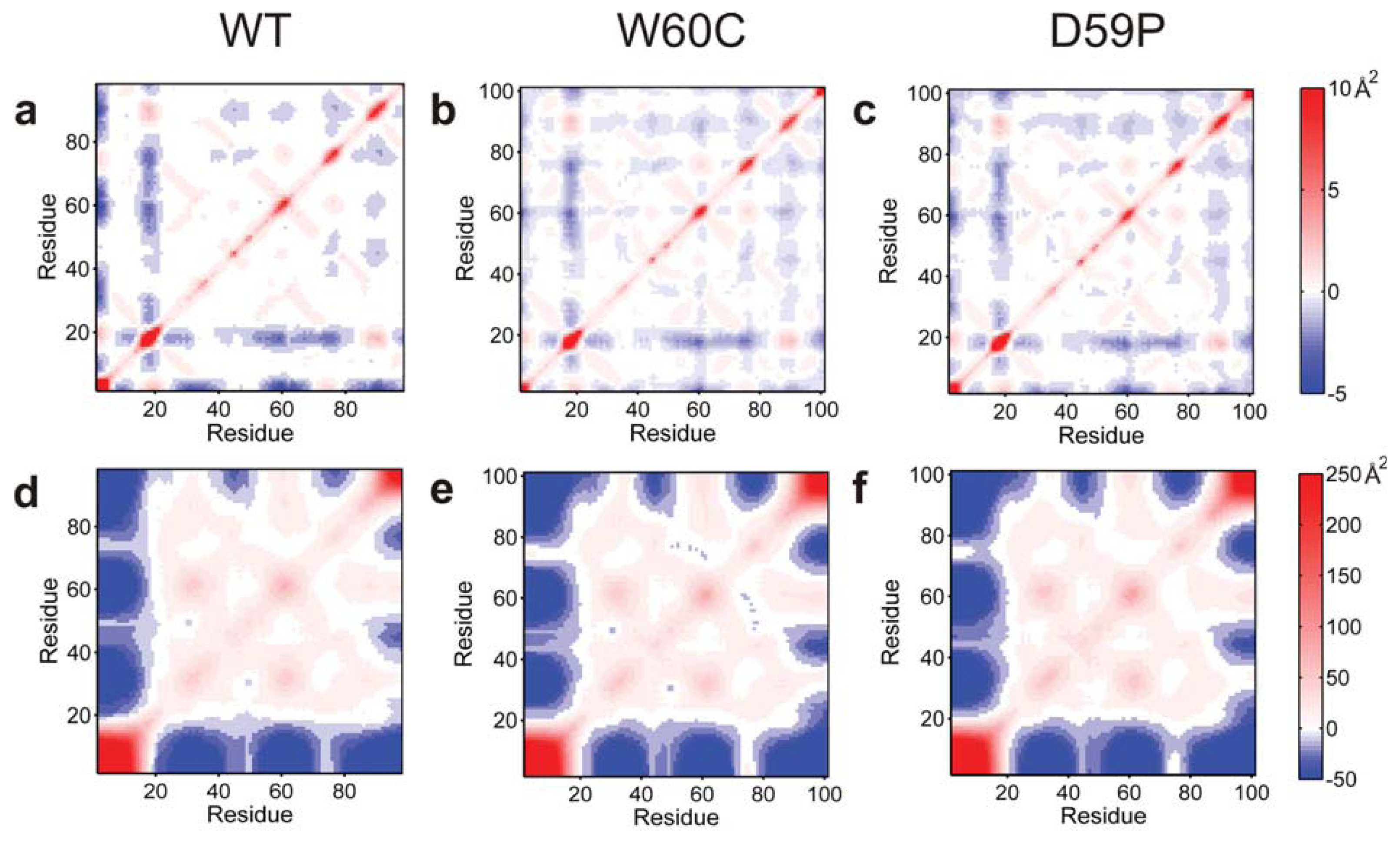
2.6. Large Displacements of the Termini Dominate the Intermediate State’s Dynamical Repertoire
2.7. Is the Predicted Intermediate State Compatible with a Molten Globule Identified for Hβ2m at pH 4?
3. Experimental Section
3.1. Gō Model and Discrete Molecular Dynamics Simulations
3.2. Structural Clustering
3.3. Principal Component Analysis
4. Conclusions
Supplementary Information
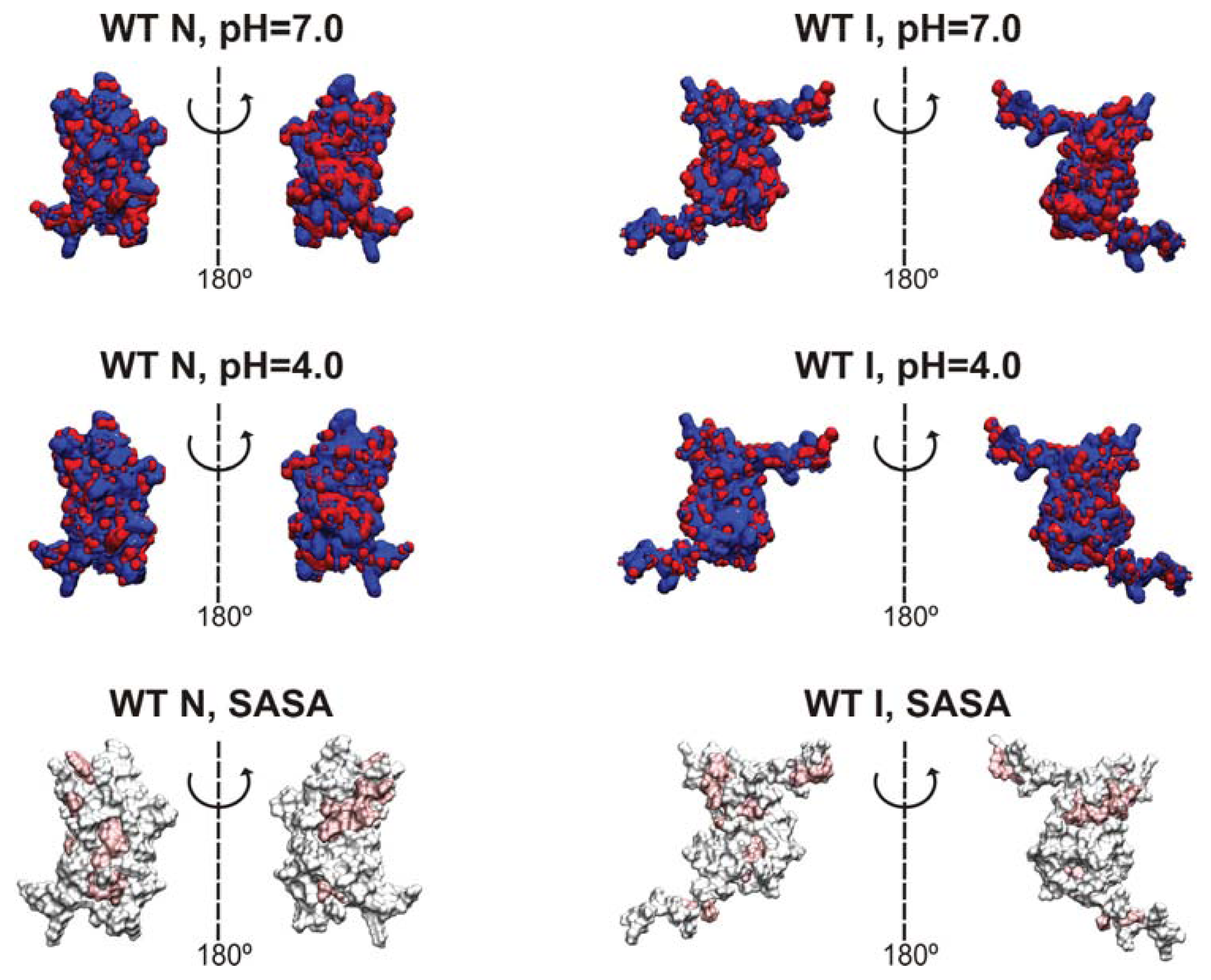
Electrostatic isocontours of the WT native (N)—X-ray structure—and intermediate (I)—clustering representative—states in acidic (4.0) and neutral pH (7.0) conditions. Coloring from red to blue corresponds to electrostatic potentials of −5 to +5 kBT. The 21 hydrophobic core amino acids are represented in pink in the SASA representation/depiction of the protein at the bottom of the figure. The protonation states at both pH values were attributed with PROPKA [36] via the webserver PDB2PQR v1.8 [54]. The Poisson-Boltzmann equation was solved using the Adaptive Poisson-Boltzmann Solver APBS v1.4 [55] in VMD v1.8.7 [56]. All isosurfaces were generated at −5 and +5 kBT.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | pH | Gsolv,polar (150 mM NaCl) | Gsolv,nonpolar |
|---|---|---|---|
| WT N | 4 (8e) | 2841 | 146 |
| 7 (−2e) | 2991 | ||
| WT I | 4 (11e) | 3263 | |
| 7 (−3e) | 3528 | 205 | |
| W60C N | 4 (11e) | 3029 | 151 |
| 7 (−2e) | 3328 | ||
| W60C I | 4 (11e) | 3676 | 222 |
| 7 (−2e) | 3990 | ||
| D59P N | 4 (9e) | 3075 | 156 |
| 7 (−1e) | 3347 | ||
| D59P I | 4 (9e) | 3769 | 217 |
| 7 (−1e) | 4112 | ||
Acknowledgments
Conflicts of Interest
References
- Trinh, C.H.; Smith, D.P.; Kalverda, A.P.; Phillips, S.E.V.; Radford, S.E. Crystal structure of monomeric human β-2-microglobulin reveals clues to its amyloidogenic properties. Proc. Natl. Acad. Sci. USA 2002, 99, 9771–9776. [Google Scholar]
- Hasegawa, K.; Ohhashi, Y.; Yamaguchi, I.; Takahashi, N.; Tsutsumi, S.; Goto, Y.; Gejyo, F.; Naiki, H. Amyloidogenic synthetic peptides of β2-microglobulin—A role of the disulfide bond. Biochem. Biophys. Res. Commun 2003, 304, 101–106. [Google Scholar]
- Yamamoto, K.; Yagi, H.; Ozawa, D.; Sasahara, K.; Naiki, H.; Goto, Y. Thiol compounds inhibit the formation of amyloid fibrils by β2-microglobulin at neutral pH. J. Mol. Biol 2008, 376, 258–268. [Google Scholar]
- Bellotti, V.; Gallieni, M.; Giorgetti, S.; Brancaccio, D. Dynamic of β2-microglobulin fibril formation and reabsorption: The role of proteolysis. Semin. Dial 2001, 14, 117–122. [Google Scholar]
- Eakin, C.M.; Miranker, A.D. From chance to frequent encounters: Origins of β2-microglobulin fibrillogenesis. BBA-Proteins Proteom 2005, 1753, 92–99. [Google Scholar]
- Platt, G.W.; Radford, S.E. Glimpses of the molecular mechanisms of β2-microglobulin fibril formation in vitro: Aggregation on a complex energy landscape. FEBS Lett 2009, 583, 2623–2629. [Google Scholar]
- Smith, D.P.; Jones, S.; Serpell, L.C.; Sunde, M.; Radford, S.E. A systematic investigation into the effect of protein destabilisation on beta 2-microglobulin amyloid formation. J. Mol. Biol 2003, 330, 943–954. [Google Scholar]
- Ohhashi, Y.; Kihara, M.; Naiki, H.; Goto, Y. Ultrasonication-induced amyloid fibril formation of β2-microglobulin. J. Biol. Chem 2005, 280, 32843–32848. [Google Scholar]
- Calabrese, M.F.; Eakin, C.M.; Wang, J.M.; Miranker, A.D. A regulatable switch mediates self-association in an immunoglobulin fold. Nat. Struct. Mol. Biol 2008, 15, 965–971. [Google Scholar]
- Hodkinson, J.P.; Radford, S.E.; Ashcroft, A.E. The role of conformational flexibility in β2-microglobulin amyloid fibril formation at neutral pH. Rapid Commun. Mass Spectrom 2012, 26, 1783–1792. [Google Scholar]
- Chiti, F.; Mangione, P.; Andreola, A.; Giorgetti, S.; Stefani, M.; Dobson, C.M.; Bellotti, V.; Taddei, N. Detection of two partially structured species in the folding process of the amyloidogenic protein [beta]2-microglobulin. J. Mol. Biol. 2001, 307, 379–391. [Google Scholar]
- Chiti, F.; de Lorenzi, E.; Grossi, S.; Mangione, P.; Giorgetti, S.; Caccialanza, G.; Dobson, C.M.; Merlini, G.; Ramponi, G.; Bellotti, V. A partially structured species of [beta]2-microglobulin is significantly populated under physiological conditions and involved in fibrillogenesis. J. Biol. Chem 2001, 276, 46714–46721. [Google Scholar]
- Jahn, T.R.; Parker, M.J.; Homans, S.W.; Radford, S.E. Amyloid formation under physiological conditions proceeds via a native-like folding intermediate. Nat. Struct. Mol. Biol 2006, 13, 195–201. [Google Scholar]
- Eichner, T.; Radford, S.E. A generic mechanism of β2-microglobulin amyloid assembly at neutral pH involving a specific proline switch. J. Mol. Biol 2009, 386, 1312–1326. [Google Scholar]
- Esposito, G.; Michelutti, R.; Verdone, G.; Viglino, P.; Hernández, H.H.; Robinson, C.V.; Amoresano, A.; Piaz, F.D.; Monti, M.; Pucci, P.; et al. Removal of the N-terminal hexapeptide from human β2-microglobulin facilitates protein aggregation and fibril formation. Protein Sci 2000, 9, 831–845. [Google Scholar]
- Eichner, T.; Kalverda, A.P.; Thompson, G.S.; Homans, S.W.; Radford, S.E. Conformational conversion during amyloid formation at atomic resolution. Mol. Cell 2011, 41, 161–172. [Google Scholar]
- Routledge, K.E.; Tartaglia, G.G.; Platt, G.W.; Vendruscolo, M.; Radford, S.E. Competition between intramolecular and intermolecular interactions in an amyloid-forming protein. J. Mol. Biol 2009, 389, 776–786. [Google Scholar]
- Mendoza, V.L.; Antwi, K.; Barón-Rodríguez, M.A.; Blanco, C.; Vachet, R.W. Structure of the preamyloid dimer of β-2-microglobulin from covalent labeling and mass spectrometry. Biochemistry 2010, 49, 1522–1532. [Google Scholar]
- Colombo, M.; de Rosa, M.; Bellotti, V.; Ricagno, S.; Bolognesi, M. A recurrent D-strand association interface is observed in β-2 microglobulin oligomers. FEBS J 2012, 279, 1131–1143. [Google Scholar]
- Esposito, G.; Ricagno, S.; Corazza, A.; Rennella, E.; Gümral, D.; Mimmi, M.C.; Betto, E.; Pucillo, C.E.M.; Fogolari, F.; Viglino, P.; et al. The controlling roles of Trp60 and Trp95 in β2-microglobulin function, folding and amyloid aggregation properties. J. Mol. Biol 2008, 378, 887–897. [Google Scholar]
- Santambrogio, C.; Ricagno, S.; Colombo, M.; Barbiroli, A.; Bonomi, F.; Bellotti, V.; Bolognesi, M.; Grandori, R. DE-loop mutations affect beta2 microglobulin stability, oligomerization, and the low-pH unfolded form. Protein Sci 2010, 19, 1386–1394. [Google Scholar]
- Ricagno, S.; Colombo, M.; de Rosa, M.; Sangiovanni, E.; Giorgetti, S.; Raimondi, S.; Bellotti, V.; Bolognesi, M. DE loop mutations affect β2-microglobulin stability and amyloid aggregation. Biochem. Biophys. Res. Commun 2008, 377, 146–150. [Google Scholar]
- Raimondi, S.; Barbarini, N.; Mangione, P.; Esposito, G.; Ricagno, S.; Bolognesi, M.; Zorzoli, I.; Marchese, L.; Soria, C.; Bellazzi, R.; et al. The two tryptophans of beta2-microglobulin have distinct roles in function and folding and might represent two independent responses to evolutionary pressure. BMC Evol. Biol 2011, 11, 159. [Google Scholar]
- Kihara, M.; Chatani, E.; Iwata, K.; Yamamoto, K.; Matsuura, T.; Nakagawa, A.; Naiki, H.; Goto, Y. Conformation of amyloid fibrils of β2-microglobulin probed by tryptophan mutagenesis. J. Biol. Chem 2006, 281, 31061–31069. [Google Scholar]
- Estácio, S.G.; Fernandes, C.S.; Krobath, H.; Faísca, P.F.N.; Shakhnovich, E.I. Robustness of atomistic Gō models in predicting native-like folding intermediates. J. Chem. Phys. 2012, 137. [Google Scholar]
- Esposito, G.; Bellotti, V. Emerging Molecular Targets in the Therapy of Dialysis-related Amyloidosis. In Protein Misfolding Diseases; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2010; pp. 843–865. [Google Scholar]
- Richardson, J.S.; Richardson, D.C. Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. USA 2002, 99, 2754–2759. [Google Scholar]
- Martinez, J.C.; Pisabarro, M.T.; Serrano, L. Obligatory steps in protein folding and the conformational diversity of the transition state. Nat. Struct. Mol. Biol 1998, 5, 721–729. [Google Scholar]
- Ohgushi, M.; Wada, A. “Molten-globule state”: A compact form of globular proteins with mobile side-chains. FEBS Lett 1983, 164, 21–24. [Google Scholar]
- Chiti, F. Relative Importance of Hydrophobicity, Net Charge, and Secondary Structure Propensities in Protein Aggregation. In Protein Misfolding, Aggregation, and Conformational Diseases—Protein Reviews; Springer: New York, NY, USA, 2006; Volume 4, pp. 43–59. [Google Scholar]
- McParland, V.J.; Kalverda, A.P.; Homans, S.W.; Radford, S.E. Structural properties of an amyloid precursor of [beta]2-microglobulin. Nat. Struct. Biol 2002, 9, 326–331. [Google Scholar]
- Mukaiyama, A.; Nakamura, T.; Makabe, K.; Maki, K.; Goto, Y.; Kuwajima, K. The molten globule of β2-microglobulin accumulated at pH 4 and its role in protein folding. J. Mol. Biol 2013, 425, 273–291. [Google Scholar]
- Mukaiyama, A.; Nakamura, T.; Makabe, K.; Maki, K.; Goto, Y.; Kuwajima, K. Native-state heterogeneity of β2-microglobulin as revealed by kinetic folding and real-time NMR experiments. J. Mol. Biol 2013, 425, 257–272. [Google Scholar]
- McParland, V.; Kad, N.; Kalverda, A.; Brown, A.; Kirwin-Jones, P.; Hunter, M.; Sunde, M.; Radford, S. Partially unfolded states of β2-microglobulin and amyloid formation in vitro. Biochemistry 2000, 39, 8735–8746. [Google Scholar]
- Platt, G.W.; McParland, V.J.; Kalverda, A.P.; Homans, S.W.; Radford, S.E. Dynamics in the unfolded state of [beta]2-microglobulin studied by NMR. J. Mol. Biol 2005, 346, 279–294. [Google Scholar]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins: Struct. Funct. Bioinf 2005, 61, 704–721. [Google Scholar]
- Krobath, H.; Estácio, S.G.; Faísca, P.F.N.; Shakhnovich, E.I. Identification of a conserved aggregation-prone intermediate state in the folding pathways of Spc-SH3 amyloidogenic variants. J. Mol. Biol 2012, 422, 705–722. [Google Scholar]
- Huang, L.; Shakhnovich, E.I. Is there an en route folding intermediate for cold shock proteins? Protein Sci 2012, 21, 677–685. [Google Scholar]
- Tsai, J.; Levitt, M.; Baker, D. Hierarchy of structure loss in MD simulations of src SH3 domain unfolding. J. Mol. Biol 1999, 291, 215–225. [Google Scholar]
- Gō, N.; Abe, H. Noninteracting local-structure model of folding and unfolding transition in globular proteins. I. Formulation. Biopolymers 1981, 20, 991–1011. [Google Scholar]
- Shimada, J.; Kussell, E.L.; Shakhnovich, E.I. The folding thermodynamics and kinetics of crambin using an all-atom Monte Carlo simulation. J. Mol. Biol 2001, 308, 79–95. [Google Scholar]
- Dokholyan, N.V.; Buldyrev, S.V.; Stanley, H.E.; Shakhnovich, E.I. Discrete molecular dynamics studies of the folding of a protein-like model. Fold. Des 1998, 3, 577–587. [Google Scholar]
- Smith, S.W.; Hall, C.K.; Freeman, B.D. Molecular dynamics for polymeric fluids using discontinuous potentials. J. Comput. Phys 1997, 134, 16–30. [Google Scholar]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett 1999, 314, 141–151. [Google Scholar]
- Chodera, J.D.; Swope, W.C.; Pitera, J.W.; Seok, C.; Dill, K.A. Use of the weighted histogram analysis method for the analysis of simulated and parallel tempering simulations. J. Chem. Theor. Comput 2007, 3, 26–41. [Google Scholar]
- Feig, M.; Karanicolas, J.; Brooks, C.L., III. MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph. Model. 2004, 22, 377–395. [Google Scholar]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar]
- Neudecker, P.; Robustelli, P.; Cavalli, A.; Walsh, P.; Lundström, P.; Zarrine-Afsar, A.; Sharpe, S.; Vendruscolo, M.; Kay, L.E. Structure of an intermediate state in protein folding and aggregation. Science 2012, 336, 362–366. [Google Scholar]
- Chiti, F.; Dobson, C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol 2009, 5, 15–22. [Google Scholar]
- Jones, S.; Smith, D.P.; Radford, S.E. Role of the N and C-terminal strands of [beta]2-microglobulin in amyloid formation at neutral pH. J. Mol. Biol 2003, 330, 935–941. [Google Scholar]
- Hinsen, K.; Kneller, G.R. Solvent effects in the slow dynamics of proteins. Proteins Struct. Funct. Bioinf 2008, 70, 1235–1242. [Google Scholar]
- Hughson, F.; Wright, P.; Baldwin, R. Structural characterization of a partly folded apomyoglobin intermediate. Science 1990, 249, 1544–1548. [Google Scholar]
- Park, S.H. Hydrophobic core variant ubiquitin forms a molten globule conformation at acidic pH. J. Biochem. Mol. Biol 2004, 37, 676–683. [Google Scholar]
- Dolinsky, T.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res 2004, 35, W522–W525. [Google Scholar]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph 1996, 14, 33–38. [Google Scholar]
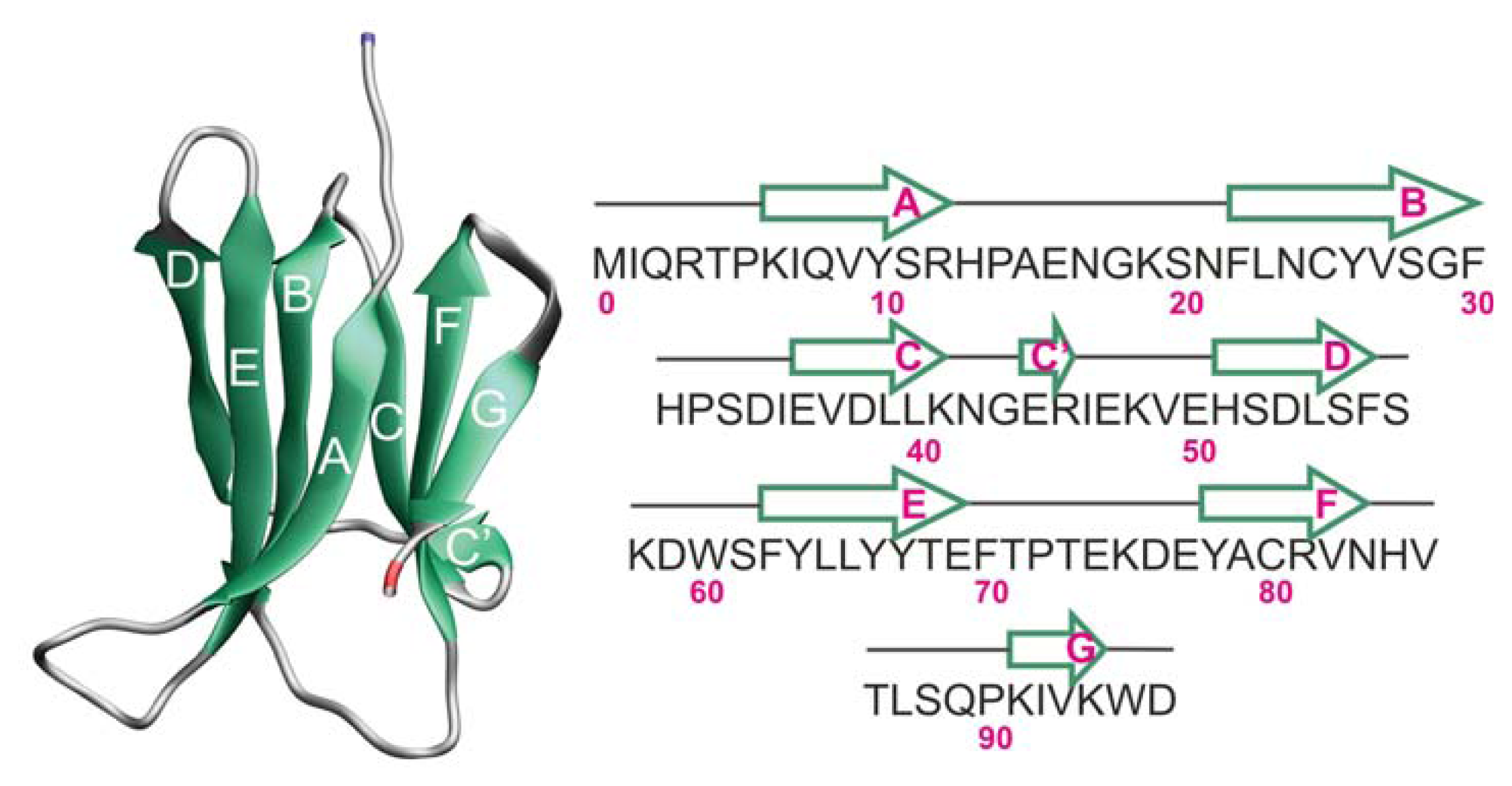

| β-strand | WT | W60C | D59P |
|---|---|---|---|
| A (6–12) | 140 | 149 (+6.4%) | 136 (−2.8%) |
| B (22–30) | 319 | 330 (+3.4%) | 305 (−4.4%) |
| C (36–41) | 187 | 195 (+4.3%) | 187 |
| C′ (44–45) | 35 | 39 (+11.4%) | 32 (−8.6%) |
| D (51–56) | 108 | 108 | 102 (−5.6%) |
| E (62–69) | 324 | 325 (+0.3%) | 323 (−0.3%) |
| F (78–83) | 204 | 197 (−3.4%) | 199 (−2.4%) |
| G (91–94) | 72 | 63 (−12.5%) | 66 (−8.3%) |
| Loop | WT | W60C | D59P |
| AB (13–21) | 89 | 87 (−2.2%) | 80 (−10.1%) |
| BC (31–35) | 102 | 92 (−9.8%) | 97 (−4.9%) |
| DE (57–61) | 44 | 38 (−13.6%) | 46 (+4.5%) |
| EF (70–77) | 111 | 100 (−9.9%) | 108 (−2.7%) |
| FG (84–90) | 95 | 94 (−1.0%) | 88 (−7.4%) |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Estácio, S.G.; Shakhnovich, E.I.; Faísca, P.F.N. Assessing the Effect of Loop Mutations in the Folding Space of β2-Microglobulin with Molecular Dynamics Simulations. Int. J. Mol. Sci. 2013, 14, 17256-17278. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms140917256
Estácio SG, Shakhnovich EI, Faísca PFN. Assessing the Effect of Loop Mutations in the Folding Space of β2-Microglobulin with Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2013; 14(9):17256-17278. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms140917256
Chicago/Turabian StyleEstácio, Sílvia G., Eugene I. Shakhnovich, and Patrícia F. N. Faísca. 2013. "Assessing the Effect of Loop Mutations in the Folding Space of β2-Microglobulin with Molecular Dynamics Simulations" International Journal of Molecular Sciences 14, no. 9: 17256-17278. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms140917256