
Site-Specific PEGylation of Therapeutic Proteins

Abstract
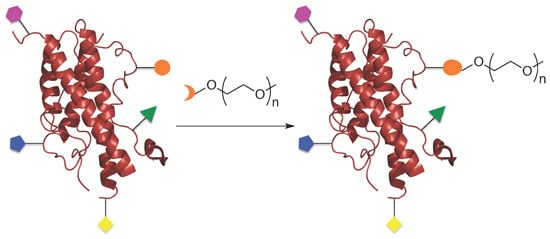
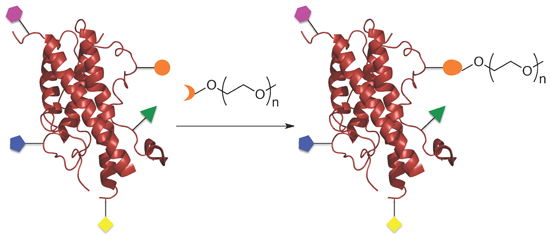
:
1. Introduction


{kind=link}
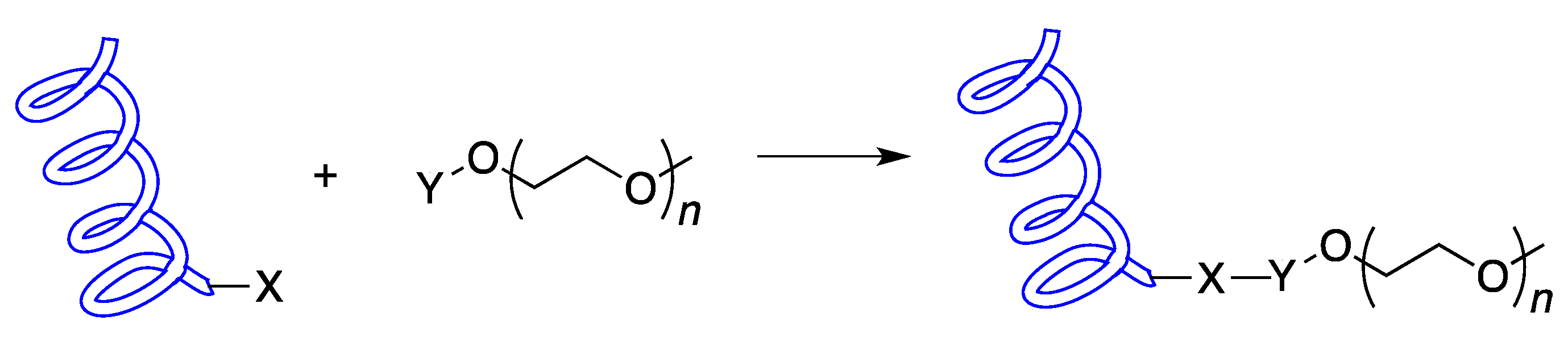{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Protein | Protein Size (kDa) | PEG Size (kDa) | Functional Group on PEG | Site of Attachment | Site-Specific | Year of Approval | Use | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Adagen® | Adenosine deaminase | 40 | 5 | Succinimidyl ester | Lysines, serines, tyrosines, histidines | No | 1990 | Severe combined immunodeficiency disease | [18] |
| Oncaspar® | Asparaginase | 31 | 5 | Succinimidyl ester | Lysines, serines, tyrosines, histidines | No | 1994 | Leukemia | [23] |
| PegIntron® | Interferon-α-2b | 19.2 | 12 | Succinimidyl ester | Lysines, serines, tyrosines, histidines,cysteines | No, but 47.8% one isomer | 2000 | Hepatitis C | [26] |
| Pegasys® | Interferon-α-2a | 19.2 | 40 | Succinimidyl ester | Lysines | No | 2001 | Hepatitis C | [25] |
| Neulasta® | Granulocyte colony-stimulating factor (G-CSF) | 18.8 | 20 | Aldehyde | N-Terminal amine | Yes | 2002 | Neutropenia | [30] |
| Somavert® | Human growth hormone (hGH) | 22 | 5 | Succinimidyl ester | Lysines, N-terminus, phenylalanine | No | 2003 | Acromegaly | [31] |
| Mircera® | Erythropoietin | 30 (18 unglycosylated) | 40 | Succinimidyl ester | Lysines | No | 2007 | Anemia | [32] |
| Cimzia® | Anti-tumor necrosis factor (TNF) α Fab’ | 51 | 40 | Maleimide | C-Terminal cysteine | Yes | 2008 | Rheumatoid arthritis, Crohn disease, psoriatic arthritis | [33] |
| Krystexxa® | Urate oxidase | 34 | 10 | p-Nitrophenyl carbonate ester | Lysines | No | 2010 | Gout | [34] |
| Omontys | Synthetic, dimeric peptide (erythropoiesis stimulating agent) | 4.9 | 40 (2 20 kDa chains) | Succinimidyl ester (added during chemical synthesis of the peptide) | Lysines | Yes | 2012 (Recalled 2014) | Anemia in chronic kidney disease | [35] |
2. Chemical Synthesis
3. Amino Acid Labeling
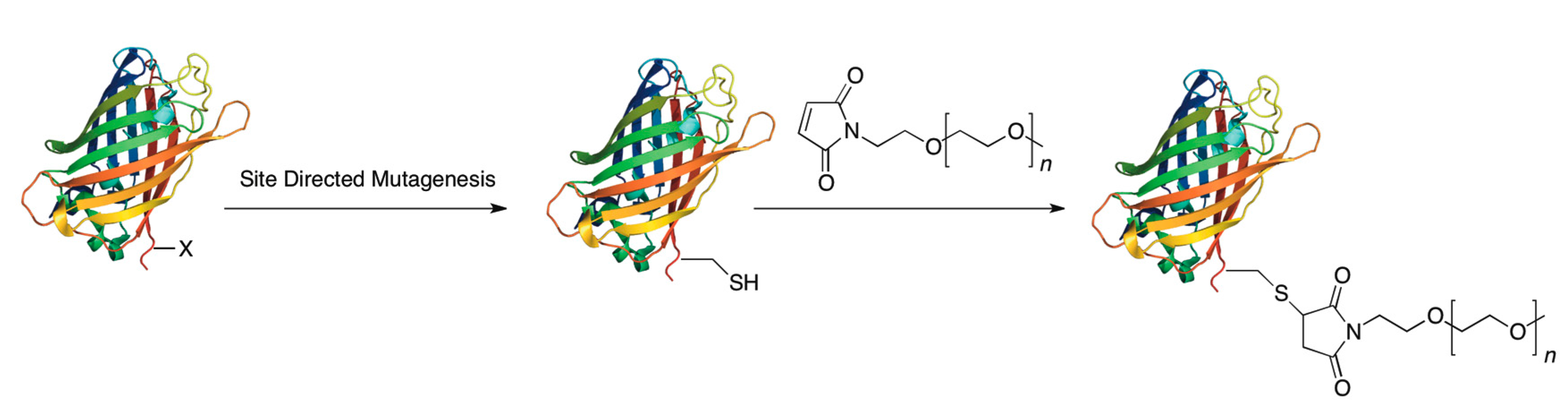
3.1. Cysteine Tagging

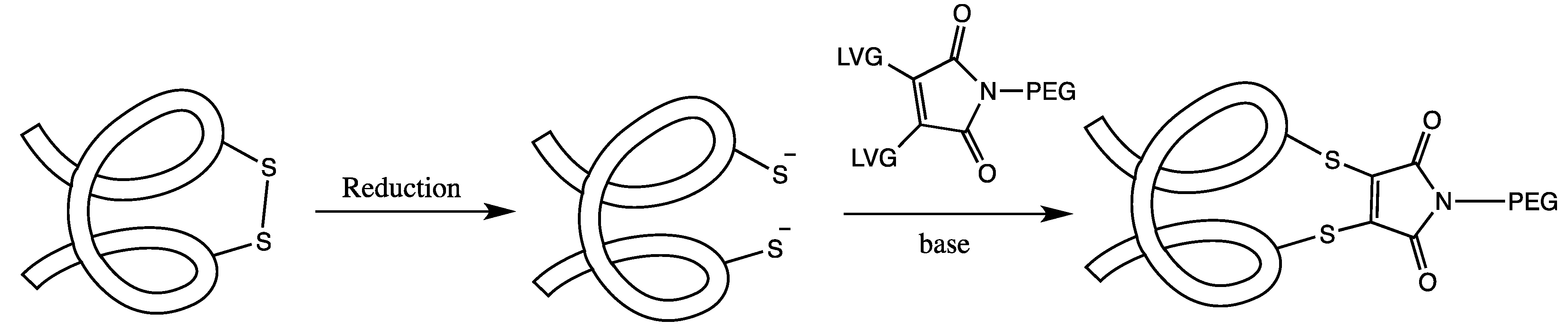
3.2. Cysteine Disulfide (Cystine) Labeling

3.3. Tyrosine Tagging
3.4. Serine and Threonine Reactions
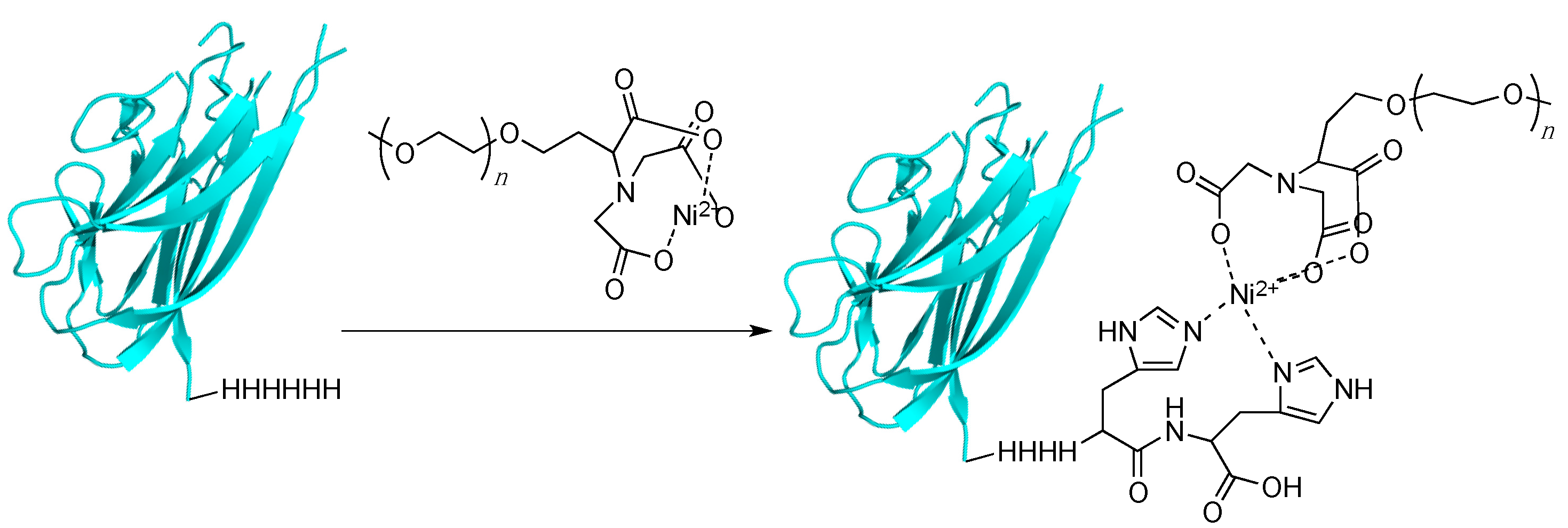
3.5. Histidine Tagging and Related Multiresidue Tags

4. Bio-Orthogonal Labeling
4.1. Incorporation via Auxotrophic Strains
4.2. Nonsense Suppression Methods
5. Enzymatic Labeling
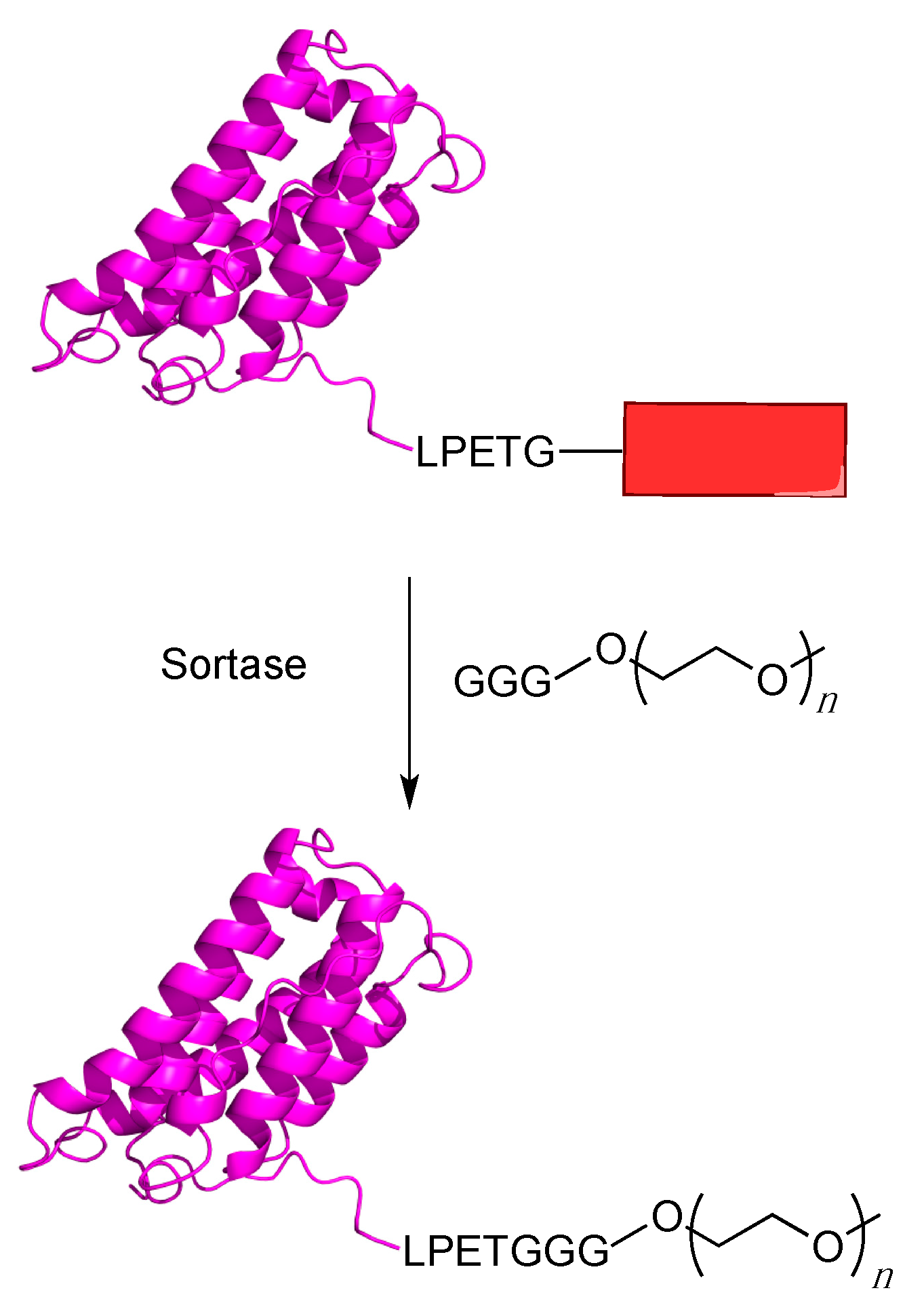
5.1. Sortagging

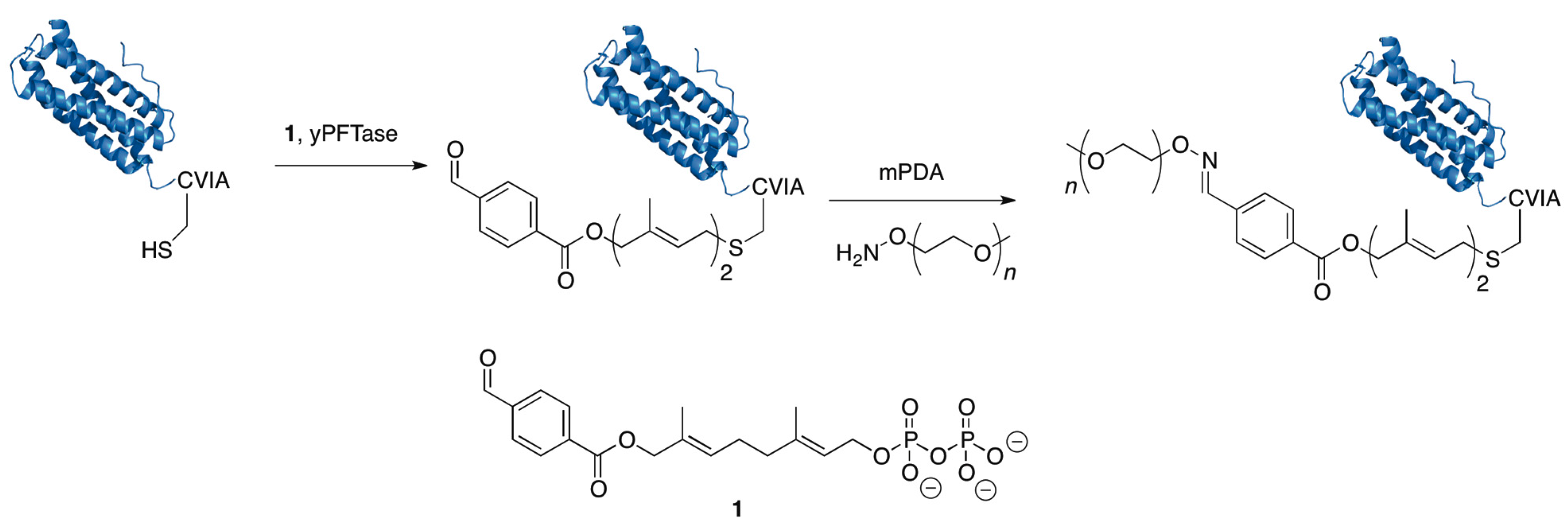
5.2. Protein Farnesyltransferase

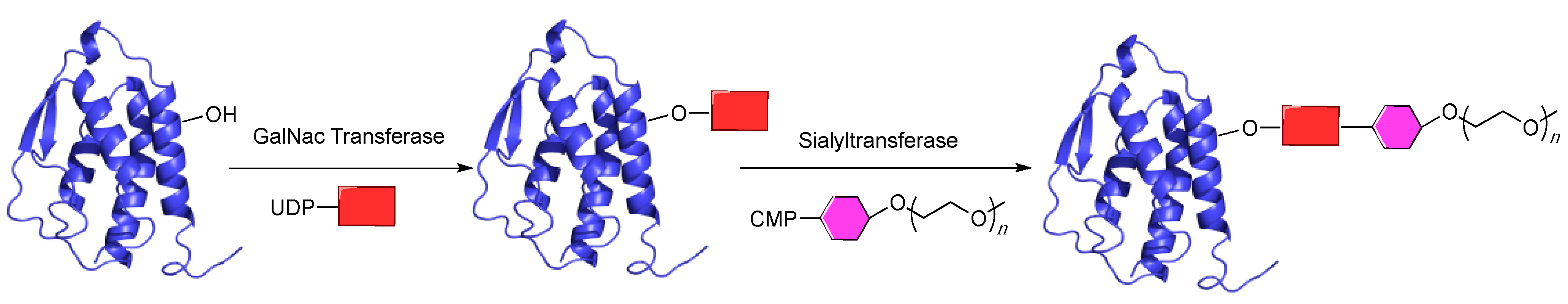
5.3. GlycoPEGylation

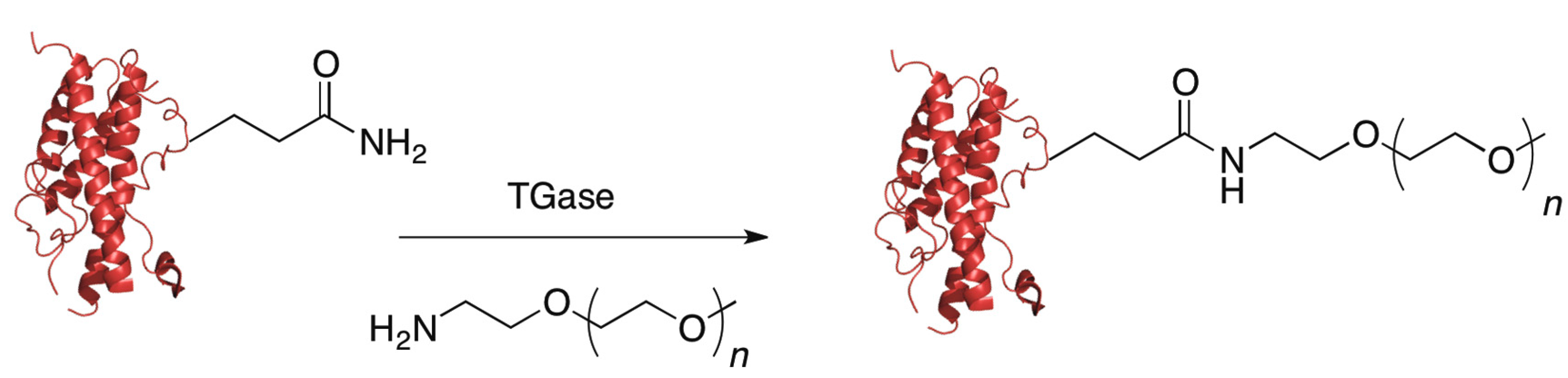
5.4. Transglutaminase

5.5. Formylglycine-Generating Enzyme

6. Perspectives and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J. Introduction to current and future protein therapeutics: A protein engineering perspective. Exp. Cell Res. 2011, 317, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Szymkowski, D.E. Creating the next generation of protein therapeutics through rational drug design. Curr. Opin. Drug Discov. Dev. 2005, 8, 590–600. [Google Scholar]
- De Groot, A.S.; Scott, D.W. Immunogenicity of protein therapeutics. Trends Immunol. 2007, 28, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Kromminga, A.; Schellekens, H. Antibodies against erythropoietin and other protein-based therapeutics: An overview. Ann. N. Y. Acad. Sci. 2005, 1050, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Pastan, I. Removal of B cell epitopes as a practical approach for reducing the immunogenicity of foreign protein-based therapeutics. Adv. Drug Deliv. Rev. 2009, 61, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Szlachcic, A.; Zakrzewska, M.; Otlewski, J. Longer action means better drug: Tuning up protein therapeutics. Biotechnol. Adv. 2011, 29, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Constantinou, A.; Chester, K.A.; Vyas, B.; Canis, K.; Haslam, S.M.; Dell, A.; Epenetos, A.A.; Deonarain, M.P. Glycoengineering approach to half-life extension of recombinant biotherapeutics. Bioconjug. Chem. 2012, 23, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Hutt, M.; Färber-Schwarz, A.; Unverdorben, F.; Richter, F.; Kontermann, R.E. Plasma half-life extension of small recombinant antibodies by fusion to immunoglobulin-binding domains. J. Biol. Chem. 2012, 287, 4462–4469. [Google Scholar] [CrossRef] [PubMed]
- Trüssel, S.; Dumelin, C.; Frey, K.; Villa, A.; Buller, F.; Neri, D. New strategy for the extension of the serum half-life of antibody fragments. Bioconjug. Chem. 2009, 20, 2286–2292. [Google Scholar] [CrossRef] [PubMed]
- Gaberc-Porekar, V.; Zore, I.; Podobnik, B.; Menart, V. Obstacles and pitfalls in the PEGylation of therapeutic proteins. Curr. Opin. Drug Discov. Dev. 2008, 11, 242–250. [Google Scholar]
- Veronese, F.M.; Mero, A. The impact of PEGylation on biological therapies. BioDrugs 2008, 22, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Abuchowski, A.; van Es, T.; Palczuk, N.C.; Davis, F.F. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J. Biol. Chem. 1977, 252, 3578–3581. [Google Scholar] [PubMed]
- Abuchowski, A.; McCoy, J.R.; Palczuk, N.C.; van Es, T.; Davis, F.F. Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J. Biol. Chem. 1977, 252, 3582–3586. [Google Scholar] [PubMed]
- Chen, R.H.L.; Abuchowski, A.; van Es, T.; Palczuk, N.C.; Davis, F.F. Properties of two urate oxidases modified by the covalent attachment of poly(ethylene glycol). Biochim. Biophys. Acta Enzymol. 1981, 660, 293–298. [Google Scholar] [CrossRef]
- Savoca, K.V.; Abuchowski, A.; van Es, T.; Davis, F.F.; Palczuk, N.C. Preparation of a non-immunogenic arginase by the covalent attachment of polyethylene glycol. Biochim. Biophys. Acta Protein Struct. 1979, 578, 47–53. [Google Scholar] [CrossRef]
- Levy, Y.; Hershfield, M.S.; Fernandez-Mejia, C.; Polmar, S.H.; Scudiery, D.; Berger, M.; Sorensen, R.U. Adenosine deaminase deficiency with late onset of recurrent infections: Response to treatment with polyethylene glycol-modified adenosine deaminase. J. Pediatr. 1988, 113, 312–317. [Google Scholar] [CrossRef]
- Hershfield, M.S.; Buckley, R.H.; Greenberg, M.L.; Melton, A.L.; Schiff, R.; Hatem, C.; Kurtzberg, J.; Markert, M.L.; Kobayashi, R.H.; Kobayashi, A.L. Treatment of adenosine deaminase deficiency with polyethylene glycol-modified adenosine deaminase. N. Engl. J. Med. 1987, 316, 589–596. [Google Scholar] [CrossRef] [PubMed]
- McHutchison, J.G.; Lawitz, E.J.; Shiffman, M.L.; Muir, A.J.; Galler, G.W.; McCone, J.; Nyberg, L.M.; Lee, W.M.; Ghalib, R.H.; Schiff, E.R.; et al. Peginterferon α-2b or α-2a with ribavirin for treatment of hepatitis c infection. N. Engl. J. Med. 2009, 361, 580–593. [Google Scholar] [CrossRef] [PubMed]
- Lau, G.K.K.; Piratvisuth, T.; Luo, K.X.; Marcellin, P.; Thongsawat, S.; Cooksley, G.; Gane, E.; Fried, M.W.; Chow, W.C.; Paik, S.W.; et al. Peginterferon α-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis b. N. Engl. J. Med. 2005, 352, 2682–2695. [Google Scholar] [CrossRef] [PubMed]
- Wursthorn, K.; Lutgehetmann, M.; Dandri, M.; Volz, T.; Buggisch, P.; Zollner, B.; Longerich, T.; Schirmacher, P.; Metzler, F.; Zankel, M.; et al. Peginterferon α-2b plus adefovir induce strong cccDNA decline and HBsAg reduction in patients with chronic hepatitis b. Hepatology 2006, 44, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.L. Pegaspargase: A review of clinical studies. Adv. Drug Deliv. Rev. 2003, 55, 1293–1302. [Google Scholar] [CrossRef]
- Sherman, M.R.; Saifer, M.G.P.; Perez-Ruiz, F. Peg-uricase in the management of treatment-resistant gout and hyperuricemia. Adv. Drug Deliv. Rev. 2008, 60, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Foser, S.; Schacher, A.; Weyer, K.A.; Brugger, D.; Dietel, E.; Marti, S.; Schreitmüller, T. Isolation, structural characterization, and antiviral activity of positional isomers of monoPEGylated interferon α-2a (pegasys). Protein Expr. Purif. 2003, 30, 78–87. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Youngster, S.; Grace, M.; Bausch, J.; Bordens, R.; Wyss, D.F. Structural and biological characterization of PEGylated recombinant interferon α-2b and its therapeutic implications. Adv. Drug Deliv. Rev. 2002, 54, 547–570. [Google Scholar] [CrossRef]
- Finn, R.F. PEGylation of human growth hormone: Strategies and properties. In PEGylated Protein Drugs: Basic Science and Clinical Applications; Birkhauser Verlag: Birkhauser Verlag, Basel, Switzerland, 2009; pp. 187–203. [Google Scholar]
- Fishburn, C.S. The pharmacology of PEGylation: Balancing PD with PK to generate novel therapeutics. J. Pharm. Sci. 2008, 97, 4167–4183. [Google Scholar] [CrossRef] [PubMed]
- Alconcel, S.N.; Baas, A.S.; Maynard, H.D. FDA-approved poly (ethylene glycol)-protein conjugate drugs. Polym. Chem. 2011, 2, 1442–1448. [Google Scholar] [CrossRef]
- Piedmonte, D.M.; Treuheit, M.J. Formulation of neulasta® (pegfilgrastim). Adv. Drug Deliv. Rev. 2008, 60, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Pradhananga, S.; Wilkinson, I.; Ross, R. Pegvisomant: Structure and function. J. Mol. Endocrinol. 2002, 29, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Macdougall, I.C.; Eckardt, K. Novel strategies for stimulating erythropoiesis and potential new treatments for anaemia. Lancet 2006, 368, 947–953. [Google Scholar] [CrossRef]
- Blick, S.K.A.; Curran, M.P. Certolizumab pegol. BioDrugs 2007, 21, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, N.; Yasothan, U.; Kirkpatrick, P. Pegloticase. Nat. Rev. Drug Discov. 2011, 10, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Woodburn, K.W.; Fong, K.-L.; Wilson, S.D.; Sloneker, S.; Strzemienski, P.; Solon, E.; Moriya, Y.; Tagawa, Y. Peginesatide clearance, distribution, metabolism, and excretion in monkeys following intravenous administration. Drug Metab. Dispos. 2013, 41, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Melmed, G.Y.; Targan, S.R.; Yasothan, U.; Hanicq, D.; Kirkpatrick, P. Certolizumab pegol. Nat. Rev. Drug Discov. 2008, 7, 641–642. [Google Scholar] [CrossRef] [PubMed]
- Ordas, I.; Mould, D.R.; Feagan, B.G.; Sandborn, W.J. Anti-TNF monoclonal antibodies in inflammatory bowel disease: Pharmacokinetics-based dosing paradigms. Clin. Pharmacol. Ther. 2012, 91, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Goel, N.; Stephens, S. Certolizumab pegol. MAbs 2010, 2, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Kinstler, O.; Brems, D.; Lauren, S.; Paige, A.; Hamburger, J.; Treuheit, M. Characterization and stability of N-terminally PEGylated rhG-CSF. Pharm. Res. 1996, 13, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Molineux, G. Pegfilgrastim: Using PEGylation technology to improve neutropenia support in cancer patients. Anticancer Drugs 2003, 14, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Holmes, F.A.; O’Shaughnessy, J.A.; Vukelja, S.; Jones, S.E.; Shogan, J.; Savin, M.; Glaspy, J.; Moore, M.; Meza, L.; Wiznitzer, I.; et al. Blinded, randomized, multicenter study to evaluate single administration pegfilgrastim once per cycle versus daily filgrastim as an adjunct to chemotherapy in patients with high-risk stage II or stage III/IV breast cancer. J. Clin. Oncol. 2002, 20, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Green, M.D.; Koelbl, H.; Baselga, J.; Galid, A.; Guillem, V.; Gascon, P.; Siena, S.; Lalisang, R.I.; Samonigg, H.; Clemens, M.R.; et al. A randomized double-blind multicenter phase III study of fixed-dose single-administration pegfilgrastim vs. daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann. Oncol. 2003, 14, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W. The top prescription drugs of 2012 globally: Biologics dominate, but small molecule CNS drugs hold on to top spots. ACS Chem. Neurosci. 2013, 4, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Veronese, F.M. State of the art in PEGylation: The great versatility achieved after forty years of research. J. Control. Release 2012, 161, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Jevševar, S.; Kunstelj, M.; Porekar, V.G. PEGylation of therapeutic proteins. Biotechnol. J. 2010, 5, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Kling, J. PEGylation of biologics: A multipurpose solution. BioProc. Int. 2013, 11, 3. [Google Scholar]
- Roberts, M.J.; Bentley, M.D.; Harris, J.M. Chemistry for peptide and protein PEGylation. Adv. Drug Deliv. Rev. 2012, 64, 116–127. [Google Scholar] [CrossRef]
- Jølck, R.I.; Berg, R.H.; Andresen, T.L. Solid-phase synthesis of PEGylated lipopeptides using click chemistry. Bioconjug. Chem. 2010, 21, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Pandey, B.K.; Smith, M.S.; Torgerson, C.; Lawrence, P.B.; Matthews, S.S.; Watkins, E.; Groves, M.L.; Prigozhin, M.B.; Price, J.L. Impact of site-specific PEGylation on the conformational stability and folding rate of the Pin WW domain depends strongly on PEG oligomer length. Bioconjug. Chem. 2013, 24, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Powers, E.T.; Kelly, J.W. N-PEGylation of a reverse turn is stabilizing in multiple sequence contexts, unlike N-glcnacylation. ACS Chem. Biol. 2011, 6, 1188–1192. [Google Scholar] [CrossRef] [PubMed]
- Zalipsky, S.; Menon-Rudolph, S. Hydrazide Derivatives of Poly(Ethylene Glycol) and Their Bioconjugates; ACS Publications: Washington, DC, USA, 1997; pp. 318–341. [Google Scholar]
- Kinstler, O.; Molineux, G.; Treuheit, M.; Ladd, D.; Gegg, C. Mono-N-terminal poly(ethylene glycol)-protein conjugates. Adv. Drug Deliv. Rev. 2002, 54, 477–485. [Google Scholar] [CrossRef]
- Lee, H.; Jang, I.; Ryu, S.; Park, T. N-terminal site-specific mono-PEGylation of epidermal growth factor. Pharm. Res. 2003, 20, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Unterweger, B.; Stoisser, T.; Leitgeb, S.; Birner-Grünberger, R.; Nidetzky, B. Engineering of aerococcus viridansl-lactate oxidase for site-specific PEGylation: Characterization and selective bioorthogonal modification of a S218C mutant. Bioconjug. Chem. 2012, 23, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Tong, Y.; Gao, X.; Yao, W. Development of a C-terminal site-specific PEGylated analog of GLP-1 with improved anti-diabetic effects in diabetic mice. Drug Dev. Res. 2013, 74, 186–193. [Google Scholar] [CrossRef]
- Qiu, H.; Boudanova, E.; Park, A.; Bird, J.J.; Honey, D.M.; Zarazinski, C.; Greene, B.; Kingsbury, J.S.; Boucher, S.; Pollock, J.; et al. Site-specific PEGylation of human thyroid stimulating hormone to prolong duration of action. Bioconjug. Chem. 2013, 24, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Jiang, H.H.; Lim, S.M.; Youn, Y.S.; Choi, K.Y.; Lee, S.; Chen, X.; Byun, Y.; Lee, K.C. Site-specific PEGylated exendin-4 modified with a high molecular weight trimeric PEG reduces steric hindrance and increases type 2 antidiabetic therapeutic effects. Bioconjug. Chem. 2012, 23, 2214–2220. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Bussiere, J.; Yie, J.; Sickmier, A.; An, P.; Belouski, E.; Stanislaus, S.; Walker, K.W. Polyethylene glycol modified FGF21 engineered to maximize potency and minimize vacuole formation. Bioconjug. Chem. 2013. [Google Scholar] [CrossRef] [PubMed]
- Danial, M.; van Dulmen, T.H.H.; Aleksandrowicz, J.; Pötgens, A.J.G.; Klok, H. Site-specific PEGylation of HR2 peptides: Effects of PEG conjugation position and chain length on HIV-1 membrane fusion inhibition and proteolytic degradation. Bioconjug. Chem. 2012, 23, 1648–1660. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Wang, H.; Lai, J.; Xu, Y.; Zhang, C.; Chen, S. Site-specific PEGylation of a mutated-cysteine residue and its effect on tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL). Biomaterials 2013, 34, 9115–9123. [Google Scholar] [CrossRef] [PubMed]
- Balan, S.; Choi, J.-W.; Godwin, A.; Teo, I.; Laborde, C.M.; Heidelberger, S.; Zloh, M.; Shaunak, S.; Brocchini, S. Site-specific PEGylation of protein disulfide bonds using a three-carbon bridge. Bioconjug. Chem. 2006, 18, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, F.F.; Nobles, M.; Ryan, C.P.; Smith, M.E.B.; Tinker, A.; Caddick, S.; Baker, J.R. In situ maleimide bridging of disulfides and a new approach to protein PEGylation. Bioconjug. Chem. 2011, 22, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Robin, M.P.; Wilson, P.; Mabire, A.B.; Kiviaho, J.K.; Raymond, J.E.; Haddleton, D.M.; O’Reilly, R.K. Conjugation-induced fluorescent labeling of proteins and polymers using dithiomaleimides. J. Am. Chem. Soc. 2013, 135, 2875–2878. [Google Scholar] [CrossRef] [PubMed]
- Badescu, G.; Bryant, P.; Bird, M.; Henseleit, K.; Swierkosz, J.; Parekh, V.; Tommasi, R.; Pawlisz, E.; Jurlewicz, K.; Farys, M.; et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug. Chem. 2014, 25, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Gavrilyuk, J.; Barbas, C.F. Tyrosine bioconjugation through aqueous ene-type reactions: A click-like reaction for tyrosine. J. Am. Chem. Soc. 2010, 132, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Nagano, M.; Gavrilyuk, J.; Hakamata, W.; Inokuma, T.; Barbas, C.F. Facile and stabile linkages through tyrosine: Bioconjugation strategies with the tyrosine-click reaction. Bioconjug. Chem. 2013, 24, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, H.F.; Offord, R.E. Site-specific attachment of functionalized poly(ethylene glycol) to the amino terminus of proteins. Bioconjug. Chem. 1996, 7, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Levine, P.M.; Craven, T.W.; Bonneau, R.; Kirshenbaum, K. Intrinsic bioconjugation for site-specific protein PEGylation at N-terminal serine. Chem. Commun. 2014, 50, 6909–6912. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.C.F.; Wong, J.H.; Ng, T.B. Immobilized metal ion affinity chromatography: A review on its applications. Appl. Microbiol. Biot. 2012, 96, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Terpe, K. Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biot. 2003, 60, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Swierczewska, M.; Oh, Y.; Kim, A.; Jo, D.G.; Park, J.H.; Byun, Y.; Sadegh-Nasseri, S.; Pomper, M.G.; Lee, K.C.; et al. Mix to validate: A facile, reversible PEGylation for fast screening of potential therapeutic proteins in vivo. Angew. Chem. Int. Ed. 2013, 52, 6880–6884. [Google Scholar]
- Cong, Y.; Pawlisz, E.; Bryant, P.; Balan, S.; Laurine, E.; Tommasi, R.; Singh, R.; Dubey, S.; Peciak, K.; Bird, M.; et al. Site-specific PEGylation at histidine tags. Bioconjug. Chem. 2012, 23, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Thom, J.; Anderson, D.; McGregor, J.; Cotton, G. Recombinant protein hydrazides: Application to site-specific protein PEGylation. Bioconjug. Chem. 2011, 22, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, A.J.; Kooijman, M.; Hennink, W.E.; Mastrobattista, E. Nonnatural amino acids for site-specific protein conjugation. Bioconjug. Chem. 2009, 20, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Cazalis, C.S.; Haller, C.A.; Sease-Cargo, L.; Chaikof, E.L. C-Terminal site-specific PEGylation of a truncated thrombomodulin mutant with retention of full bioactivity. Bioconjug. Chem. 2004, 15, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Nairn, N.W.; Shanebeck, K.D.; Wang, A.; Graddis, T.J.; VanBrunt, M.P.; Thornton, K.C.; Grabstein, K. Development of copper-catalyzed azide-alkyne cycloaddition for increased in vivo efficacy of interferon β-1b by site-specific PEGylation. Bioconjug. Chem. 2012, 23, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, J.; Schultz, P.G. Expanding the genetic code. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Axup, J.Y.; Schultz, P.G. Protein conjugation with genetically encoded unnatural amino acids. Curr. Opin. Chem. Biol. 2013, 17, 412–419. [Google Scholar] [PubMed]
- Chin, J.W.; Santoro, S.W.; Martin, A.B.; King, D.S.; Wang, L.; Schultz, P.G. Addition of p-azido-l-phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc. 2002, 124, 9026–9027. [Google Scholar] [CrossRef] [PubMed]
- Deiters, A.; Cropp, T.A.; Summerer, D.; Mukherji, M.; Schultz, P.G. Site-specific PEGylation of proteins containing unnatural amino acids. Bioorg. Med. Chem. Lett. 2004, 14, 5743–5745. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.; Spicer, C.D.; Gao, Z.; Takehana, T.; Lin, Y.A.; Yasukohchi, T.; Davis, B.G. Self-liganded Suzuki–Miyaura coupling for site-selective protein PEGylation. Angew. Chem. Int. Ed. 2013, 52, 3916–3921. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, M.; Huang, Y.; Song, X.; Liu, L.; Chen, P.R. Genetically encoded alkenyl-pyrrolysine analogues for thiol-ene reaction mediated site-specific protein labeling. Chem. Sci. 2012, 3, 2766–2770. [Google Scholar] [CrossRef]
- Cho, H.; Daniel, T.; Buechler, Y.J.; Litzinger, D.C.; Maio, Z.; Putnam, A.-M.H.; Kraynov, V.S.; Sim, B.-C.; Bussell, S.; Javahishvili, T. Optimized clinical performance of growth hormone with an expanded genetic code. Proc. Natl. Acad. Sci. USA 2011, 108, 9060–9065. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Pinkstaff, J.; Li, Z.; Skidmore, L.; Li, N.; Myler, H.; Dallas-Yang, Q.; Putnam, A.-M.; Yao, J.; Bussell, S.; et al. FGF21 analogs of sustained action enabled by orthogonal biosynthesis demonstrate enhanced antidiabetic pharmacology in rodents. Diabetes 2012, 61, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; Dozier, J.K.; Distefano, M.D. Enzymatic labeling of proteins: Techniques and approaches. Bioconjug. Chem. 2013, 24, 1277–1294. [Google Scholar] [CrossRef] [PubMed]
- Tsukiji, S.; Nagamune, T. Sortase-mediated ligation: A gift from gram-positive bacteria to protein engineering. ChemBioChem 2009, 10, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Antos, J.M.; Grotenbreg, G.M.; Spooner, E.; Ploegh, H.L. Sortagging: A versatile method for protein labeling. Nat. Chem. Biol. 2007, 3, 707–708. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, R.; Subramanian, S.; Boder, E.T. Sortase A as a novel molecular “stapler” for sequence-specific protein conjugation. Bioconjug. Chem. 2007, 18, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Dougan, S.K.; Chuang, T.-Y.; Spooner, E.; Ploegh, H.L. Sortase-catalyzed transformations that improve the properties of cytokines. Proc. Natl. Acad. Sci. USA 2011, 108, 3169–3174. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.K.M.; Hagemeyer, C.E.; Johnston, A.P.R.; Gonzales, C.; Kamphuis, M.M.J.; Ardipradja, K.; Such, G.K.; Peter, K.; Caruso, F. Bio-click chemistry: Enzymatic functionalization of PEGylated capsules for targeting applications. Angew. Chem. Int. Ed. 2012, 124, 7244–7248. [Google Scholar] [CrossRef]
- Tomita, U.; Yamaguchi, S.; Maeda, Y.; Chujo, K.; Minamihata, K.; Nagamune, T. Protein cell-surface display through in situ enzymatic modification of proteins with a poly(ethylene glycol)-lipid. Biotechnol. Bioeng. 2013, 110, 2785–2789. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-M.; Li, Y.-T.; Pan, M.; Kong, X.-Q.; Huang, Y.-C.; Hong, Z.-Y.; Liu, L. Irreversible site-specific hydrazinolysis of proteins by use of sortase. Angew. Chem. 2014, 126, 2230–2234. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Distefano, M.D. Solid-phase synthesis of C-terminal peptide libraries for studying the specificity of enzymatic protein prenylation. Chem. Commun. 2012, 48, 8228–8230. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, B.P.; Xu, J.; Taton, T.A.; Guo, A.; Distefano, M.D. Site-specific, covalent attachment of proteins to a solid surface. Bioconjug. Chem. 2006, 17, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, B.P.; Zhang, Z.; Hosokawa, A.; Distefano, M.D. Selective labeling of proteins by using protein farnesyltransferase. ChemBioChem 2007, 8, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; Dozier, J.K.; Lenevich, S.; Distefano, M.D. Selective labeling of polypeptides using protein farnesyltransferase via rapid oxime ligation. Chem. Commun. 2010, 46, 8998–9000. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; Mahmoodi, M.M.; Shah, R.; Dozier, J.K.; Wagner, C.R.; Distefano, M.D. A highly efficient catalyst for oxime ligation and hydrazone-oxime exchange suitable for bioconjugation. Bioconjug. Chem. 2013, 24, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Dursina, B.; Reents, R.; Delon, C.; Wu, Y.; Kulharia, M.; Thutewohl, M.; Veligodsky, A.; Kalinin, A.; Evstifeev, V.; Ciobanu, D.; et al. Identification and specificity profiling of protein prenyltransferase inhibitors using new fluorescent phosphoisoprenoids. J. Am. Chem. Soc. 2006, 128, 2822–2835. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, M.A.; Wiemer, A.J.; Kuder, C.H.; Hohl, R.J.; Wiemer, D.F. Synthesis of fluorescently tagged isoprenoid bisphosphonates that inhibit protein geranylgeranylation. Bioorgan. Med. Chem. 2007, 15, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Turek, T.C.; Gaon, I.; Distefano, M.D.; Strickland, C.L. Synthesis of farnesyl diphosphate analogues containing ether-linked photoactive benzophenones and their application in studies of protein prenyltransferases. J. Org. Chem. 2001, 66, 3253–3264. [Google Scholar] [CrossRef] [PubMed]
- Völkert, M.; Uwai, K.; Tebbe, A.; Popkirova, B.; Wagner, M.; Kuhlmann, J.; Waldmann, H. Synthesis and biological activity of photoactivatable N-Ras peptides and proteins. J. Am. Chem. Soc. 2003, 125, 12749–12758. [Google Scholar] [CrossRef] [PubMed]
- Hovlid, M.L.; Edelstein, R.L.; Henry, O.; Ochocki, J.; DeGraw, A.; Lenevich, S.; Talbot, T.; Young, V.G.; Hruza, A.W.; Lopez-Gallego, F.; et al. Synthesis, properties, and applications of diazotrifluropropanoyl-containing photoactive analogs of farnesyl diphosphate containing modified linkages for enhanced stability. Chem. Biol. Drug Des. 2010, 75, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; Song, J.M.; Pricer, R.E.; Distefano, M.D. Chemoenzymatic reversible immobilization and labeling of proteins without prior purification. J. Am. Chem. Soc. 2012, 134, 8455–8467. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hapka, D.; Chen, H.; Vallera, D.A.; Wagner, C.R. Self-assembly of antibodies by chemical induction. Angew. Chem. Int. Ed. 2008, 47, 10179–10182. [Google Scholar] [CrossRef] [PubMed]
- Volpato, J.P.; Mayotte, N.; Fossati, E.; Guerrero, V.; Sauvageau, G.; Pelletier, J.N. Selectively weakened binding of methotrexate by human dihydrofolate reductase allows rapid ex vivo selection of mammalian cells. J. Mol. Recognit. 2011, 24, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Dozier, J.K.; Khatwani, S.L.; Wollack, J.W.; Wang, Y.-C.; Schmidt-Dannert, C.; Distefano, M.D. Engineering protein farnesyltransferase for enzymatic protein labeling applications. Bioconjug. Chem. 2014, 25, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- DeFrees, S.; Wang, Z.-G.; Xing, R.; Scott, A.E.; Wang, J.; Zopf, D.; Gouty, D.L.; Sjoberg, E.R.; Panneerselvam, K.; Brinkman-van der Linden, E.C.M.; et al. GlycoPEGylation of recombinant therapeutic proteins produced in Escherichia coli. Glycobiology 2006, 16, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Stennicke, H.R.; Østergaard, H.; Bayer, R.J.; Kalo, M.S.; Kinealy, K.; Holm, P.K.; Sørensen, B.B.; Zopf, D.; Bjørn, S.E. Generation and biochemical characterization of glycoPEGylated factor VIIa derivatives. Thromb. Haemost. 2008, 100, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Plesner, B.; Westh, P.; Nielsen, A.D. Biophysical characterisation of glycoPEGylated recombinant human factor VIIa. Int. J. Pharm. 2011, 406, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Østergaard, H.; Bjelke, J.R.; Hansen, L.; Petersen, L.C.; Pedersen, A.A.; Elm, T.; Møller, F.; Hermit, M.B.; Holm, P.K.; Krogh, T.N.; et al. Prolonged half-life and preserved enzymatic properties of factor ix selectively PEGylated on native N-glycans in the activation peptide. Blood 2011, 118, 2333–2341. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.W.; Moss, J.; Knobe, K.; Groth, A.; Colberg, T.; Watson, E. Population pharmacokinetic modeling for dose setting of nonacog beta pegol (N9-GP), a glycoPEGylated recombinant factor IX. J. Thromb. Haemost. 2012, 10, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Stennicke, H.R.; Kjalke, M.; Karpf, D.M.; Balling, K.W.; Johansen, P.B.; Elm, T.; Øvlisen, K.; Möller, F.; Holmberg, H.L.; Gudme, C.N.; et al. A novel B-domain O-glycoPEGylated FVIII (N8-GP) demonstrates full efficacy and prolonged effect in hemophilic mice models. Blood 2013, 121, 2108–2116. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Honey, D.M.; Hou, L.; Bird, J.J.; Zarazinski, C.; Searles, M.; Braithwaite, C.; Kingsbury, J.S.; Kyazike, J.; Culm-Merdek, K.; et al. Carbohydrate-mediated polyethylene glycol conjugation of TSH improves its pharmacological properties. Endocrinology 2013, 154, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Mero, A.; Schiavon, M.; Veronese, F.M.; Pasut, G. A new method to increase selectivity of transglutaminase mediated PEGylation of salmon calcitonin and human growth hormone. J. Control. Release 2011, 154, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Ikeda, M.; Suzuki, K.; Hirayama, K. Site-specific modification of interleukin-2 by the combined use of genetic engineering techniques and transglutaminase. Biochemistry 1996, 35, 13072–13080. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Hayashi, E.; Yamada, N.; Yatagai, M.; Takahara, Y. Further studies on the site-specific protein modification by microbial transglutaminase. Bioconjug. Chem. 2001, 12, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Mero, A.; Spolaore, B.; Veronese, F.M.; Fontana, A. Transglutaminase-mediated PEGylation of proteins: Direct identification of the sites of protein modification by mass spectrometry using a novel monodisperse PEG. Bioconjug. Chem. 2009, 20, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; Spolaore, B.; Mero, A.; Veronese, F.M. Site-specific modification and PEGylation of pharmaceutical proteins mediated by transglutaminase. Adv. Drug Deliv. Rev. 2008, 60, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Mariniello, L.; Porta, R.; Sorrentino, A.; Giosafatto, C.V.L.; Rossi Marquez, G.; Esposito, M.; di Pierro, P. Transglutaminase-mediated macromolecular assembly: Production of conjugates for food and pharmaceutical applications. Amino Acids 2014, 46, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Rachel, N.; Pelletier, J. Biotechnological applications of transglutaminases. Biomolecules 2013, 3, 870–888. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Freitas, D.; Mero, A.; Pasut, G. Chemical and enzymatic site specific PEGylation of hgh. Bioconjug. Chem. 2013, 24, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Maullu, C.; Raimondo, D.; Caboi, F.; Giorgetti, A.; Sergi, M.; Valentini, M.; Tonon, G.; Tramontano, A. Site-directed enzymatic PEGylation of the human granulocyte colony-stimulating factor. FEBS J. 2009, 276, 6741–6750. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Shaw, A.C.; Wang, J.; Chang, C.; Deng, J.; Su, J. A novel high-throughput screening method for microbial transglutaminases with high specificity toward Gln141 of human growth hormone. J. Biomol. Screen. 2010, 15, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Dierks, T.; Schmidt, B.; Borissenko, L.V.; Peng, J.; Preusser, A.; Mariappan, M.; von Figura, K. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human cα-formylglycine generating enzyme. Cell 2003, 113, 435–444. [Google Scholar] [CrossRef]
- Carrico, I.S.; Carlson, B.L.; Bertozzi, C.R. Introducing genetically encoded aldehydes into proteins. Nat. Chem. Biol. 2007, 3, 321–322. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dozier, J.K.; Distefano, M.D. Site-Specific PEGylation of Therapeutic Proteins. Int. J. Mol. Sci. 2015, 16, 25831-25864. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161025831
Dozier JK, Distefano MD. Site-Specific PEGylation of Therapeutic Proteins. International Journal of Molecular Sciences. 2015; 16(10):25831-25864. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161025831
Chicago/Turabian StyleDozier, Jonathan K., and Mark D. Distefano. 2015. "Site-Specific PEGylation of Therapeutic Proteins" International Journal of Molecular Sciences 16, no. 10: 25831-25864. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161025831