
Computational Prediction of RNA-Binding Proteins and Binding Sites

Abstract
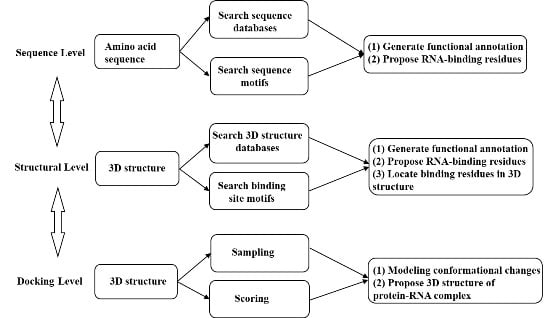
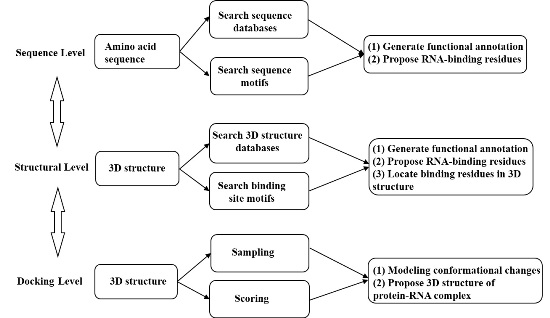
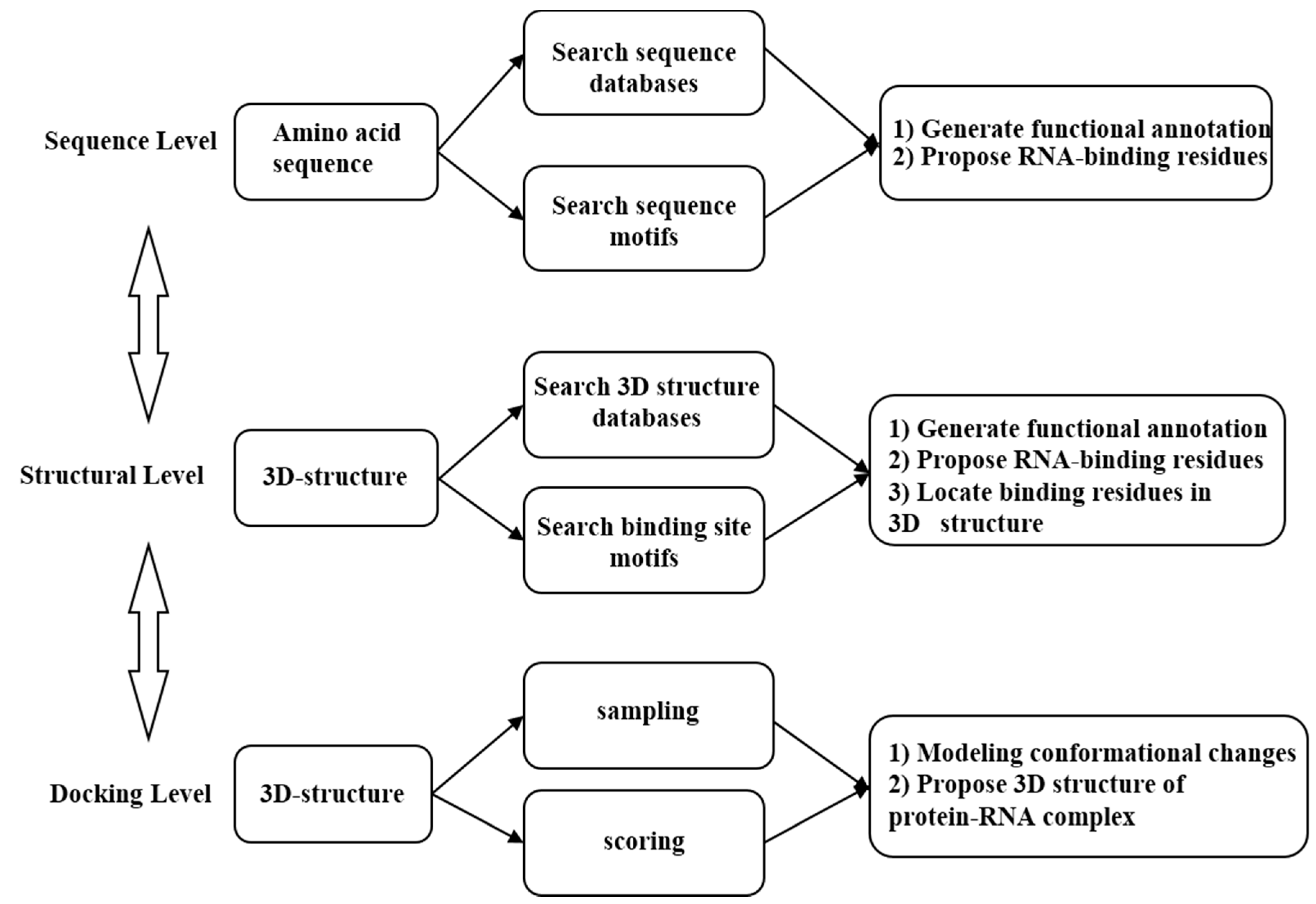
:
1. Introduction

2. Development of Computational Methods for Prediction of RNA-Binding Site
2.1. Data Set

{kind=link}
{kind=link}
| ID | Reference | Publication Year | Notes |
|---|---|---|---|
| PRIPU dataset | [27] | 2015 | The dataset contains positive and unlabeled examples, which is an innovation because previous ones usually have negative samples. Such negative samples are not real negative samples, some even may be unknown positive samples |
| a RB344 | [26] | 2015 | 344 RNA binding proteins, almost entirely non-redundant at 30% sequence identity |
| RB172 | [28] | 2014 | 172 protein entries with sequence identity of less than 25% |
| RB75 | [8] | 2012 | 75 RNP complexes released between 1 January and 28 April 2011 from PDB database b, non-redundant at 40% sequence identity |
| RB199 | [25,29] | 2011 | Extracted dataset (May 2010) from PDB database. Proteins with >30% sequence identity or structures with resolution worse than 3.5 Å were removed |
| RB164 | [30] | 2010 | The data were downloaded from RsiteDB. After removing protein and RNA chains with sequence identity above 25% and 60%, respectively, 205 non-redundant protein–RNA chains in 164 complexes were obtained |
| RB86 | [31] | 2008 | 86 RNA-binding protein chains were collected for training and fivefold cross validation |
| RB147 | [32] | 2007 | Adding novel RNA-binding complexes since 2006, based on RB109 |
| RB109 | [33] | 2006 | 109 RNA–protein complexes extracted from structures of known RNA–protein complexes solved by X-ray crystallography in the PDB. Proteins with >30% sequence identity or structures with resolution worse than 3.5 Å were removed |
2.2. Feature Selection for RNA-Binding Residues and Protein Predictors
2.2.1. Sequence-Based Features
Amino Acid Composition
Sequence Similarity
Evolutionary Information
2.2.2. Structure-Based Features
The Secondary Structure (SS)
Accessible Surface Area (ASA)
2.2.3. Chemical and Physical Features
Hydrophobicity
Electrostatic Patches
Cleft Size
2.3. Prediction Methods
2.3.1. Sequences-Based Methods
Sequence-Based Methods for RNA-Binding Site Prediction
Sequence-Based Methods for RNA-Binding Proteins (RBPs) Prediction
2.3.2. Structure-Based Methods
Structure-Based Methods for RNA-Binding Site Prediction
Structure-Based Methods for RBP Prediction
2.3.3. Protein–RNA Complex Docking
2.4. Prediction Algorithms
2.5. Evaluation and Performance of Various Predictors
2.5.1. Performance Measures
2.5.2. Comparison of Various Prediction Methods
2.5.3. Collection of Web Servers of RBPs and RNA-Binding Site Predictors
3. Conclusions and Future Perspectives
| Methods | URLs | References | Available | Seq/Struc/Docking | Sites/Protein |
|---|---|---|---|---|---|
| PRIPU | http://admis.fudan.edu.cn/projects/pripu.htm | Cheng et al. (2015) [27] | ○ | seq | site |
| RNABindRPlus | http://einstein.cs.iastate.edu/RNABindRPlus/ | Walia et al. (2014) [58] | ○ | site | |
| CatRAPID omics | http://s.tartaglialab.com/catrapid/omics | Agostini et al. (2013) [81] | ○ | site | |
| SRCPred | http://tardis.nibio.go.jp/netasa/srcpred | Fernandez et al. (2011) [29] | ○ | site | |
| SPOT | http://sparks.informatics.iupui.edu | Zhao et al. (2011) [15] | X | protein | |
| PRBR | http://www.cbi.seu.edu.cn/PRBR/ | Ma et al. (2011) [12] | ○ | site | |
| RNAPred | http://www.imtech.res.in/raghava/rnapred/ | Kumar et al. (2011) [60] | ○ | protein | |
| RPISeq | http://pridb.gdcb.iastate.edu/RPISeq/ | Muppirala et al. (2011) [82] | ○ | site | |
| BindN+ | http://bioinfo.ggc.org/bindn+/ | Wang et al. (2010) [11] | ○ | site | |
| NAPS | http://prediction.bioengr.uic.edu/ | Carson et al. (2010) [81] | X | site | |
| PiRaNhA | http://bioinformatics.sussex.ac.uk/PIRANHA/ | Murakami et al. (2010) [10] | ○ | site | |
| PRNA | http://www.sysbio.ac.cn/datatools.asp | Liu et al. (2010) [56] | X | site | |
| RNA | http://mcgill.3322.org/RNA/ | Li et al. (2010) [55] | X | site | |
| RISP | http://grc.seu.edu.cn/RISP | Tong et al. (2008) [54] | X | site | |
| PRINTR | http://210.42.106.80/printr/ | Wang et al. (2008) [53] | X | site | |
| PPRInt | http://www.imtech.res.in/raghava/pprint/ | Kumar et al. (2008) [52] | ○ | site | |
| RNABindR | http://bindr2.gdcb.iastate.edu/RNABindR/ | Terribilini et al. (2007) [32] | ○ | site | |
| BindN | http://bioinfo.ggc.org/bindn/ | Wang and Brown (2006) [9] | ○ | site | |
| SVMProt | http://jing.cz3.nus.edu.sg/cgi-bin/svmprot.cgi | Han et al. (2004) [36] | X | protein | |
| RBPDetector | http://ibi.hzau.edu.cn/rbrdetector | Yang et al. (2014) [64] | ○ | struc | site |
| SPOT-Seq-RNA | http://sparks-lab.org/server/SPOT-Seq-RNA/ | Yang et al. (2014) [65] | X | protein | |
| DRNA | http://sparks.informatics.iupui.edu/yueyang/DFIRE/dRdR-DB-service | Zhao et al. (2011) [15] | X | protein | |
| OPRA | Program available upon request from the authors | Perez-Cano and Fernandez-Recio (2010) [14] | ○ | site | |
| PRIP | http://www.qfab.org/PRIP | Maetschke et al. (2009) [62] | X | site | |
| KYG | http://cib.cf.ocha.ac.jp/KYG/ | Kim et al. (2006) [13] | X | protein | |
| DARS-RNP and QUASI-RNP | http://www.genesilico.pl/RNP/ | Tuszynska and Bujnicki (2011) [66] | ○ | docking | complex |
| PatchDock | http://bioinfo3d.cs.tau.ac.il/PatchDock/index.html | Schneidman-Duhovny et al. (2005) [21] | ○ | complex | |
| Haddock | http://www.nmr.chem.uu.nl/haddock/; http://haddock.science.uu.nl/services/HADDOCK | Dominguez et al. (2003) [18] | ○ | complex | |
| Hex | http://hex.loria.fr/; http://hexserver.loria.fr/ | Ritchie and Kemp (2000) [20] | ○ | complex | |
| FTDock (3D-Dock) | http://www.sbg.bio.ic.ac.uk/docking/ | Gabb et al. (1997) [22] | ○ | complex | |
| GRAMM | http://vakser.bioinformatics.ku.edu/main/resources_gramm1.03.php | Katchalski-Katzir et al. (1992) [19] | ○ | complex |
| Parameter | Meaning | Expression |
|---|---|---|
| Accuracy (ACC) | Percentage of correct prediction | a |
| Sensitivity | Percentage of correctly predicted positive | |
| Specificity | Percentage of correctly predicted negative | |
| Strength | Mean value of the sum of sensitivity and specificity | |
| MCC | Matthews correlation coefficient | |
| Precision | Positive predictive rate | |
| F-measure | The harmonic mean of sensitivity and specificity | |
| AUC b | Probability that a classifier will rank a randomly chosen positive instance higher than a randomly chosen negative one |
| Methods | Data Set | Performance | Reference | Feature | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | SEN | SPE | AUC | MCC | Strength | F-Measure | Precision | ||||
| PiRaNhA | RB75 | - | - | - | 0.822 | 0.435 | - | - | - | [8] | Sequence-based |
| PPRInt | RB75 | - | - | - | 0.779 | 0.339 | - | - | - | [8] | |
| RB172 | 0.71 | - | 0.25 | 0.66 | - | - | [28] | ||||
| RB344 | 0.70 | 0.45 | 0.82 | 0.68 | 0.28 | - | 0.49 | 0.53 | [26] | ||
| BindN | RB75 | - | - | - | 0.733 | 0.297 | - | - | - | [8] | |
| RB172 | 0.75 | - | - | - | 0.23 | 0.64 | - | - | [28] | ||
| BindN+ | RB75 | - | - | - | 0.821 | 0.397 | - | - | - | [8] | |
| RB172 | 0.79 | - | - | - | 0.34 | 0.71 | - | - | [28] | ||
| RB344 | 0.72 | 0.32 | 0.89 | 0.68 | 0.26 | - | 0.41 | 0.56 | [26] | ||
| RNABindR | RB75 | - | - | - | 0.708 | 0.317 | - | - | - | [8] | |
| RNABindR v2.0 | RB172 | 0.66 | - | - | - | 0.27 | 0.69 | - | - | [28] | |
| PRBR | RB75 | - | - | - | N/A a | 0.294 | - | - | - | [8] | |
| NAPS | RB75 | - | - | - | 0.679 | 0.215 | - | - | - | [8] | |
| RB172 | 0.66 | - | - | - | 0.17 | 0.61 | - | - | [28] | ||
| RNAProB | RB172 | 0.82 | - | - | - | 0.22 | 0.60 | - | - | [28] | |
| KYG * | RB75 | - | - | - | N/A | 0.382 | - | - | - | [8] | Structure-based |
| DRNA * | RB75 | - | - | - | N/A | 0.382 | - | - | - | [8] | |
| RB344 | 0.75 | 0.21 | 0.94 | N/A | 0.22 | - | 0.31 | 0.54 | [26] | ||
| OPRA * | RB75 | - | - | - | N/A | 0.296 | - | - | - | [8] | |
| Ren’s method | RB344 | 0.68 | 0.48 | 0.76 | 0.68 | 0.26 | - | 0.48 | 0.48 | [26,83] | |
| Meta-predictor b | RB75 | - | - | - | 0.835 | 0.460 | - | - | - | [8,34] | |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jacobs Anderson, J.S.; Parker, R. Computational identification of cis-acting elements affecting post-transcriptional control of gene expression in Saccharomyces cerevisiae. Nucleic Acids Res. 2000, 28, 1604–1617. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Kuwano, Y.; Kim, H.H.; Gorospe, M. Posttranscriptional gene regulation by RNA-binding proteins during oxidative stress: Implications for cellular senescence. Biol. Chem. 2008, 389, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Saunus, J.M.; French, J.D.; Edwards, S.L.; Beveridge, D.J.; Hatchell, E.C.; Wagner, S.A.; Stein, S.R.; Davidson, A.; Simpson, K.J.; Francis, G.D.; et al. Posttranscriptional regulation of the breast cancer susceptibility gene BRCA1 by the RNA binding protein HuR. Cancer Res. 2008, 68, 9469–9478. [Google Scholar] [CrossRef] [PubMed]
- Noller, H.F. RNA structure: Reading the ribosome. Science 2005, 309, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Orengo, C.A.; Michie, A.D.; Jones, S.; Jones, D.T.; Swindells, M.B.; Thornton, J.M. CATH—A hierarchic classification of protein domain structures. Structure 1997, 5, 1093–1108. [Google Scholar] [CrossRef]
- Ponting, C.P.; Schultz, J.; Milpetz, F.; Bork, P. SMART: Identification and annotation of domains from signalling and extracellular protein sequences. Nucleic Acids Res. 1999, 27, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Puton, T.; Kozlowski, L.; Tuszynska, I.; Rother, K.; Bujnicki, J.M. Computational methods for prediction of protein–RNA interactions. J. Struct. Biol. 2012, 179, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brown, S.J. BindN: A web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 2006, 34, W243–W248. [Google Scholar] [CrossRef]
- Murakami, Y.; Spriggs, R.V.; Nakamura, H.; Jones, S. PiRaNhA: A server for the computational prediction of RNA-binding residues in protein sequences. Nucleic Acids Res. 2010, 38, W412–W416. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, C.; Yang, M.Q.; Yang, J.Y. BindN+ for accurate prediction of DNA and RNA-binding residues from protein sequence features. BMC Syst. Biol. 2010, 4 (Suppl. S1). [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Guo, J.; Wu, J.; Liu, H.; Yu, J.; Xie, J.; Sun, X. Prediction of RNA-binding residues in proteins from primary sequence using an enriched random forest model with a novel hybrid feature. Proteins 2011, 79, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.T.; Yura, K.; Go, N. Amino acid residue doublet propensity in the protein-RNA interface and its application to RNA interface prediction. Nucleic Acids Res. 2006, 34, 6450–6460. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cano, L.; Solernou, A.; Pons, C.; Fernández-Recio, J. Structural prediction of protein-RNA interaction by computational docking with propensity-based statistical potentials. Pac. Symp. Biocomput. 2010, 2010, 293–301. [Google Scholar]
- Zhao, H.; Yang, Y.; Zhou, Y. Structure-based prediction of RNA-binding domains and RNA-binding sites and application to structural genomics targets. Nucleic Acids Res. 2011, 39, 3017–3025. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Protein–protein docking dealing with the unknown. J. Comput. Chem. 2010, 31, 317–342. [Google Scholar] [CrossRef] [PubMed]
- Tuszynska, I.; Matelska, D.; Magnus, M.; Chojnowski, G.; Kasprzak, J.M.; Kozlowski, L.P.; Dunin-Horkawicz, S.; Bujnicki, J.M. Computational modeling of protein–RNA complex structures. Methods 2014, 65, 310–319. [Google Scholar] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein–protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Katchalski-Katzir, E.; Shariv, I.; Eisenstein, M.; Friesem, A.A.; Aflalo, C.; Vakser, I.A. Molecular surface recognition: Determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. USA 1992, 89, 2195–2199. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.W.; Kemp, G.J. Protein docking using spherical polar Fourier correlations. Proteins 2000, 39, 178–194. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Gabb, H.A.; Jackson, R.M.; Sternberg, M.J. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J. Mol. Biol. 1997, 272, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Zhao, R.; Wu, R. An overview of the prediction of protein DNA-binding sites. Int. J. Mol. Sci. 2015, 16, 5194–5215. [Google Scholar] [CrossRef] [PubMed]
- Wichadakul, D.; McDermott, J.; Samudrala, R. Prediction and integration of regulatory and protein-protein interactions. Methods Mol. Biol. 2009, 541, 101–143. [Google Scholar] [PubMed]
- Lewis, B.A.; Walia, R.R.; Terribilini, M.; Ferguson, J.; Zheng, C.; Honavar, V.; Dobbs, D.S. PRIDB: A Protein–RNA interface database. Nucleic Acids Res. 2011, 39, D277–D282. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Shen, Y. RNA-binding residues prediction using structural features. BMC Bioinform. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zhou, S.; Guan, J. Computationally predicting protein-RNA interactions using only positive and unlabeled examples. J. Bioinf. Comput. Biol. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, R.; Gromiha, M.M. Prediction of RNA binding residues: An extensive analysis based on structure and function to select the best predictor. PLoS ONE 2014, 9, e91140. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Kumagai, Y.; Standley, D.M.; Sarai, A.; Mizuguchi, K.; Ahmad, S. Prediction of dinucleotide-specific RNA-binding sites in proteins. BMC Bioinform. 2011, 12 (Suppl. S13). [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-W.; Su, E.C.-Y.; Hwang, J.-K.; Sung, T.-Y.; Hsu, W.-L. Predicting RNA-binding sites of proteins using support vector machines and evolutionary information. BMC Bioinform. 2008, 9 (Suppl. S12). [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Sarai, A. Analysis of electric moments of RNA-binding proteins: Implications for mechanism and prediction. BMC Struct. Biol. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Terribilini, M.; Sander, J.D.; Lee, J.H.; Zaback, P.; Jernigan, R.L.; Honavar, V.; Dobbs, D. RNABindR: A server for analyzing and predicting RNA-binding sites in proteins. Nucleic Acids Res. 2007, 35, W578–W584. [Google Scholar] [CrossRef] [PubMed]
- Petrey, D.; Honig, B. GRASP2: Visualization, surface properties, and electrostatics of macromolecular structures and sequences. Methods Enzymol. 2003, 374, 492–509. [Google Scholar] [PubMed]
- Si, J.; Zhang, Z.; Lin, B.; Schroeder, M.; Huang, B. MetaDBSite: A meta approach to improve protein DNA-binding sites prediction. BMC Syst. Biol. 2011, 5 (Suppl. S1). [Google Scholar] [CrossRef] [PubMed]
- Bartlett, G.J.; Porter, C.T.; Borkakoti, N.; Thornton, J.M. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 2002, 324, 105–121. [Google Scholar] [CrossRef]
- Han, L.Y.; Cai, C.Z.; Lo, S.L.; Chung, M.C.; Chen, Y.Z. Prediction of RNA-binding proteins from primary sequence by a support vector machine approach. RNA 2004, 10, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.; Andersen, C.A.; Rost, B. DSSPcont: Continuous secondary structure assignments for proteins. Nucleic Acids Res. 2003, 31, 3293–3295. [Google Scholar] [CrossRef] [PubMed]
- Si, J.N.; Yan, R.X.; Wang, C.; Zhang, Z.; Su, X.D. TIM-Finder: A new method for identifying TIM-barrel proteins. BMC Struct. Biol. 2009, 9. [Google Scholar] [CrossRef] [PubMed]
- Karypis, G. YASSPP: Better kernels and coding schemes lead to improvements in protein secondary structure prediction. Proteins 2006, 64, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.R.; Liu, L.; Niu, B.; Lv, Y.L.; Li, M.J.; Yuan, Y.L.; Zhu, Y.B.; Lu, W.C.; Cai, Y.D. Prediction of RNA-binding proteins by voting systems. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Cao, J.; Cai, Y.; Shi, T.; Li, Y. Predicting rRNA-, RNA-, and DNA-binding proteins from primary structure with support vector machines. J. Theor. Biol. 2006, 240, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.J.; Thornton, J.M. NACCESS Computer Program; Department of Biochemistry and Molecular Biology, University College of London: London, UK, 1993. [Google Scholar]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Stawiski, E.W.; Gregoret, L.M.; Mandel-Gutfreund, Y. Annotating nucleic acid-binding function based on protein structure. J. Mol. Biol. 2003, 326, 1065–1079. [Google Scholar] [CrossRef]
- Nayal, M.; Hitz, B.C.; Honig, B. GRASS: A server for the graphical representation and analysis of structures. Protein Sci. 1999, 8, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Shazman, S.; Celniker, G.; Haber, O.; Glaser, F.; Mandel-Gutfreund, Y. Patch Finder Plus (PFplus): A web server for extracting and displaying positive electrostatic patches on protein surfaces. Nucleic Acids Res. 2007, 35, W526–W530. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Luscombe, N.M.; Swindells, M.B.; Thornton, J.M. Protein clefts in molecular recognition and function. Protein Sci. 1996, 5, 2438–2452. [Google Scholar] [PubMed]
- Jeong, E.; Chung, I.F.; Miyano, S. A neural network method for identification of RNA-interacting residues in protein. Genome Inform. 2004, 15, 105–116. [Google Scholar] [PubMed]
- Terribilini, M.; Lee, J.H.; Yan, C.; Jernigan, R.L.; Honavar, V.; Dobbs, D. Prediction of RNA binding sites in proteins from amino acid sequence. RNA 2006, 12, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gromiha, M.M.; Raghava, G.P. Prediction of RNA binding sites in a protein using SVM and PSSM profile. Proteins 2008, 71, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xue, Z.; Shen, G.; Xu, J. PRINTR: Prediction of RNA binding sites in proteins using SVM and profiles. Amino Acids 2008, 35, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Jiang, P.; Lu, Z.H. RISP: A web-based server for prediction of RNA-binding sites in proteins. Comput. Methods Programs Biomed. 2008, 90, 148–453. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cao, Z.; Liu, H. Improve the prediction of RNA-binding residues using structural neighbours. Protein Pept. Lett. 2010, 17, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.P.; Wu, L.Y.; Wang, Y.; Zhang, X.S.; Chen, L. Prediction of protein–RNA binding sites by a random forest method with combined features. Bioinformatics 2010, 26, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, H.; Chen, K.; Ruan, J.; Shen, S.; Kurgan, L. Analysis and prediction of RNA-binding residues using sequence, evolutionary conservation, and predicted secondary structure and solvent accessibility. Curr. Protein Pept. Sci. 2010, 11, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Walia, R.R.; Xue, L.C.; Wilkins, K.; El-Manzalawy, Y.; Dobbs, D.; Honavar, V. RNABindRPlus: A predictor that combines machine learning and sequence homology-based methods to improve the reliability of predicted RNA-binding residues in proteins. PLoS ONE 2014, 9, e97725. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Tian, Y.; Wu, L.; Wang, Y.; Jing, L.; Deng, N. Predicting DNA- and RNA-binding proteins from sequences with kernel methods. J. Theor. Biol. 2009, 258, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gromiha, M.M.; Raghava, G.P. SVM based prediction of RNA-binding proteins using binding residues and evolutionary information. J. Mol. Recognit. 2011, 24, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Lim, C. Predicting RNA-binding sites from the protein structure based on electrostatics, evolution and geometry. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef] [PubMed]
- Maetschke, S.R.; Yuan, Z. Exploiting structural and topological information to improve prediction of RNA–protein binding sites. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Towfic, F.; Caragea, C.; Gemperline, D.C.; Dobbs, D.; Honavar, V. Struct-NB: Predicting protein–RNA binding sites using structural features. Int. J. Data Min. Bioinform. 2010, 4, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.X.; Deng, Z.L.; Liu, R. RBRDetector: Improved prediction of binding residues on RNA-binding protein structures using complementary feature- and template-based strategies. Proteins 2014, 82, 2455–2471. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhao, H.; Wang, J.; Zhou, Y. SPOT-Seq-RNA: Predicting protein-RNA complex structure and RNA-binding function by fold recognition and binding affinity prediction. Methods Mol. Biol. 2014, 1137, 119–130. [Google Scholar] [PubMed]
- Tuszynska, I.; Bujnicki, J.M. DARS-RNP and QUASI-RNP: New statistical potentials for protein-RNA docking. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Han, K. Prediction of RNA-binding amino acids from protein and RNA sequences. BMC Bioinform. 2011, 12 (Suppl. S13). [Google Scholar] [CrossRef] [PubMed]
- Walia, R.R.; Caragea, C.; Lewis, B.A.; Towfic, F.; Terribilini, M.; El-Manzalawy, Y.; Dobbs, D.; Honavar, V. Protein-RNA interface residue prediction using machine learning: An assessment of the state of the art. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Zhu, L.; Fan, Y.X.; Yan, J. Predicting protein–RNA interaction amino acids using random forest based on submodularity subset selection. Comput. Biol. Chem. 2014, 53, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, Y.; Zhou, Y. Prediction of RNA binding proteins comes of age from low resolution to high resolution. Mol. Biosyst. 2013, 9, 2417–2425. [Google Scholar] [CrossRef] [PubMed]
- Denesyuk, N.A.; Thirumalai, D. Coarse-grained model for predicting RNA folding thermodynamics. J. Phys. Chem. B 2013, 117, 4901–4911. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Sharma, S.; Chalasani, P.; Demidov, V.V.; Broude, N.E.; Dokholyan, N.V. Ab initio RNA folding by discrete molecular dynamics: From structure prediction to folding mechanisms. RNA 2008, 14, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Baker, D. Automated de novo prediction of native-like RNA tertiary structures. Proc. Natl. Acad. Sci. USA 2007, 104, 14664–14669. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.S.; Lee, H.M.; Yang, F.; Che, C.M.; Wong, C.C.; Abagyan, R.; Leung, C.H.; Ma, D.L. Structure-based discovery of natural-product-like TNF-α inhibitors. Angew. Chem. Int. Ed. Engl. 2010, 49, 2860–2864. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.H.; Zhong, H.J.; Yang, H.; Cheng, Z.; Chan, D.S.; Ma, V.P.; Abagyan, R.; Wong, C.Y.; Ma, D.L. A metal-based inhibitor of tumor necrosis factor-α. Angew. Chem. Int. Ed. Engl. 2012, 51, 9010–9014. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.L.; Lin, S.; Leung, K.H.; Zhong, H.J.; Liu, L.J.; Chan, D.S.; Bourdoncle, A.; Mergny, J.L.; Wang, H.M.; Leung, C.H. An oligonucleotide-based label-free luminescent switch-on probe for RNA detection utilizing a G-quadruplex-selective iridium(III) complex. Nanoscale 2014, 6, 8489–8494. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.L.; Liu, L.J.; Leung, K.H.; Chen, Y.T.; Zhong, H.J.; Chan, D.S.; Wang, H.M.; Leung, C.H. Antagonizing STAT3 dimerization with a rhodium(III) complex. Angew. Chem. Int. Ed. Engl. 2014, 53, 9178–9182. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.J.; Lu, L.; Leung, K.H.; Wong, C.C.L.; Peng, C.; Yan, S.C.; Leung, C.H. An iridium(III)-based irreversible protein–protein interaction inhibitor of BRD4 as a potent anticancer agent. Chem. Sci. 2015, 6, 5400–5408. [Google Scholar] [CrossRef]
- Rother, K.; Rother, M.; Boniecki, M.; Puton, T.; Bujnicki, J.M. RNA and protein 3D structure modeling: Similarities and differences. J. Mol. Model. 2011, 17, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Agostini, F.; Zanzoni, A.; Klus, P.; Marchese, D.; Cirillo, D.; Tartaglia, G.G. catRAPID omics: A web server for large-scale prediction of protein-RNA interactions. Bioinformatics 2013, 29, 2928–2930. [Google Scholar] [CrossRef] [PubMed]
- Muppirala, U.K.; Honavar, V.G.; Dobbs, D. Predicting RNA-protein interactions using only sequence information. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.B.; Langlois, R.; Lu, H. NAPS: A residue-level nucleic acid-binding prediction server. Nucleic Acids Res. 2010, 38, W431–W455. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Si, J.; Cui, J.; Cheng, J.; Wu, R. Computational Prediction of RNA-Binding Proteins and Binding Sites. Int. J. Mol. Sci. 2015, 16, 26303-26317. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161125952
Si J, Cui J, Cheng J, Wu R. Computational Prediction of RNA-Binding Proteins and Binding Sites. International Journal of Molecular Sciences. 2015; 16(11):26303-26317. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161125952
Chicago/Turabian StyleSi, Jingna, Jing Cui, Jin Cheng, and Rongling Wu. 2015. "Computational Prediction of RNA-Binding Proteins and Binding Sites" International Journal of Molecular Sciences 16, no. 11: 26303-26317. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161125952