
Single-Cell Isolation and Gene Analysis: Pitfalls and Possibilities

Abstract
:1. Introduction
2. Historical Background—Nucleic Acid Amplification
3. Single-Cell Isolation and Harvesting Strategies
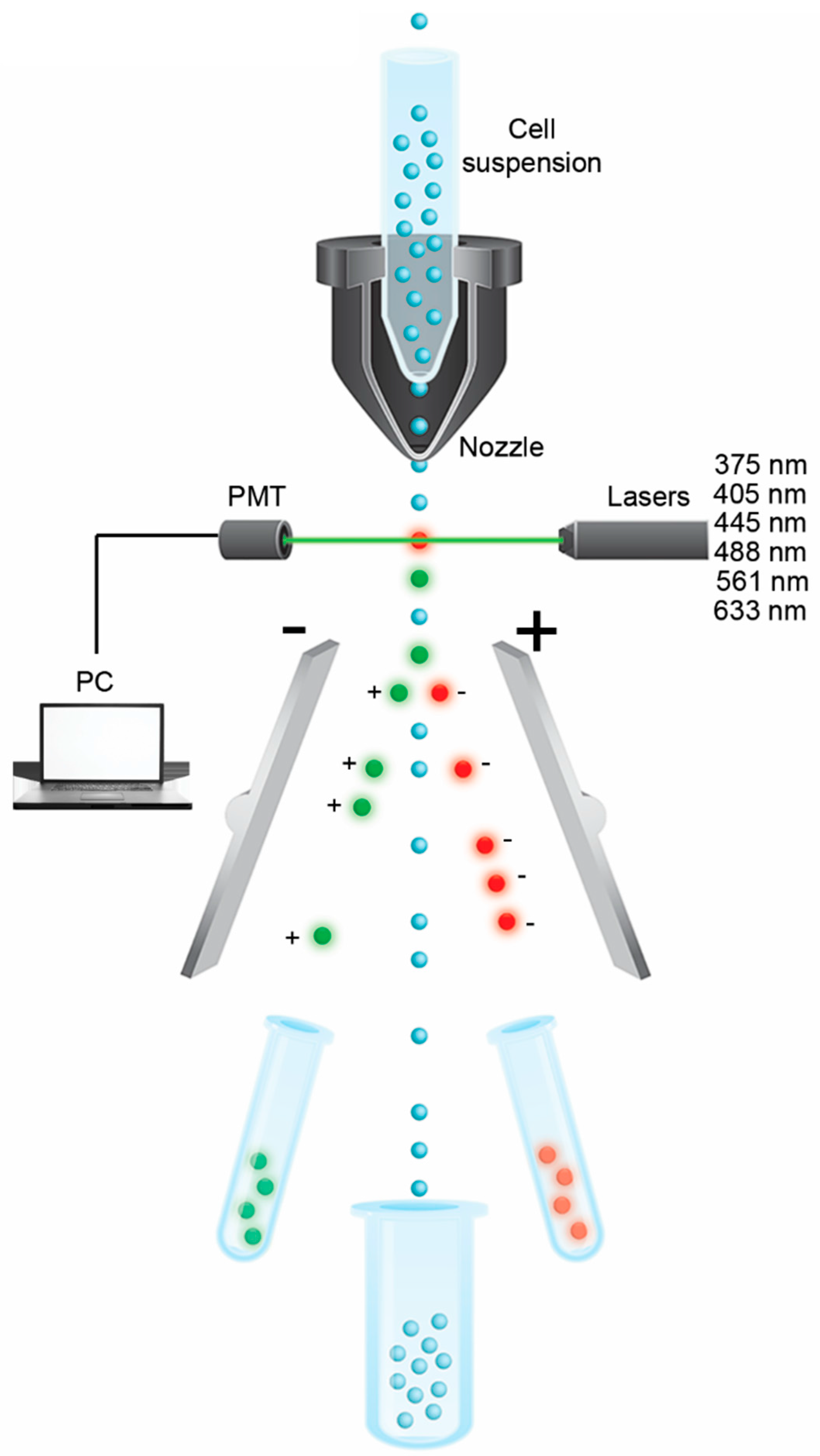
Fluidics Technology

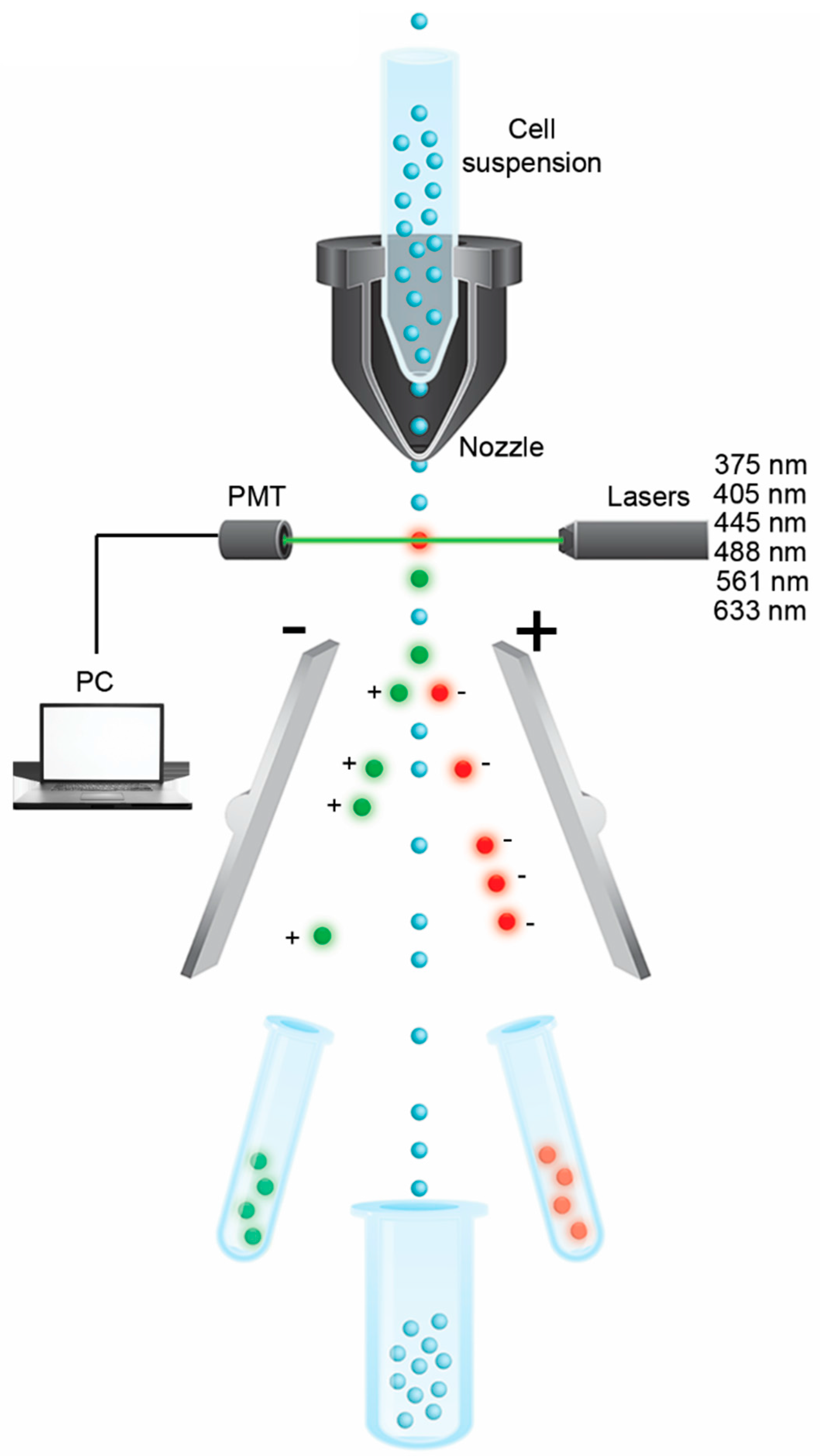{kind=link}
{kind=link}
{kind=link}
{kind=link}
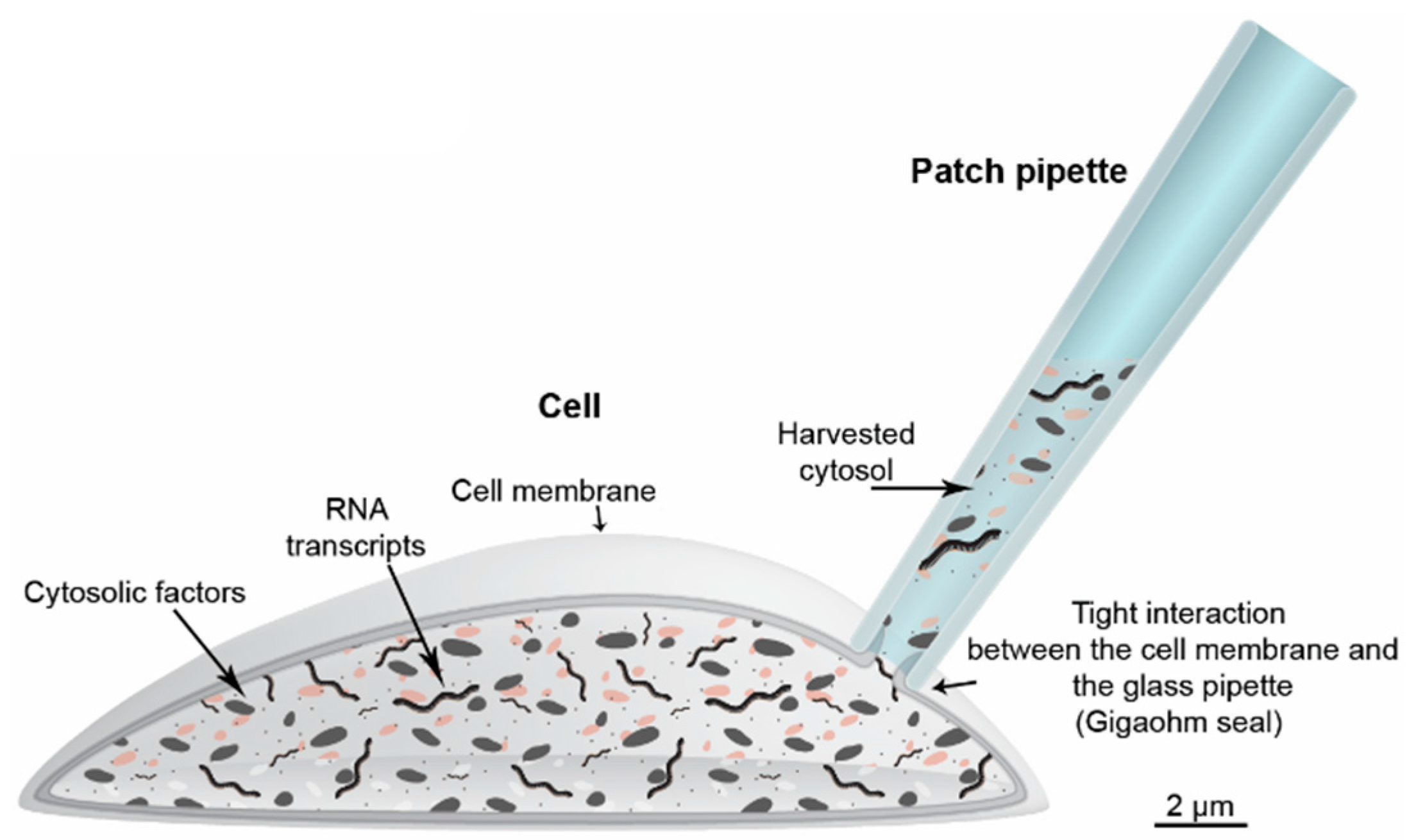{kind=link}
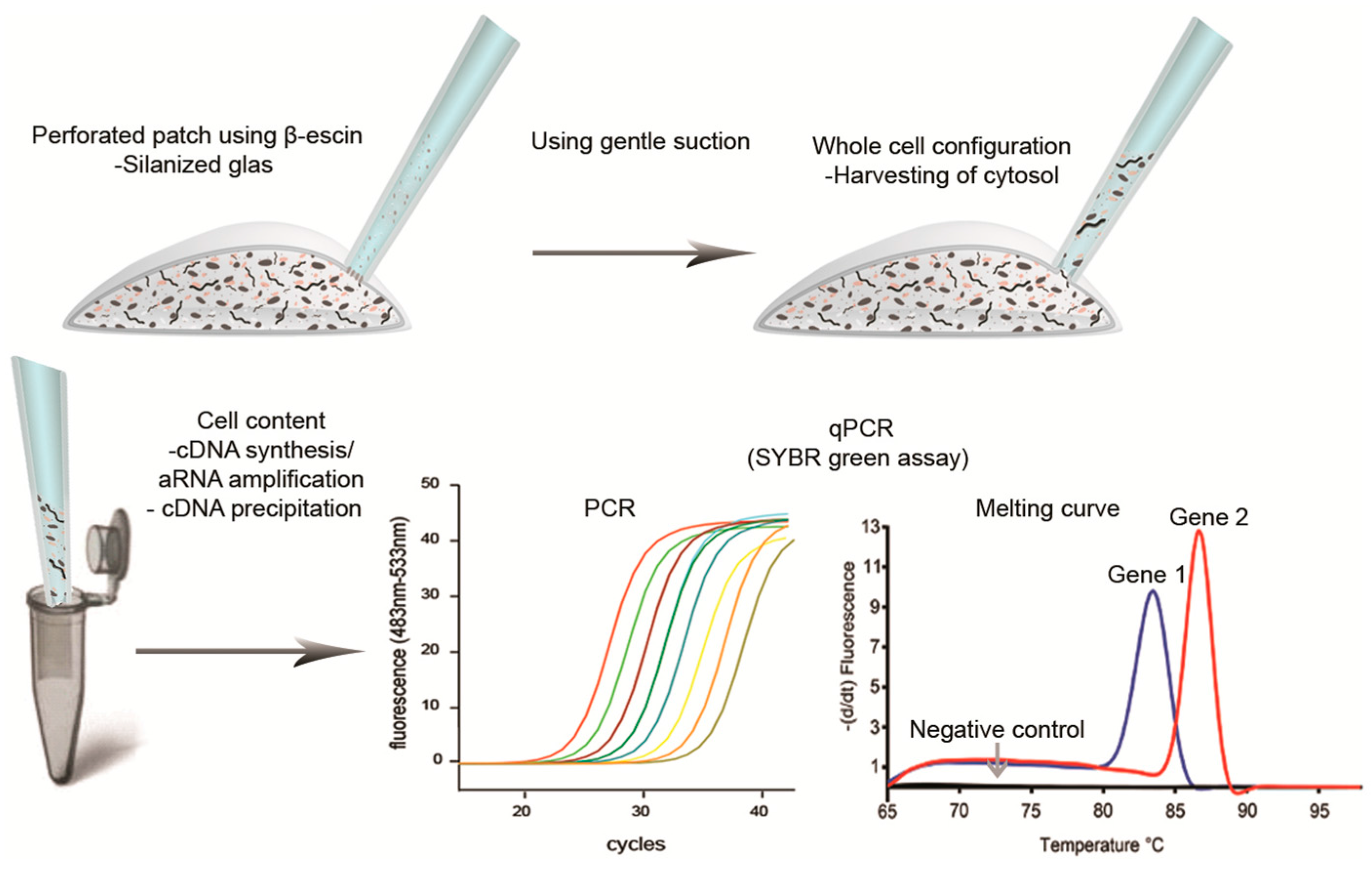{kind=link}
| Method | Equipment Costs | Laboratory Skills | Throuhgput | Tissue |
|---|---|---|---|---|
| FACA | High | Normal | High | Dissociated cells (in vitro) |
| Microfluidics | High | Normal | High | Dissociated cells (in vitro) |
| Laser assisted microdissection | High | High | Low | Intact fixed and live tissue (in vitro/ex vivo) |
| Whole cell harvesting | Low | Normal | Medium | Dissociated cells (in vitro) |
| Harvesting of cytosol using patch pipette | High | High | Low | Intact live tissue (in vitro/ex vivo) |

4. Single-Cell Laser-Assisted Microdissection

5. Harvesting Cells or Cytosol through Glass Capillary
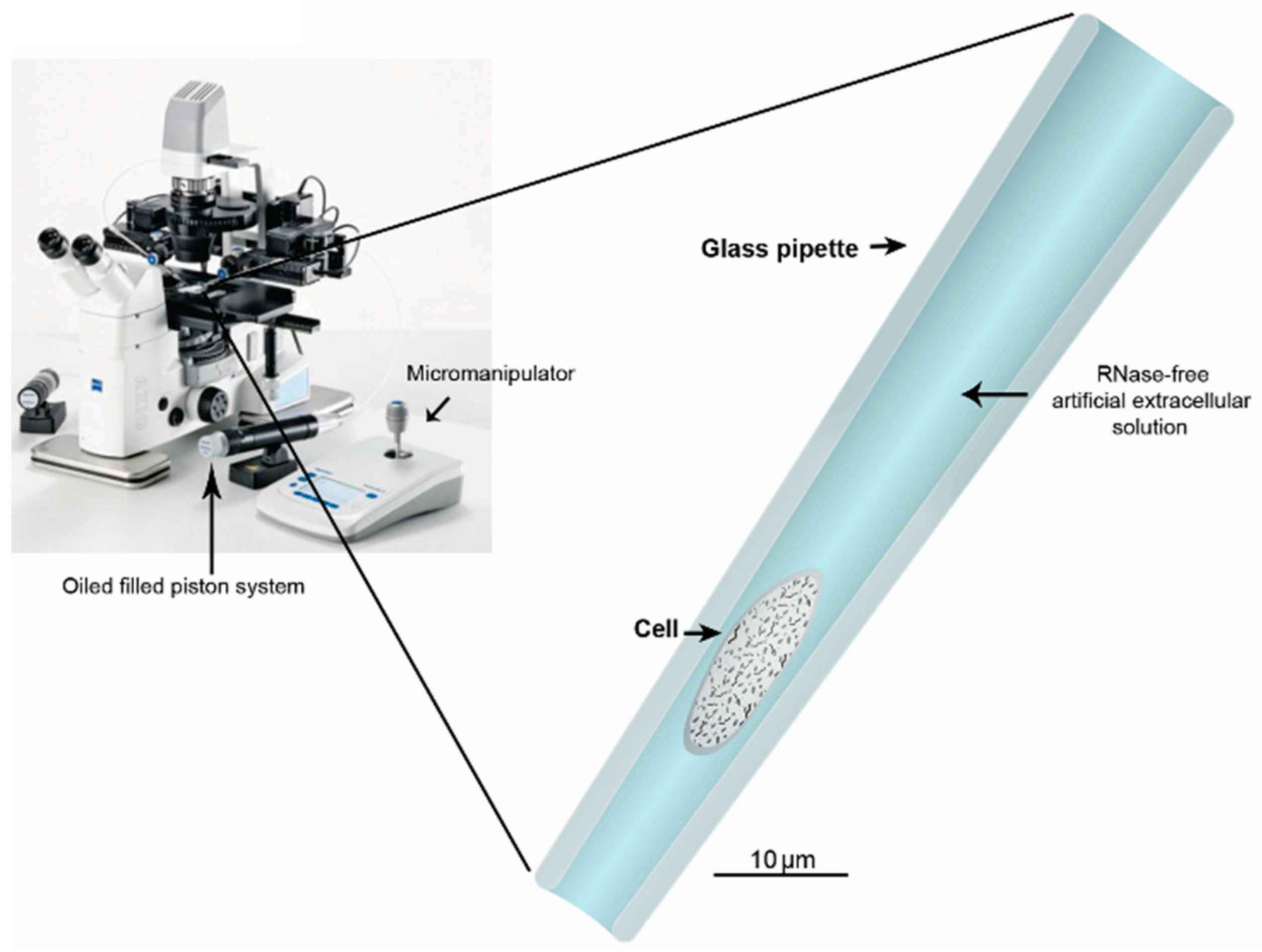

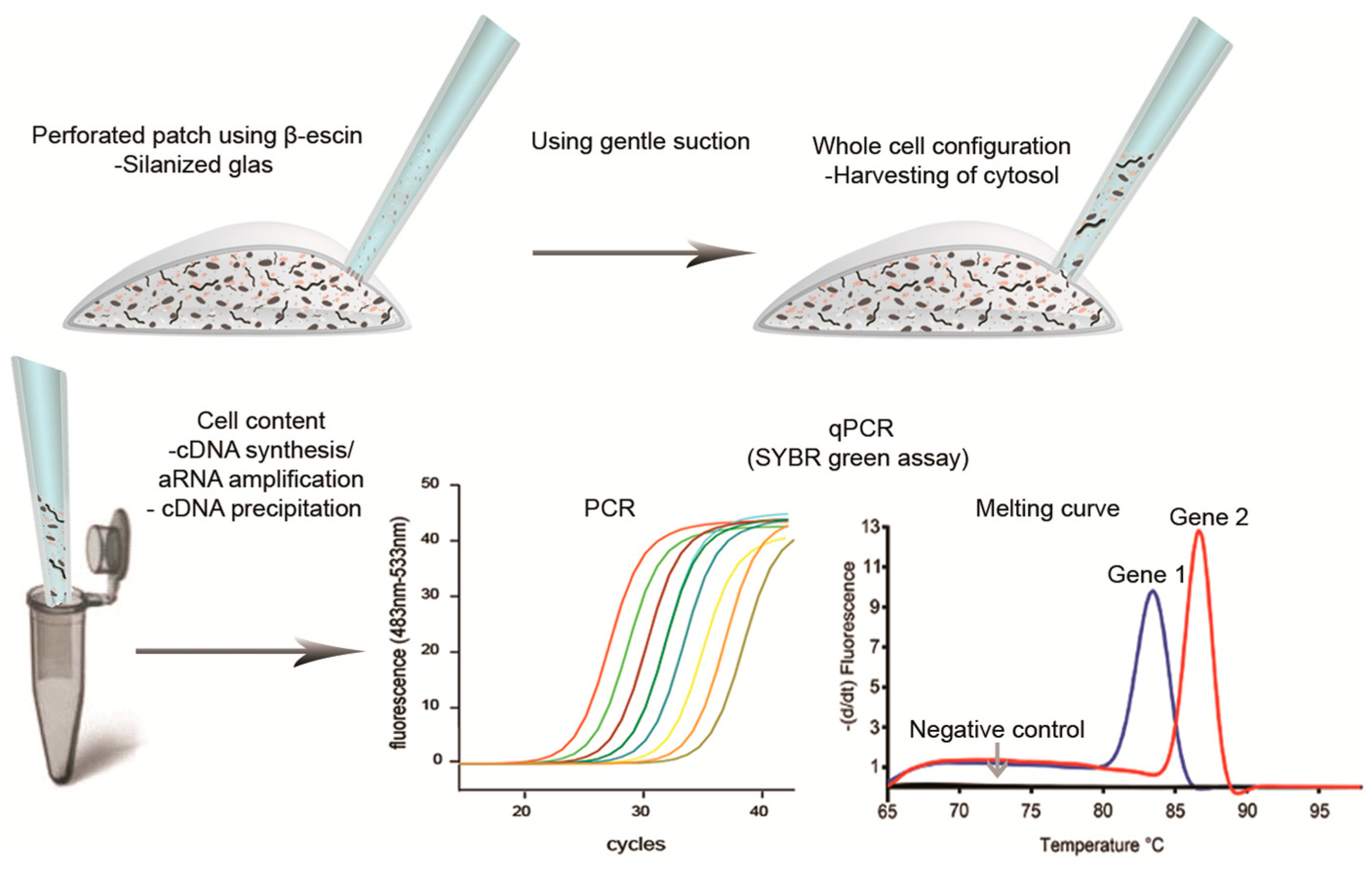
6. Lysis and Securing the RNA
7. Reverse Transcription
8. qPCR
9. Quantitative Gene Analysis
10. Future Possibilities and Challenges
11. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ko, M.S.H.; Nakauchi, H.; Takahashi, N. The dose dependence of glucocorticoid-inducible gene-expression results from changes in the number of transcriptionally active templates. EMBO J. 1990, 9, 2835–2842. [Google Scholar] [PubMed]
- Walters, M.C.; Fiering, S.; Eidemiller, J.; Magis, W.; Groudine, M.; Martin, D.I.K. Enhancers increase the probability but not the level of gene-expression. Proc. Natl. Acad. Sci. USA 1995, 92, 7125–7129. [Google Scholar] [CrossRef] [PubMed]
- McAdams, H.H.; Arkin, A. Stochastic mechanisms in gene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic gene expression in a single cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.J.; Stirland, J.A.; McFerran, D.W.; Seymour, Z.C.; Spiller, D.G.; Loudon, A.S.I.; White, M.R.H.; Davis, J.R.E. Dynamic patterns of growth hormone gene transcription in individual living pituitary cells. Mol. Endocrinol. 2003, 17, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, M.; Stahlberg, A.; Rorsman, P.; Kubista, M. Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Res. 2005, 15, 1388–1392. [Google Scholar] [CrossRef] [PubMed]
- Chubb, J.R.; Trcek, T.; Shenoy, S.M.; Singer, R.H. Transcriptional pulsing of a developmental gene. Curr. Biol. 2006, 16, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; Peskin, C.S.; Tranchina, D.; Vargas, D.Y.; Tyagi, S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006, 4, 1707–1719. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; van Oudenaarden, A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Method of the Year 2013. Methods to sequence the DNA and RNA of single cells are poised to transform many areas of biology and medicine. Nat. Methods 2014, 11, 1. [CrossRef] [PubMed]
- Malnic, B.; Hirono, J.; Sato, T.; Buck, L.B. Combinatorial receptor codes for odors. Cell 1999, 96, 713–723. [Google Scholar] [CrossRef]
- Raghunathan, A.; Ferguson, H.R.; Bornarth, C.J.; Song, W.M.; Driscoll, M.; Lasken, R.S. Genomic DNA amplification from a single bacterium. Appl. Environ. Microbiol. 2005, 71, 3342–3347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Martiny, A.C.; Reppas, N.B.; Barry, K.W.; Malek, J.; Chisholm, S.W.; Church, G.M. Sequencing genomes from single cells by polymerase cloning. Nat. Biotechnol. 2006, 24, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Zong, C.H.; Lu, S.J.; Chapman, A.R.; Xie, X.S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef]
- Kleppe, K.; Ohtsuka, E.; Kleppe, R.; Molineux, I.; Khorana, H.G. Studies on polynucleotides. XCVI. Repair replications of short synthetic DNA’s as catalyzed by DNA polymerases. J. Mol. Biol. 1971, 56, 341–361. [Google Scholar] [CrossRef]
- Rabinow, P. Making PCR: A Story of Biotechnology; University of Chicago Press: Chicago, IL, USA, 1996. [Google Scholar]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle-cell anemia. Science 1985, 230, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific enzymatic amplification of DNA in vitro—The polymerase chain-reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA-polymerase. Science 1988, 239, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Chien, A.; Edgar, D.B.; Trela, J.M. Deoxyribonucleic-acid polymerase from extreme thermophile Thermus aquaticus. J. Bacteriol. 1976, 127, 1550–1557. [Google Scholar] [PubMed]
- Brock, T.D.; Freeze, H. Thermus aquaticus gen. n. and sp. n., a nonsporulating extreme thermophile. J. Bacteriol. 1969, 98, 289–297. [Google Scholar] [PubMed]
- Baltimore, D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 1970, 226, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M.; Mizutani, S. Viral RNA-dependent DNA polymerase: RNA-dependent DNA polymerase in virions of rous sarcoma virus. Rev. Med. Virol. 1998, 8, 3–11. [Google Scholar] [CrossRef]
- Verma, I.M.; Temple, G.F.; Baltimor, D.; Fan, H. In vitro synthesis of DNA complementary to rabbit reticulocyte 10S RNA. Nat. New Biol. 1972, 235, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Efstratiadis, A.; Kafatos, F.C.; Maxam, A.M.; Maniatis, T. Enzymatic in vitro synthesis of globin genes. Cell 1976, 7, 279–288. [Google Scholar] [CrossRef]
- Fisher, M.P.; Dingman, C.W. Role of molecular conformation in determining electrophoretic properties of polynucleotides in agarose-acrylamide composite gels. Biochemistry 1971, 10, 1895–1899. [Google Scholar] [CrossRef] [PubMed]
- Aaij, C.; Borst, P. The gel-electrophoresis of DNA. Biochim. Biophys. Acta 1972, 269, 192–200. [Google Scholar] [CrossRef]
- Sharp, P.A.; Sugden, B.; Sambrook, J. Detection of two restriction endonuclease activities in haemophilus-parainfluenzae using analytical agarose-ethidium bromide electrophoresis. Biochemistry 1973, 12, 3055–3063. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gyllensten, U.B.; Cui, X.; Saiki, R.K.; Erlich, H.A.; Arnheim, N. Amplification and analysis of DNA sequences in single human sperm and diploid cells. Nature 1988, 335, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Brady, G.; Barbara, M.; Iscove, N.N. Representative in vitro cDNA amplification from individual hemopoietic cells and colonies. Methods Mol. Cell. Biol. 1990, 2, 17–25. [Google Scholar]
- Vangelder, R.N.; Vonzastrow, M.E.; Yool, A.; Dement, W.C.; Barchas, J.D.; Eberwine, J.H. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc. Natl. Acad. Sci. USA 1990, 87, 1663–1667. [Google Scholar] [CrossRef]
- Eberwine, J.; Yeh, H.; Miyashiro, K.; Cao, Y.X.; Nair, S.; Finnell, R.; Zettel, M.; Coleman, P. Analysis of gene-expression in single live neurons. Proc. Natl. Acad. Sci. USA 1992, 89, 3010–3014. [Google Scholar] [CrossRef] [PubMed]
- Lambolez, B.; Audinat, E.; Bochet, P.; Crepel, F.; Rossier, J. AMPA receptor subunits expressed by single Purkinje cells. Neuron 1992, 9, 247–258. [Google Scholar] [CrossRef]
- Higuchi, R.; Dollinger, G.; Walsh, P.S.; Griffith, R. Simultaneous amplification and detection of specific DNA sequences. Bio/Technology 1992, 10, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR analysis: Real-time monitoring of DNA amplification reactions. Bio/Technology 1993, 11, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- McPherson, M.J.; Hames, B.D.; Taylor, G.R. PCR 2: A Practical Approach; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Kainz, P. The PCR plateau phase—Towards an understanding of its limitations. Biochim. Biophys. Acta 2000, 1494, 23–27. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Zakrajsek, B.A.; Mills, A.G.; Gorn, V.; Singer, M.J.; Reed, M.W. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: Comparison of endpoint and real-time methods. Anal. Biochem. 2000, 285, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): Trends and problems. J. Mol. Endocrinol. 2002, 29, 17. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, F.; Steen, H.B.; Nesland, J.M. SYBR green I DNA staining increases the detection sensitivity of viruses by polymerase chain-reaction. J. Virol. Methods 1995, 55, 153–156. [Google Scholar] [CrossRef]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, C.T.; Ririe, K.M.; Andrew, R.V.; David, D.A.; Gundry, R.A.; Balis, U.J. The LightCycler: A microvolume multisample fluorimeter with rapid temperature control. Biotechniques 1997, 22, 176–181. [Google Scholar] [PubMed]
- Holland, P.M.; Abramson, R.D.; Watson, R.; Gelfand, D.H. Detection of specific polymerase chain-reaction product by utilizing the 5ʹ–3ʹ exonuclease activity of Thermus aquaticus DNA-polymerase. Proc. Natl. Acad. Sci. USA 1991, 88, 7276–7280. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Mathies, R.A. Integrated microfluidic systems for high-performance genetic analysis. Trends Biotechnol. 2009, 27, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, N.; Pop, M. Sequence assembly demystified. Nat. Rev. Genet. 2013, 14, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, P.; Voet, T. Single cell analysis of cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Achim, K.; Pettit, J.-B.; Saraiva, L.R.; Gavriouchkina, D.; Larsson, T.; Arendt, D.; Marioni, J.C. High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat. Biotechnol. 2015, 33, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.A.; Marioni, J.C.; Stegie, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Crosetto, N.; Bienko, M.; van Oudenaarden, A. Spatially resolved transcriptomics and beyond. Nat. Rev. Genet. 2015, 16, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Zhang, Y.; Enge, M.; Caneda, C.; Shuer, L.M.; Gephart, M.G.H.; Barres, B.A.; Quake, S.R. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA 2015, 112, 7285–7290. [Google Scholar] [CrossRef] [PubMed]
- Stegle, O.; Teichmann, S.A.; Marioni, J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef] [PubMed]
- EmmertBuck, M.R.; Bonner, R.F.; Smith, P.D.; Chuaqui, R.F.; Zhuang, Z.P.; Goldstein, S.R.; Weiss, R.A.; Liotta, L.A. Laser capture microdissection. Science 1996, 274, 998–1001. [Google Scholar] [CrossRef]
- Bonner, R.F.; EmmertBuck, M.; Cole, K.; Pohida, T.; Chuaqui, R.; Goldstein, S.; Liotta, L.A. Laser capture microdissection: Molecular analysis of tissue. Science 1997, 278, 1481–1483. [Google Scholar] [CrossRef] [PubMed]
- Schutze, K.; Lahr, G. Identification of expressed genes by laser-mediated manipulation of single cells. Nat. Biotechnol. 1998, 16, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Podgorny, O.V. Live cell isolation by laser microdissection with gravity transfer. J. Biomed. Opt. 2013, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Herzenberg, L.A.; Sweet, R.G. Fluorescence-activated cell sorting. Sci. Am. 1976, 234, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Herzenberg, L.A.; Parks, D.; Sahaf, B.; Perez, O.; Roederer, M. The history and future of the fluorescence activated cell sorter and flow cytometry: A view from Stanford. Clin. Chem. 2002, 48, 1819–1827. [Google Scholar] [PubMed]
- Wheeler, A.R.; Throndset, W.R.; Whelan, R.J.; Leach, A.M.; Zare, R.N.; Liao, Y.H.; Farrell, K.; Manger, I.D.; Daridon, A. Microfluidic device for single-cell analysis. Anal. Chem. 2003, 75, 3581–3586. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.A.; Hong, J.W.; Quake, S.R.; Leadbetter, J.R. Microfluidic digital PCR enables multigene analysis of individual environmental bacteria. Science 2006, 314, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Bryder, D.; Weissman, I.L.; Quake, S.R. Transcription factor profiling in individual hematopoietic progenitors by digital RT-PCR. Proc. Natl. Acad. Sci. USA 2006, 103, 17807–17812. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.R.; Neff, N.F.; Kalisky, T.; Dalerba, P.; Treutlein, B.; Rothenberg, M.E.; Mburu, F.M.; Mantalas, G.L.; Sim, S.; Clarke, M.F.; Quake, S.R. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods 2014, 11, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.A.; Chou, H.P.; Thorsen, T.; Scherer, A.; Quake, S.R. Monolithic microfabricated valves and pumps by multilayer soft lithography. Science 2000, 288, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Joensson, H.N.; Svahn, H.A. Droplet microfluidics—A tool for single-cell analysis. Angew. Chem. Int. Ed. 2012, 51, 12176–12192. [Google Scholar] [CrossRef] [PubMed]
- Seemann, R.; Brinkmann, M.; Pfohl, T.; Herminghaus, S. Droplet based microfluidics. Rep. Prog. Phys. 2012, 75, 016601. [Google Scholar] [CrossRef] [PubMed]
- Pantoja, R.; Nagarah, J.M.; Starace, D.M.; Melosh, N.A.; Blunck, R.; Bezanilla, F.; Heath, J.R. Silicon chip-based patch-clamp electrodes integrated with PDMS microfluidics. Biosens. Bioelectron. 2004, 20, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.W.; Studer, V.; Hang, G.; Anderson, W.F.; Quake, S.R. A nanoliter-scale nucleic acid processor with parallel architecture. Nat. Biotechnol. 2004, 22, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.K.; Wheeler, A.; Zare, R.N. Chemical cytometry on a picoliter-scale integrated microfluidic chip. Proc. Natl. Acad. Sci. USA 2004, 101, 12809–12813. [Google Scholar] [CrossRef] [PubMed]
- Walch, A.; Specht, K.; Smida, J.; Aubele, M.; Zitzelsberger, H.; Hofler, H.; Werner, M. Tissue microdissection techniques in quantitative genome and gene expression analyses. Histochem. Cell Biol. 2001, 115, 269–276. [Google Scholar] [PubMed]
- Guo, G.J.; Huss, M.; Tong, G.Q.; Wang, C.Y.; Sun, L.L.; Clarke, N.D.; Robson, P. Resolution of Cell Fate Decisions Revealed by Single-Cell Gene Expression Analysis from Zygote to Blastocyst. Dev. Cell 2010, 18, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Hodne, K.; Haug, T.M.; Weltzien, F.A. Single-cell qPCR on dispersed primary pituitary cells—An optimized protocol. BMC Mol. Biol. 2010, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Pang, Z.P.P.; Sudhof, T.C.; Wernig, M.; Malenka, R.C. Comprehensive qPCR profiling of gene expression in single neuronal cells. Nat. Protoc. 2012, 7, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.B.; Audet, J. Current techniques for single-cell lysis. J. R. Soc. Interface 2008, 5, S131–S138. [Google Scholar] [CrossRef] [PubMed]
- Svec, D.; Andersson, D.; Pekny, M.; Sjoback, R.; Kubista, M.; Stahlberg, A. Direct cell lysis for single-cell gene expression profiling. Front. Oncol. 2013, 3, 274. [Google Scholar] [CrossRef] [PubMed]
- Stahlberg, A.; Kubista, M. The workflow of single-cell expression profiling using quantitative real-time PCR. Expert Rev. Mol. Diagn. 2014, 14, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Vonhippel, P.H.; Wong, K.Y. Neutral salts—Generality of their effects on stability of macromolecular conformations. Science 1964, 145, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, Y.; Tanford, C. Solubility of amino acids, diglycine, and triglycine in aqueous guanidine hydrochloride solutions. J. Biol. Chem. 1970, 245, 1648–1652. [Google Scholar] [PubMed]
- Gordon, J.A. Denaturation of globular proteins. Interaction of guanidinium salts with three proteins. Biochemistry 1972, 11, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, M.; Hemberg, M.; Rorsman, P.; Stahlberg, A. Quantification of mRNA in single cells and modelling of RT-qPCR induced noise. BMC Mol. Biol. 2008, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Geselowitz, D.A.; Neckers, L.M. Bovine serum-albumin is a major oligonucleotide-binding protein found on the surface of cultured-cells. Antisense Res. Dev. 1995, 5, 213–217. [Google Scholar] [PubMed]
- Kreader, C.A. Relief of amplification inhibition in PCR with bovine serum albumin or T4 gene 32 protein. Appl. Environ. Microbiol. 1996, 62, 1102–1106. [Google Scholar] [PubMed]
- Wilson, I.G. Inhibition and facilitation of nucleic acid amplification. Appl. Environ. Microbiol. 1997, 63, 3741–3751. [Google Scholar] [PubMed]
- Abu Al-Soud, W.; Radstrom, P. Effects of amplification facilitators on diagnostic PCR in the presence of blood, feces, and meat. J. Clin. Microbiol. 2000, 38, 4463–4470. [Google Scholar] [PubMed]
- Arnedo, A.; Espuelas, S.; Irache, J.M. Albumin nanoparticles as carriers for a phosphodiester oligonucleotide. Int. J. Pharm. 2002, 244, 59–72. [Google Scholar] [CrossRef]
- Farell, E.M.; Alexandre, G. Bovine serum albumin further enhances the effects of organic solvents on increased yield of polymerase chain reaction of GC-rich templates. BMC Res. Notes 2012, 5, 257. [Google Scholar] [CrossRef] [PubMed]
- Deprez, R.H.L.; Fijnvandraat, A.C.; Ruijter, J.M.; Moorman, A.F.M. Sensitivity and accuracy of quantitative real-time polymerase chain reaction using SYBR green I depends on cDNA synthesis conditions. Anal. Biochem. 2002, 307, 63–69. [Google Scholar] [CrossRef]
- Stahlberg, A.; Hakansson, J.; Xian, X.J.; Semb, H.; Kubista, M. Properties of the reverse transcription reaction in mRNA quantification. Clin. Chem. 2004, 50, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Zeisel, A.; Joost, S.; la Manno, G.; Zajac, P.; Kasper, M.; Lonnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 2014, 11, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, R.A. The efficiency of promoter clearance distinguishes T7 class-II and class-III promoters. J. Biol. Chem. 1992, 267, 11322–11328. [Google Scholar] [PubMed]
- Ikeda, R.A.; Lin, A.C.; Clarke, J. Initiation of transcription by T7-RNA polymerase at its natural promoters. J. Biol. Chem. 1992, 267, 2640–2649. [Google Scholar] [PubMed]
- Pabon, C.; Modrusan, Z.; Ruvolo, M.V.; Coleman, I.M.; Daniel, S.; Yue, H.; Arnold, L.J.; Reynolds, M.A. Optimized T7 amplification system for microarray analysis. Biotechniques 2001, 31, 874–879. [Google Scholar] [PubMed]
- Wang, J.; Hu, L.; Hamilton, S.R.; Coombes, K.R.; Zhang, W. RNA amplification strategies for cDNA microarray experiments. BioTechniques 2003, 34, 394–400. [Google Scholar] [PubMed]
- Moll, P.R.; Duschl, J.; Richter, K. Optimized RNA amplification using T7-RNA-polymerase based in vitro transcription. Anal. Biochem. 2004, 334, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Brady, G.; Billia, F.; Knox, J.; Hoang, T.; Kirsch, I.R.; Voura, E.B.; Hawley, R.G.; Cumming, R.; Buchwald, M.; Siminovitch, K.; et al. analysis of gene-expression in a complex differentiation hierarchy by global amplification of cDNA from single cells. Curr. Biol. 1995, 5, 909–922. [Google Scholar] [CrossRef]
- Iscove, N.N.; Barbara, M.; Gu, M.; Gibson, M.; Modi, C.; Winegarden, N. Representation is faithfully preserved in global cDNA amplified exponentially from sub-picogram quantities of mRNA. Nat. Biotechnol. 2002, 20, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Subkhankulova, T.; Livesey, F.J. Comparative evaluation of linear and exponential amplification techniques for expression profiling at the single-cell level. Genome Biol. 2006, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.E.; Magbanua, M.J.M.; Scott, J.H.; Makrigiorgos, G.M.; Wang, G.; Federman, S.; Esserman, L.J.; Park, J.W.; Haqq, C.M. A comparison of RNA amplification techniques at sub-nanogram input concentration. Bmc Genom. 2009, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Y.; Machleder, E.M.; Chenchik, A.; Li, R.; Siebert, P.D. Reverse transcriptase template switching: A SMART™ approach for full-length cDNA library construction. Biotechniques 2001, 30, 892–897. [Google Scholar] [PubMed]
- Tang, F.C.; Barbacioru, C.; Wang, Y.Z.; Nordman, E.; Lee, C.; Xu, N.L.; Wang, X.H.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Kjallquist, U.; Moliner, A.; Zajac, P.; Fan, J.B.; Lonnerberg, P.; Linnarsson, S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011, 21, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-cell RNA-seq by multiplexed linear amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Ramskold, D.; Luo, S.J.; Wang, Y.C.; Li, R.; Deng, Q.L.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, Y.; Nikaido, I.; Hayashi, T.; Danno, H.; Uno, K.D.; Imai, T.; Ueda, H.R. Quartz-Seq: A highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Koch, W.H. Technology platforms for pharmacogenomic diagnostic assays. Nat. Rev. Drug Discov. 2004, 3, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Persson, K.; Hamby, K.; Ugozzoli, L.A. Four-color multiplex reverse transcription polymerase chain reaction—Overcoming its limitations. Anal. Biochem. 2005, 344, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Primer3plus. Available online: http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi (accessed on 5 November 2015).
- Vector NTI. Available online: http://www.lifetechnologies.com/no/en/home/life-science/cloning/vector-nti-software.html (accessed on 5 November 2015).
- Chandler, D.P.; Wagnon, C.A.; Bolton, H. Reverse transcriptase (RT) inhibition of PCR at low concentrations of template and its implications for quantitative RT-PCR. Appl. Environ. Microbiol. 1998, 64, 669–677. [Google Scholar] [PubMed]
- Liss, B. Improved quantitative real-time RT-PCR for expression profiling of individual cells. Nucleic Acids Res. 2002, 30, 9. [Google Scholar] [CrossRef]
- Nolan, T.; Hands, R.E.; Ogunkolade, W.; Bustin, S.A. SPUD: A quantitative PCR assay for the detection of inhibitors in nucleic acid preparations. Anal. Biochem. 2006, 351, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Singhal, R.; Orynbayeva, Z.; Sundaram, R.V.K.; Niu, J.J.; Bhattacharyya, S.; Vitol, E.A.; Schrlau, M.G.; Papazoglou, E.S.; Friedman, G.; Gogotsi, Y. Multifunctional carbon-nanotube cellular endoscopes. Nat. Nanotechnol. 2011, 6, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Schrlau, M.G.; Dun, N.J.; Bau, H.H. Cell electrophysiology with carbon nanopipettes. ACS Nano 2009, 3, 563–568. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hodne, K.; Weltzien, F.-A. Single-Cell Isolation and Gene Analysis: Pitfalls and Possibilities. Int. J. Mol. Sci. 2015, 16, 26832-26849. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161125996
Hodne K, Weltzien F-A. Single-Cell Isolation and Gene Analysis: Pitfalls and Possibilities. International Journal of Molecular Sciences. 2015; 16(11):26832-26849. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161125996
Chicago/Turabian StyleHodne, Kjetil, and Finn-Arne Weltzien. 2015. "Single-Cell Isolation and Gene Analysis: Pitfalls and Possibilities" International Journal of Molecular Sciences 16, no. 11: 26832-26849. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161125996