
Leptin Promotes cPLA2 Gene Expression through Activation of the MAPK/NF-κB/p300 Cascade

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
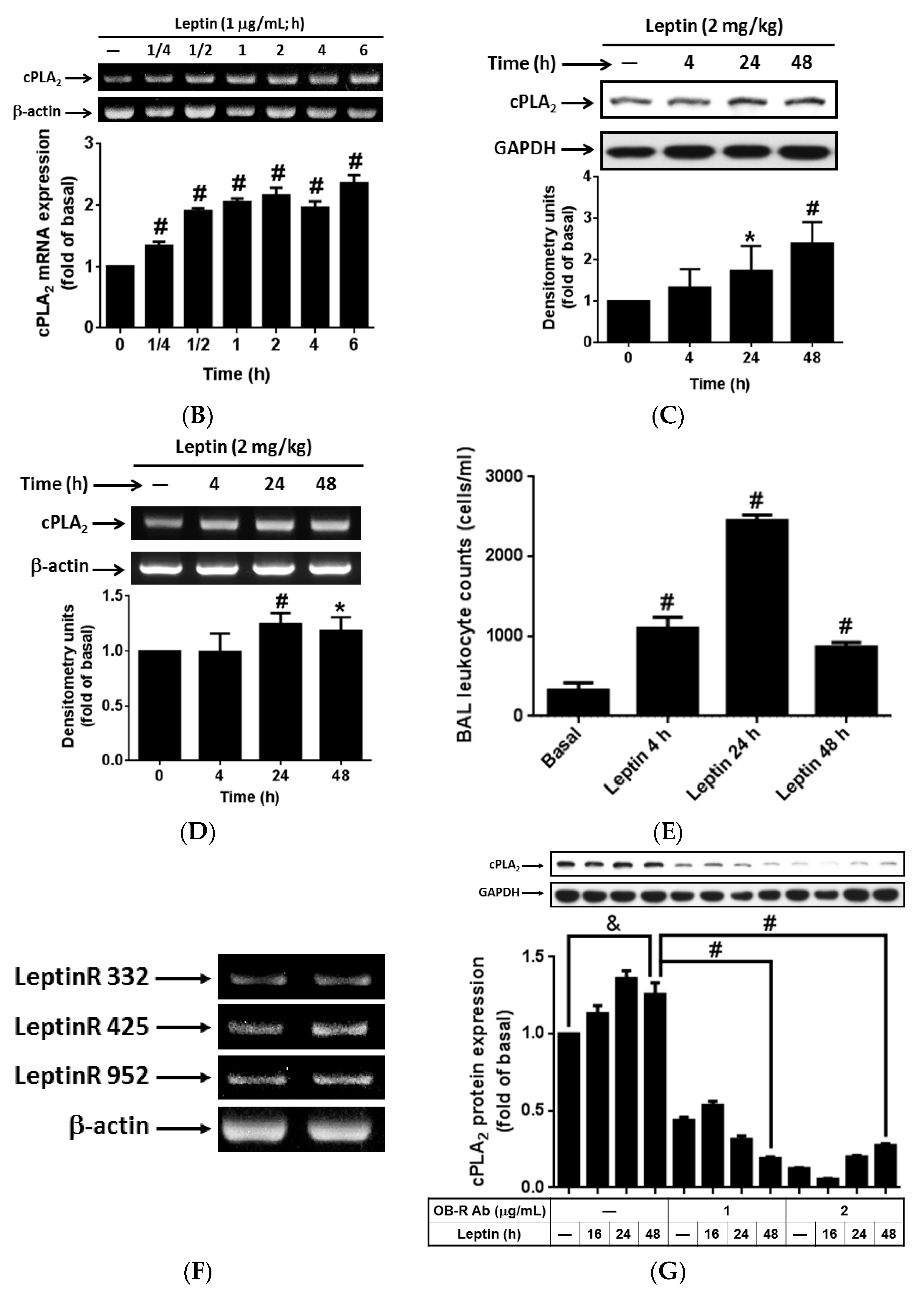
2.1. In Vitro and in Vivo Expression of cPLA2-α in Response to Leptin Stimulation


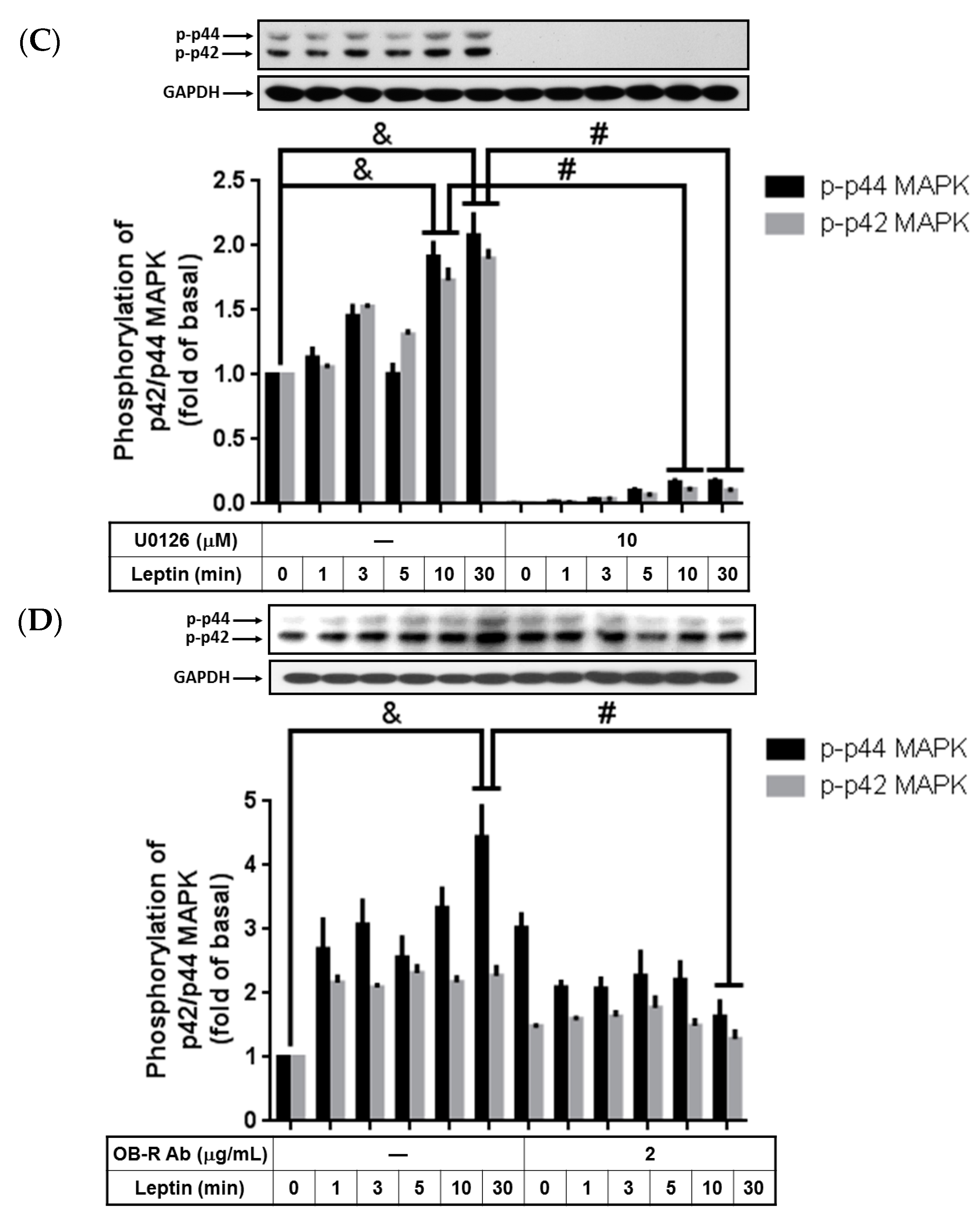
2.2. Phosphorylated p42/p44 MAPK Contributed to Leptin-Stimulated cPLA2-α Gene Expression


2.3. Activation of p38 MAPK Involved in Leptin-Stimulated cPLA2-α Expression

2.4. Participation of Activated JNK1/2 in Leptin-Upregulated cPLA2-α Expression

2.5. Leptin Stimulated cPLA2-α Expression via Activation of NF-κB

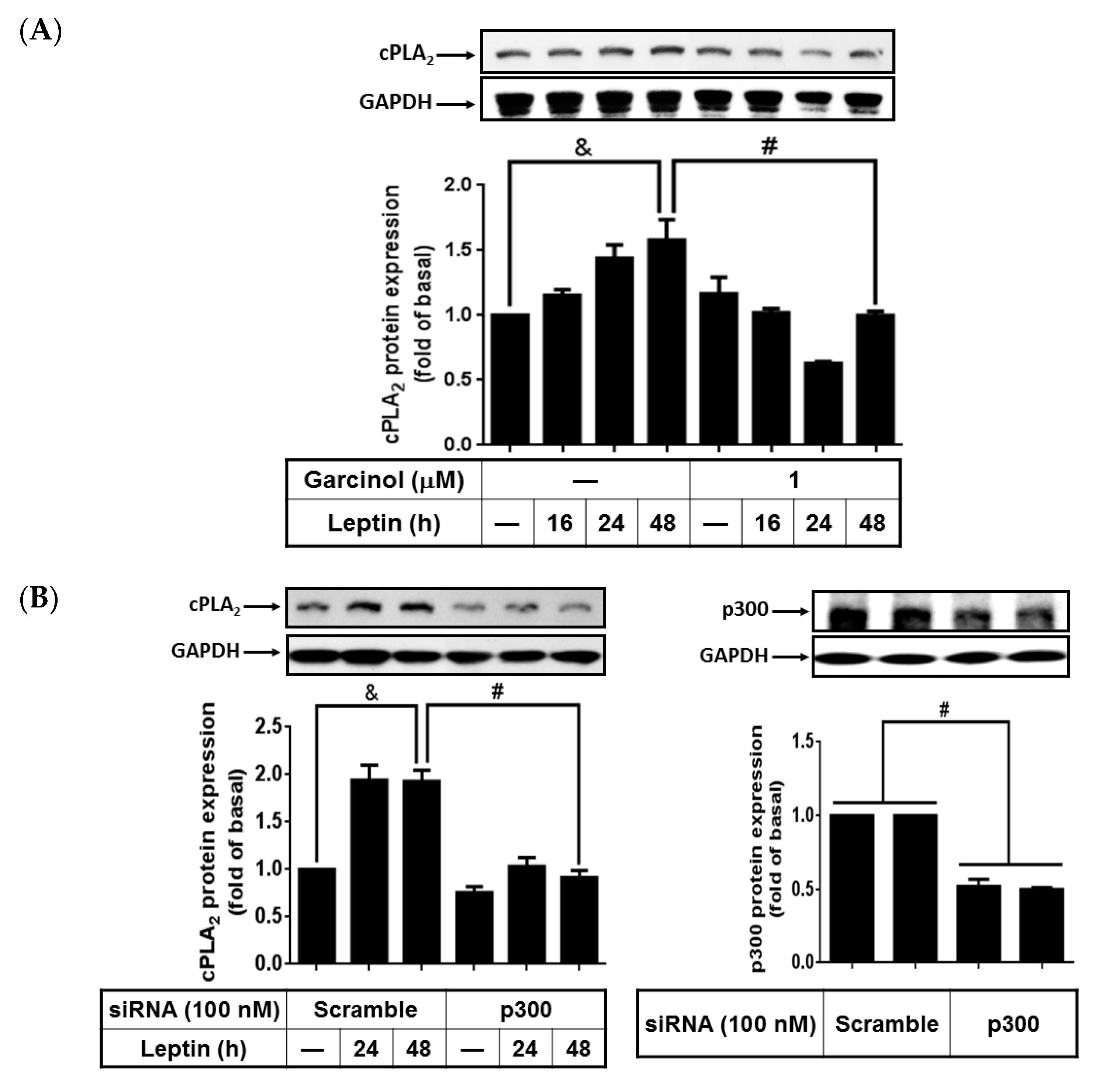
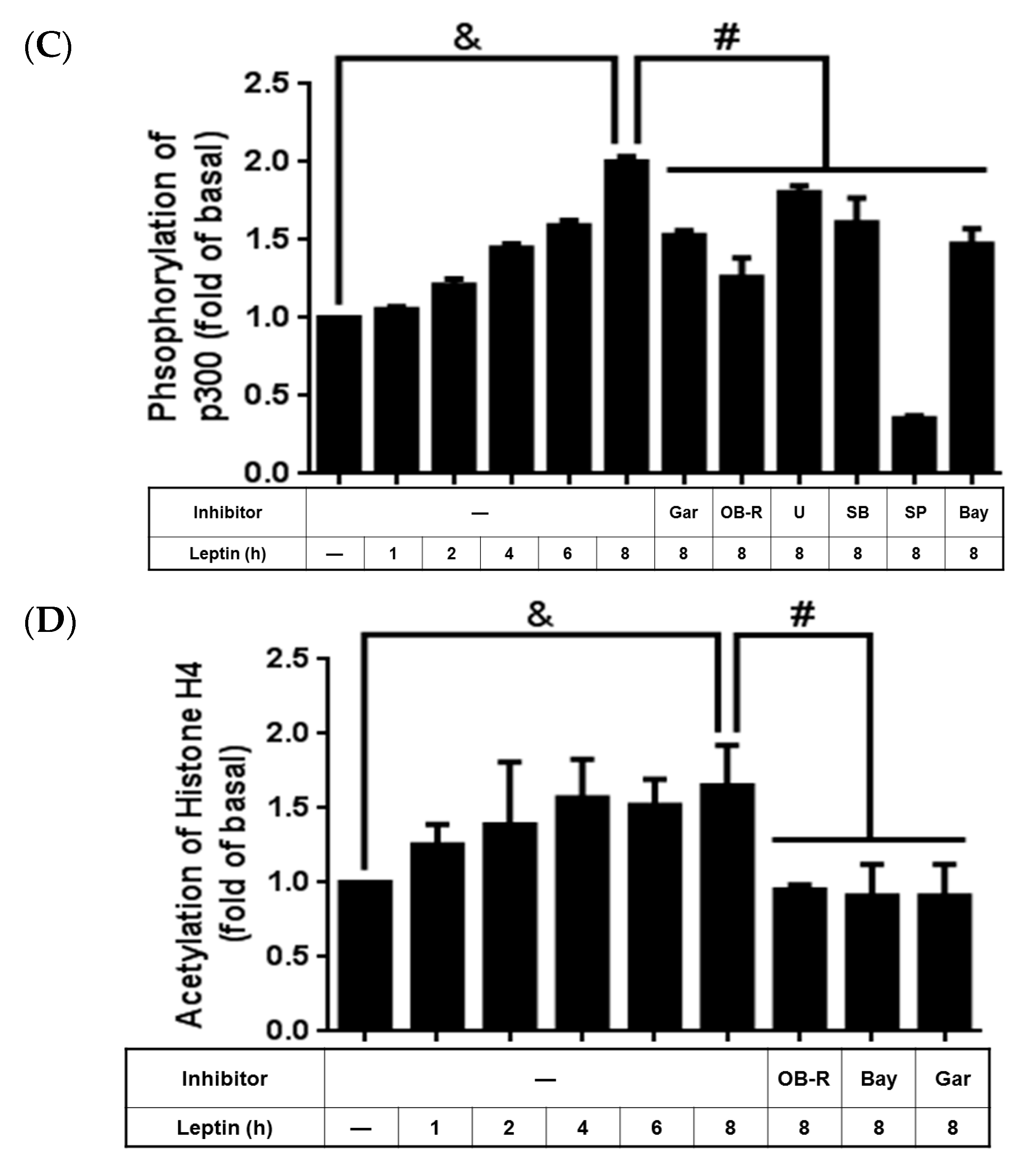
2.6. Activated p300 Contributes to Leptin-Increased cPLA2-α Expression


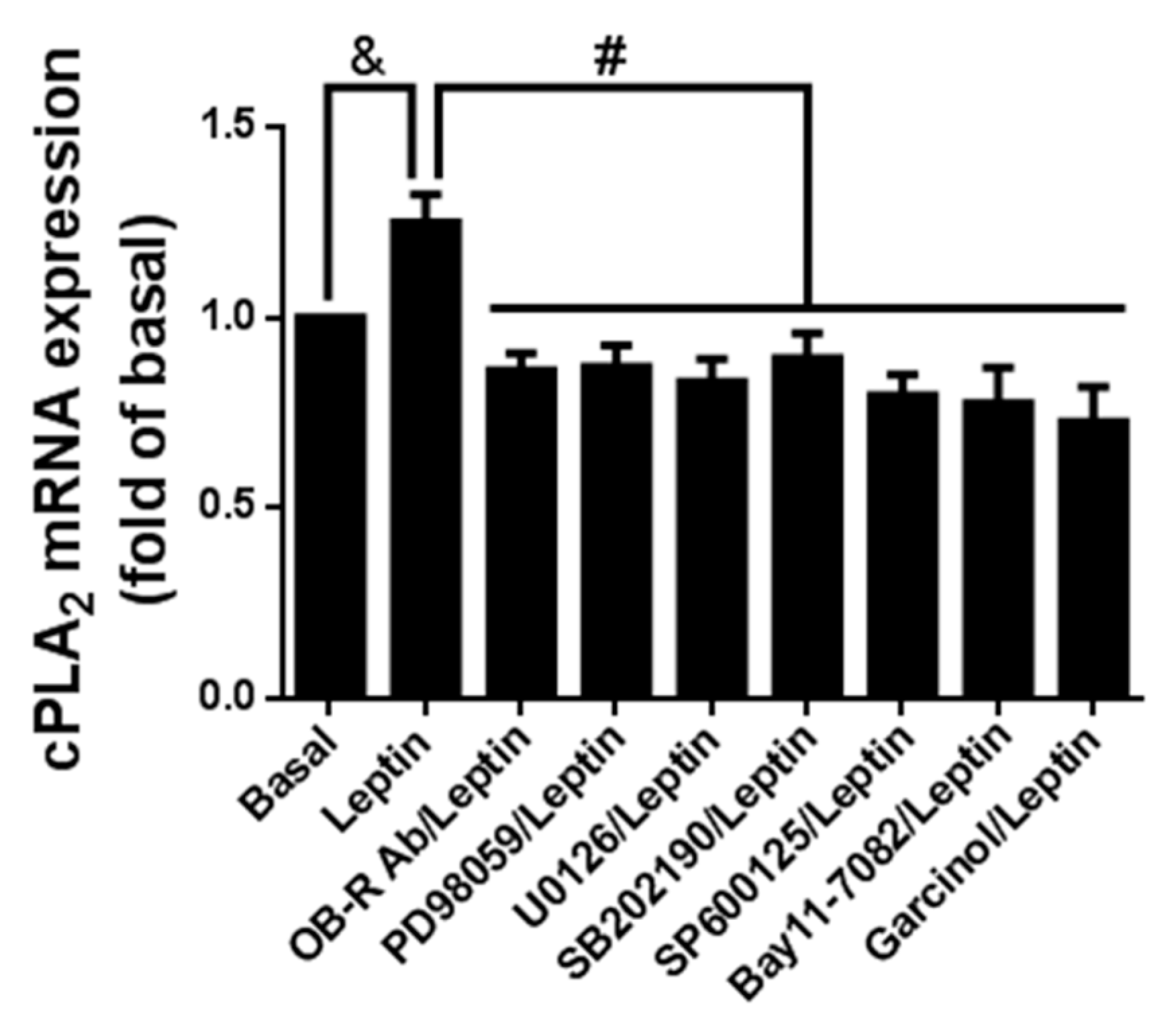
2.7. Leptin Induced cPLA2-α mRNA Expression via Ob-R/MAPK/NF-κB/p300 Cascade

3. Discussion
4. Experimental Section
4.1. Materials
4.2. Cell Culture of Human Alveolar Epithelial Cell Carcinoma (A549)
4.3. Animal Treatment
4.4. Isolation of BAL
4.5. Transfection with Small Interference RNA
4.6. Protein Extraction and Western Blot
4.7. Total RNA Extraction and Gene Expression Analysis
4.8. Statistical Analysis of Data
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McClean, K.M.; Kee, F.; Young, I.S.; Elborn, J.S. Obesity and the lung: 1. Epidemiology. Thorax 2008, 63, 649–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franssen, F.M.; O’Donnell, D.E.; Goossens, G.H.; Blaak, E.E.; Schols, A.M. Obesity and the lung: 5. Obesity and COPD. Thorax 2008, 63, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Sin, D.D.; Sutherland, E.R. Obesity and the lung: 4. Obesity and asthma. Thorax 2008, 63, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Crummy, F.; Piper, A.J.; Naughton, M.T. Obesity and the lung: 2. Obesity and sleep-disordered breathing. Thorax 2008, 63, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Konter, J.; Baez, E.; Summer, R.S. Obesity: “Priming” the lung for injury. Pulm. Pharmacol. Ther. 2013, 26, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Beuther, D.A.; Sutherland, E.R. Overweight, obesity, and incident asthma: A meta-analysis of prospective epidemiologic studies. Am. J. Respir. Crit. Care Med. 2007, 175, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, R.R.; Stenlof, K.; Hedner, J.A.; Peltonen, M.; Karason, K.; Sjostrom, L. Two year reduction in sleep apnea symptoms and associated diabetes incidence after weight loss in severe obesity. Sleep 2007, 30, 703–710. [Google Scholar] [PubMed]
- Lee, I.T.; Lee, C.W.; Tung, W.H.; Wang, S.W.; Lin, C.C.; Shu, J.C.; Yang, C.M. Cooperation of TLR2 with MyD88, PI3K, and Rac1 in lipoteichoic acid-induced cPLA2/COX-2-dependent airway inflammatory responses. Am. J. Pathol. 2010, 176, 1671–1684. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.F.; Lin, W.N.; Yang, C.M.; Lee, C.W.; Liao, C.H.; Leu, Y.L.; Hsiao, L.D. Induction of cytosolic phospholipase a2 by lipopolysaccharide in canine tracheal smooth muscle cells: Involvement of MAPKs and NF-κB pathways. Cell Signal. 2006, 18, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, L.; Medina, M.; Pernet, E.; Leduc, D.; Chignard, M.; Touqui, L.; Wu, Y. Cytosolic phospholipase A2α enhances mouse mortality induced by pseudomonas aeruginosa pulmonary infection via interleukin 6. Biochimie 2014, 107, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Nagase, T.; Uozumi, N.; Ishii, S.; Kume, K.; Izumi, T.; Ouchi, Y.; Shimizu, T. Acute lung injury by sepsis and acid aspiration: A key role for cytosolic phospholipase A2. Nat. Immunol. 2000, 1, 42–46. [Google Scholar] [PubMed]
- Lin, W.N.; Lin, C.C.; Cheng, H.Y.; Yang, C.M. Regulation of cyclooxygenase-2 and cytosolic phospholipase A2 gene expression by lipopolysaccharide through the RNA-binding protein HuR: Involvement of NADPH oxidase, reactive oxygen species and mitogen-activated protein kinases. Br. J. Pharmacol. 2011, 163, 1691–1706. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.C.; Lane, M.D. Adipose development: From stem cell to adipocyte. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Galic, S.; Oakhill, J.S.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Basinski, M.B.; Beals, J.M.; Briggs, S.L.; Churgay, L.M.; Clawson, D.K.; DiMarchi, R.D.; Furman, T.C.; Hale, J.E.; Hsiung, H.M.; et al. Crystal structure of the obese protein leptin-e100. Nature 1997, 387, 206–209. [Google Scholar] [CrossRef] [PubMed]
- La Cava, A.; Matarese, G. The weight of leptin in immunity. Nat. Rev. Immunol. 2004, 4, 371–379. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, V.; Procaccini, C.; Cali, G.; Pirozzi, G.; Fontana, S.; Zappacosta, S.; la Cava, A.; Matarese, G. A key role of leptin in the control of regulatory T cell proliferation. Immunity 2007, 26, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Watowich, S.S.; Wu, H.; Socolovsky, M.; Klingmuller, U.; Constantinescu, S.N.; Lodish, H.F. Cytokine receptor signal transduction and the control of hematopoietic cell development. Annu. Rev. Cell Dev. Biol. 1996, 12, 91–128. [Google Scholar] [CrossRef] [PubMed]
- Houseknecht, K.L.; Baile, C.A.; Matteri, R.L.; Spurlock, M.E. The biology of leptin: A review. J. Anim. Sci. 1998, 76, 1405–1420. [Google Scholar] [PubMed]
- Wang, M.Y.; Zhou, Y.T.; Newgard, C.B.; Unger, R.H. A novel leptin receptor isoform in rat. FEBS Lett. 1996, 392, 87–90. [Google Scholar] [CrossRef]
- Stofkova, A. Leptin and adiponectin: From energy and metabolic dysbalance to inflammation and autoimmunity. Endocr. Regul. 2009, 43, 157–168. [Google Scholar] [PubMed]
- Gong, Y.; Ishida-Takahashi, R.; Villanueva, E.C.; Fingar, D.C.; Munzberg, H.; Myers, M.G., Jr. The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J. Biol. Chem. 2007, 282, 31019–31027. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G., Jr. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef] [PubMed]
- Bjorbaek, C.; Buchholz, R.M.; Davis, S.M.; Bates, S.H.; Pierroz, D.D.; Gu, H.; Neel, B.G.; Myers, M.G., Jr.; Flier, J.S. Divergent roles of SHP-2 in ERK activation by leptin receptors. J. Biol. Chem. 2001, 276, 4747–4755. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Zhang, L. Leptin modulates the expression of catabolic genes in rat nucleus pulposus cells through the mitogenactivated protein kinase and janus kinase 2/signal transducer and activator of transcription 3 pathways. Mol. Med. Rep. 2015, 12, 1761–1768. [Google Scholar] [PubMed]
- Cheng, S.E.; Lin, C.C.; Lee, I.T.; Hsu, C.K.; Kou, Y.R.; Yang, C.M. Cigarette smoke extract regulates cytosolic phospholipase A2 expression via NADPH oxidase/MAPKs/AP-1 and p300 in human tracheal smooth muscle cells. J. Cell. Biochem. 2011, 112, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.L.; Luo, S.F.; Hsieh, H.L.; Lee, I.T.; Hsiao, L.D.; Chen, Y.L.; Yang, C.M. Cytosolic phospholipase A2 induction and prostaglandin E2 release by interleukin-1β via the myeloid differentiation factor 88-dependent pathway and cooperation of p300, AKT, and NF-βB activity in human rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2011, 63, 2905–2917. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Lin, W.N.; Lin, C.C.; Luo, S.F.; Wang, J.S.; Pouyssegur, J.; Yang, C.M. Transcriptional regulation of VCAM-1 expression by tumor necrosis factor-alpha in human tracheal smooth muscle cells: Involvement of MAPKs, NF-κB, p300, and histone acetylation. J. Cell. Physiol. 2006, 207, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.N.; Luo, S.F.; Lee, C.W.; Wang, C.C.; Wang, J.S.; Yang, C.M. Involvement of MAPKs and NF-κB in LPS-induced VCAM-1 expression in human tracheal smooth muscle cells. Cell Signal. 2007, 19, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Lin, W.N.; Lee, C.W.; Lin, C.C.; Luo, S.F.; Wang, J.S.; Yang, C.M. Involvement of p42/p44 mapk, p38 mapk, jnk, and nf-kappab in il-1beta-induced VCAM-1 expression in human tracheal smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L227–L237. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.L.; Chen, Y.W.; Hsiao, L.D.; Chen, Y.L.; Yang, C.M. Heme oxygenase 1 attenuates interleukin-1β-induced cytosolic phospholipase A2 expression via a decrease in NADPH oxidase/reactive oxygen species/activator protein 1 activation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2012, 64, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.P.; Zhang, L.N.; Tian, G. Perivascular adipose tissue-derived leptin promotes vascular smooth muscle cell phenotypic switching via p38 mitogen-activated protein kinase in metabolic syndrome rats. Exp. Biol. Med. 2014, 239, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, X.; Liang, J.; Wu, W.K.; Yu, J.; Shen, J. Leptin downregulates aggrecan through the p38-adamst pathway in human nucleus pulposus cells. PLoS ONE 2014, 9, e109595. [Google Scholar] [CrossRef] [PubMed]
- Shalini, V.; Jayalekshmi, A.; Helen, A. Mechanism of anti-inflammatory effect of tricin, a flavonoid isolated from Njavara rice bran in LPS induced hPBMCs and carrageenan induced rats. Mol. Immunol. 2015, 66, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Dattaroy, D.; Pourhoseini, S.; Das, S.; Alhasson, F.; Seth, R.K.; Nagarkatti, M.; Michelotti, G.A.; Diehl, A.M.; Chatterjee, S. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G298–G312. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Tu, L.; Thuillet, R.; le Hiress, M.; Phan, C.; Ricard, N.; Nadaud, S.; Fadel, E.; Humbert, M.; Guignabert, C. Leptin signalling system as a target for pulmonary arterial hypertension therapy. Eur. Respir. J. 2015, 45, 1066–1080. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.M.; Jeong, B.H.; Woo, S.Y.; Kim, S.Y.; Kim, H.; Lee, J.H.; Lim, S.Y.; Rhee, C.K.; Yoo, K.H.; Lee, J.H.; et al. Association of plasma adipokines with chronic obstructive pulmonary disease severity and progression. Ann. Am. Thorac. Soc. 2015, 12, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Mendivil, C.O.; Koziel, H.; Brain, J.D. Metabolic hormones, apolipoproteins, adipokines, and cytokines in the alveolar lining fluid of healthy adults: Compartmentalization and physiological correlates. PLoS ONE 2015, 10, e0123344. [Google Scholar] [CrossRef] [PubMed]
- Ubags, N.D.; Vernooy, J.H.; Burg, E.; Hayes, C.; Bement, J.; Dilli, E.; Zabeau, L.; Abraham, E.; Poch, K.R.; Nick, J.A.; et al. The role of leptin in the development of pulmonary neutrophilia in infection and acute lung injury. Crit. Care Med. 2014, 42, e143–e151. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, P.; Canetti, C.; Gottschalk, A.; Tithof, P.K.; Peters-Golden, M. Leptin augments alveolar macrophage leukotriene synthesis by increasing phospholipase activity and enhancing group IVC iPLA2 (cPLA2γ) protein expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L497–L502. [Google Scholar] [CrossRef] [PubMed]
- Suzukawa, M.; Koketsu, R.; Baba, S.; Igarashi, S.; Nagase, H.; Yamaguchi, M.; Matsutani, N.; Kawamura, M.; Shoji, S.; Hebisawa, A.; et al. Leptin enhances ICAM-1 expression, induces migration and cytokine synthesis, and prolongs survival of human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L801–L811. [Google Scholar] [PubMed]
- Miyazaki, T.; Bub, J.D.; Iwamoto, Y. c-Jun NH 2-terminal kinase mediates leptin-stimulated androgen-independent prostate cancer cell proliferation via signal transducer and activator of transcription 3 and Akt. Biochim. Biophys. Acta 2008, 1782, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Onuma, M.; Bub, J.D.; Rummel, T.L.; Iwamoto, Y. Prostate cancer cell-adipocyte interaction: Leptin mediates androgen-independent prostate cancer cell proliferation through c-jun NH 2-terminal kinase. J. Biol. Chem. 2003, 278, 42660–42667. [Google Scholar] [CrossRef] [PubMed]
- Somasundar, P.; Frankenberry, K.A.; Skinner, H.; Vedula, G.; McFadden, D.W.; Riggs, D.; Jackson, B.; Vangilder, R.; Hileman, S.M.; Vona-Davis, L.C. Prostate cancer cell proliferation is influenced by leptin. J. Surg. Res. 2004, 118, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Hsieh, H.L.; Sun, C.C.; Lin, C.C.; Luo, S.F.; Yang, C.M. Il-1 β induces urokinase-plasminogen activator expression and cell migration through PKC α, JNK1/2, and NF-κB in A549 cells. J. Cell. Physiol. 2009, 219, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Haijiang, W.; Yonghong, S.; Xinna, D.; Ye, S.; Chunyang, D.; Jinying, W.; Yunzhuo, R.; Ming, W.; Yanjuan, H.; Huijun, D. Inhibition of c-Src/p38 MAPK pathway ameliorates renal tubular epithelial cells apoptosis in db/db mice. Mol. Cell. Endocrinol. 2015. [Google Scholar] [CrossRef]
- Fang, Q.; Deng, L.; Wang, L.; Zhang, Y.; Weng, Q.; Yin, H.; Pan, Y.; Tong, C.; Wang, J.; Liang, G. Inhibition of MAPKs/NF-κB-dependent inflammation by a novel chalcone protects kidney from high fat diet-induced injuries in mice. J. Pharmacol. Exp. Ther. 2015, 355, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Xue, J.; Yang, Y.; Zhu, H.; Chen, F.; Wang, J.; Lou, G.; Liu, Y.; Shi, Y.; Yu, Y.; et al. Isoliquiritigenin inhibits interferon-γ-inducible genes expression in hepatocytes through down-regulating activation of JAK1/STAT1, IRF3/MyD88, ERK/MAPK, JNK/MAPK and PI3K/Akt signaling pathways. Cell. Physiol. Biochem. 2015, 37, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.S.; Hong, J.S.; Kim, S.W.; Koh, J.M.; An, C.S.; Choi, J.Y.; Cheng, S.L. Leptin induces apoptosis via ERK/cPLA2/cytochrome c pathway in human bone marrow stromal cells. J. Biol. Chem. 2003, 278, 21920–21929. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.J.; Lau, K.N.; Johnson, S.; Martinie, J.B.; Iannitti, D.A.; McKillop, I.H.; Sindram, D. Leptin inhibits hepatocellular carcinoma proliferation via p38-MAPK-dependent signalling. HPB 2011, 13, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.P.; Behar, M.; Birnbaum, H.A.; Hoffmann, A.; Wright, P.E.; Ghosh, G. Analysis of the RelA: CBP/p300 interaction reveals its involvement in NF-κB-driven transcription. PLoS Biol. 2013, 11, e1001647. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Hashimura, M.; Mastumoto, T.; Tazo, Y.; Inoue, H.; Kuwata, T.; Saegusa, M. Transcriptional upregulation of HIF-1α by NF-κB/p65 and its associations with β-catenin/p300 complexes in endometrial carcinoma cells. Lab. Investig. 2013, 93, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.H.; Kim, D.H.; Na, H.K.; Kim, J.H.; Kim, H.N.; Haegeman, G.; Surh, Y.J. Genistein inhibits phorbol ester-induced NF-κB transcriptional activity and COX-2 expression by blocking the phosphorylation of p65/RelA in human mammary epithelial cells. Mutat. Res. 2014, 768, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Lin, C.C.; Lin, W.N.; Wu, W.L.; Hsiao, L.D.; Yang, C.M. Lung inflammation caused by adenosine-5′-triphosphate is mediated via Ca2+/PKCs-dependent COX-2/PGE2 induction. Int. J. Biochem. Cell Biol. 2013, 45, 1657–1668. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.N.; Luo, S.F.; Lin, C.C.; Hsiao, L.D.; Yang, C.M. Differential involvement of PKC-dependent MAPKs activation in lipopolysaccharide-induced AP-1 expression in human tracheal smooth muscle cells. Cell Signal. 2009, 21, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, P.-S.; Wu, C.-S.; Chang, J.-F.; Lin, W.-N. Leptin Promotes cPLA2 Gene Expression through Activation of the MAPK/NF-κB/p300 Cascade. Int. J. Mol. Sci. 2015, 16, 27640-27658. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161126045
Hsu P-S, Wu C-S, Chang J-F, Lin W-N. Leptin Promotes cPLA2 Gene Expression through Activation of the MAPK/NF-κB/p300 Cascade. International Journal of Molecular Sciences. 2015; 16(11):27640-27658. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161126045
Chicago/Turabian StyleHsu, Pei-Sung, Chi-Sheng Wu, Jia-Feng Chang, and Wei-Ning Lin. 2015. "Leptin Promotes cPLA2 Gene Expression through Activation of the MAPK/NF-κB/p300 Cascade" International Journal of Molecular Sciences 16, no. 11: 27640-27658. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161126045