
Does Lysosomial Acid Lipase Reduction Play a Role in Adult Non-Alcoholic Fatty Liver Disease?

 , and
, and
Abstract
:1. Introduction
2. Non-Alcoholic Fatty Liver Disease and Cardiovascular Disease
PNPLA3 and Non-Metabolic NAFLD
3. Clinical Presentations of Genetic LAL Deficiency
4. Liver Histology in LAL Deficiency
5. The Role of LAL in Lipid Metabolism
6. The Role of LAL in Atherosclerosis
7. Who Should Be Tested for LAL Activity?

{kind=link}
| Who Should Be Tested for LAL Activity? |
|---|
| Patients with unexplained: |
| •Liver Dysfunction (≥1 of the following) |
| Persistent elevation of ALT |
| Presence of hepatomegaly |
| Hepatic steatosis |
| AND/OR |
| •Dislipidemia (≥1 of the following) |
| High LDL-C (≥160 mg/dL–4.1 mmol/L) |
| Low HDL-C (≤40 mg/dL–1.0 mmol/L in males; ≤50 mg/dL–1.3 mmol/L in females) |
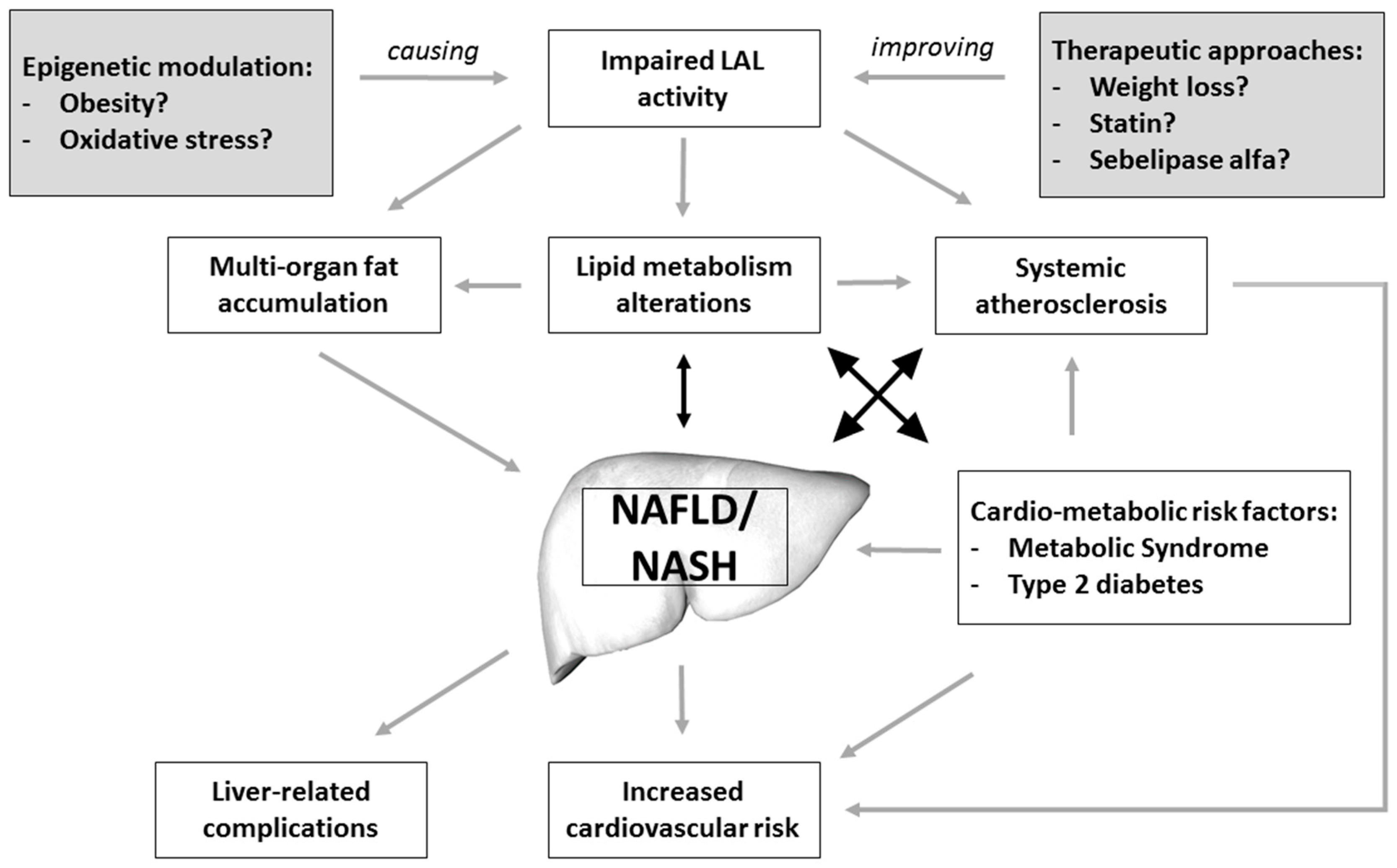
8. Current Research Status on LAL Activity and NAFLD
9. Future Directions

Author Contributions
Conflicts of Interest
References
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Baratta, F.; Carnevale, R.; Cangemi, R.; del Ben, M.; Bucci, T.; Polimeni, L.; Labbadia, G.; Nocella, C.; Scardella, L.; et al. Similar reduction of cholesterol-adjusted vitamin e serum levels in simple steatosis and non-alcoholic steatohepatitis. Clin. Transl. Gastroenterol. 2015, 6, e113. [Google Scholar] [CrossRef] [PubMed]
- Kemmer, N.; Neff, G.W.; Franco, E.; Osman-Mohammed, H.; Leone, J.; Parkinson, E.; Cece, E.; Alsina, A. Nonalcoholic fatty liver disease epidemic and its implications for liver transplantation. Transplantation 2013, 96, 860–862. [Google Scholar] [CrossRef] [PubMed]
- Soderberg, C.; Stal, P.; Askling, J.; Glaumann, H.; Lindberg, G.; Marmur, J.; Hultcrantz, R. Decreased survival of subjects with elevated liver function tests during a 28-year follow-up. Hepatology 2010, 51, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Lonardo, A.; Bonapace, S.; Byrne, C.D.; Loria, P.; Targher, G. Risk of cardiovascular, cardiac and arrhythmic complications in patients with non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1724–1745. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, L.S.; Curzen, N.P.; Calder, P.C.; Byrne, C.D. Non-alcoholic fatty liver disease: A new and important cardiovascular risk factor? Eur. Heart J. 2012, 33, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Del Ben, M.; Baratta, F.; Polimeni, L.; Angelico, F. Non-alcoholic fatty liver disease and cardiovascular disease: Epidemiological, clinical and pathophysiological evidences. Internal Emerg. Med. 2012, 7, S291–S296. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Castano, G.O.; Burgueno, A.L.; Rosselli, M.S.; Gianotti, T.F.; Mallardi, P.; Martino, J.S.; Pirola, C.J. Circulating levels and hepatic expression of molecular mediators of atherosclerosis in nonalcoholic fatty liver disease. Atherosclerosis 2010, 209, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Loffredo, L.; Perri, L.; Baratta, F.; Scardella, L.; Polimeni, L.; Pani, A.; Brancorsini, M.; Albanese, F.; Catasca, E.; et al. Relation of nonalcoholic fatty liver disease and framingham risk score to flow-mediated dilation in patients with cardiometabolic risk factors. Am. J. Cardiol. 2015, 115, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Padovani, R.; Rodella, S.; Tessari, R.; Zenari, L.; Day, C.; Arcaro, G. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 2007, 30, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Marra, F.; Marchesini, G. Increased risk of cardiovascular disease in non-alcoholic fatty liver disease: Causal effect or epiphenomenon? Diabetologia 2008, 51, 1947–1953. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Del Ben, M.; Polimeni, L.; Baratta, F.; Pastori, D.; Loffredo, L.; Angelico, F. Modern approach to the clinical management of non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 8341–8350. [Google Scholar] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Wood, K.L.; Miller, M.H.; Dillon, J.F. Systematic review of genetic association studies involving histologically confirmed non-alcoholic fatty liver disease. BMJ Open Gastroenterol. 2015, 2, e000019. [Google Scholar] [CrossRef] [PubMed]
- Del Ben, M.; Polimeni, L.; Brancorsini, M.; di Costanzo, A.; D'Erasmo, L.; Baratta, F.; Loffredo, L.; Pastori, D.; Pignatelli, P.; Violi, F.; et al. Non-alcoholic fatty liver disease, metabolic syndrome and patatin-like phospholipase domain-containing protein3 gene variants. Eur. J. Intern. Med. 2014, 25, 566–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Corte, C.; Fintini, D.; Giordano, U.; Cappa, M.; Brufani, C.; Majo, F.; Mennini, C.; Nobili, V. Fatty liver and insulin resistance in children with hypobetalipoproteinemia: The importance of aetiology. Clin. Endocrinol. 2013, 79, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Thelwall, P.E.; Smith, F.E.; Leavitt, M.C.; Canty, D.; Hu, W.; Hollingsworth, K.G.; Thoma, C.; Trenell, M.I.; Taylor, R.; Rutkowski, J.V.; et al. Hepatic cholesteryl ester accumulation in lysosomal acid lipase deficiency: Non-invasive identification and treatment monitoring by magnetic resonance. J. Hepatol. 2013, 59, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, S.; Wiebusch, H.; Jansen-Rust, M.; Rust, S.; Schulte, H.; Berger, K.; Pisciotta, L.; Bertolini, S.; Funke, H.; Seedorf, U.; et al. Heterozygosity for lysosomal acid lipase E8SJM mutation and serum lipid concentrations. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Pisciotta, L.; Fresa, R.; Bellocchio, A.; Pino, E.; Guido, V.; Cantafora, A.; di Rocco, M.; Calandra, S.; Bertolini, S. Cholesteryl ester storage disease (CESD) due to novel mutations in the LIPA gene. Mol. Genet. Metab. 2009, 97, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.L.; Hulkova, H.; Bialer, M.G.; Desnick, R.J. Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease. J. Hepatol. 2013, 58, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Z.; Guardamagna, O.; Nair, D.; Soran, H.; Hovingh, K.; Bertolini, S.; Jones, S.; Coric, M.; Calandra, S.; Hamilton, J.; et al. Lysosomal acid lipase deficiency—An under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014, 235, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Hulkova, H.; Elleder, M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology 2012, 60, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Fasano, T.; Pisciotta, L.; Bocchi, L.; Guardamagna, O.; Assandro, P.; Rabacchi, C.; Zanoni, P.; Filocamo, M.; Bertolini, S.; Calandra, S. Lysosomal lipase deficiency: Molecular characterization of eleven patients with wolman or cholesteryl ester storage disease. Mol. Genet. Metab. 2012, 105, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Dubland, J.A.; Francis, G.A. Lysosomal acid lipase: At the crossroads of normal and atherogenic cholesterol metabolism. Front. Cell Dev. Biol. 2015, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Keaney, J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Hakala, J.K.; Oksjoki, R.; Laine, P.; Du, H.; Grabowski, G.A.; Kovanen, P.T.; Pentikainen, M.O. Lysosomal enzymes are released from cultured human macrophages, hydrolyze LDL in vitro, and are present extracellularly in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.; Jones, I.; Srivastava, R.; Galloway, P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor lalistat 2. Clin. Chim. Acta 2012, 413, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Zschenker, O.; Illies, T.; Ameis, D. Overexpression of lysosomal acid lipase and other proteins in atherosclerosis. J. Biochem. 2006, 140, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Baratta, F.; Pastori, D.; del Ben, M.; Polimeni, L.; Labbadia, G.; di Santo, S.; Piemonte, F.; Tozzi, G.; Violi, F.; Angelico, F. Reduced lysosomal acid lipase activity in adult patients with non-alcoholic fatty liver disease. EBioMedicine 2015, 2, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Balwani, M.; Feillet, F.; Baric, I.; Burrow, T.A.; Camarena Grande, C.; Coker, M.; Deegan, P.; Consuelo-Sanchez, A.; di Rocco, M.; et al. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. N. Engl. J. Med. 2015, 373, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baratta, F.; Pastori, D.; Polimeni, L.; Tozzi, G.; Violi, F.; Angelico, F.; Del Ben, M. Does Lysosomial Acid Lipase Reduction Play a Role in Adult Non-Alcoholic Fatty Liver Disease? Int. J. Mol. Sci. 2015, 16, 28014-28021. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226085
Baratta F, Pastori D, Polimeni L, Tozzi G, Violi F, Angelico F, Del Ben M. Does Lysosomial Acid Lipase Reduction Play a Role in Adult Non-Alcoholic Fatty Liver Disease? International Journal of Molecular Sciences. 2015; 16(12):28014-28021. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226085
Chicago/Turabian StyleBaratta, Francesco, Daniele Pastori, Licia Polimeni, Giulia Tozzi, Francesco Violi, Francesco Angelico, and Maria Del Ben. 2015. "Does Lysosomial Acid Lipase Reduction Play a Role in Adult Non-Alcoholic Fatty Liver Disease?" International Journal of Molecular Sciences 16, no. 12: 28014-28021. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226085