
Protective Role of Proton-Sensing TDAG8 in Lipopolysaccharide-Induced Acute Lung Injury

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
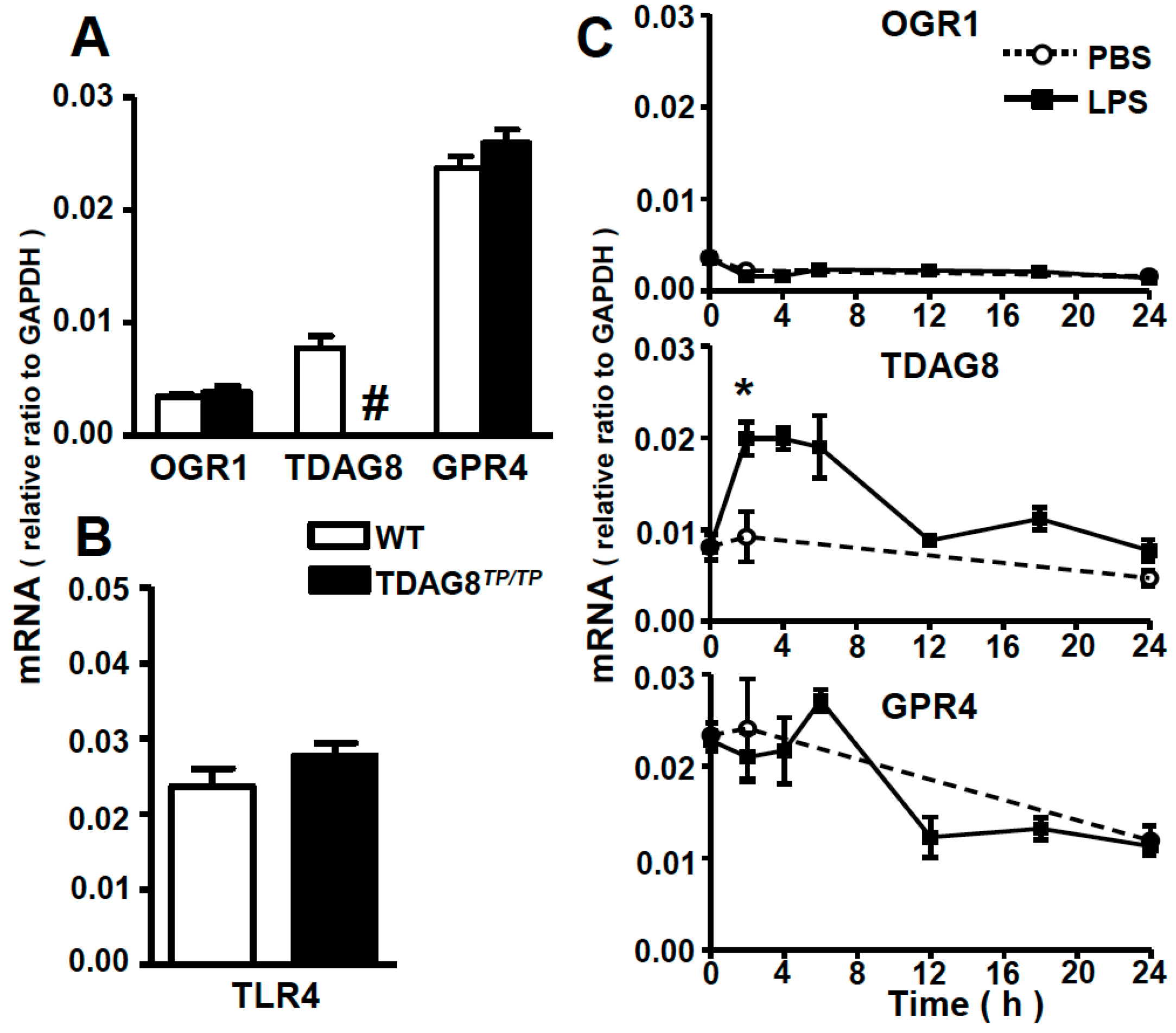
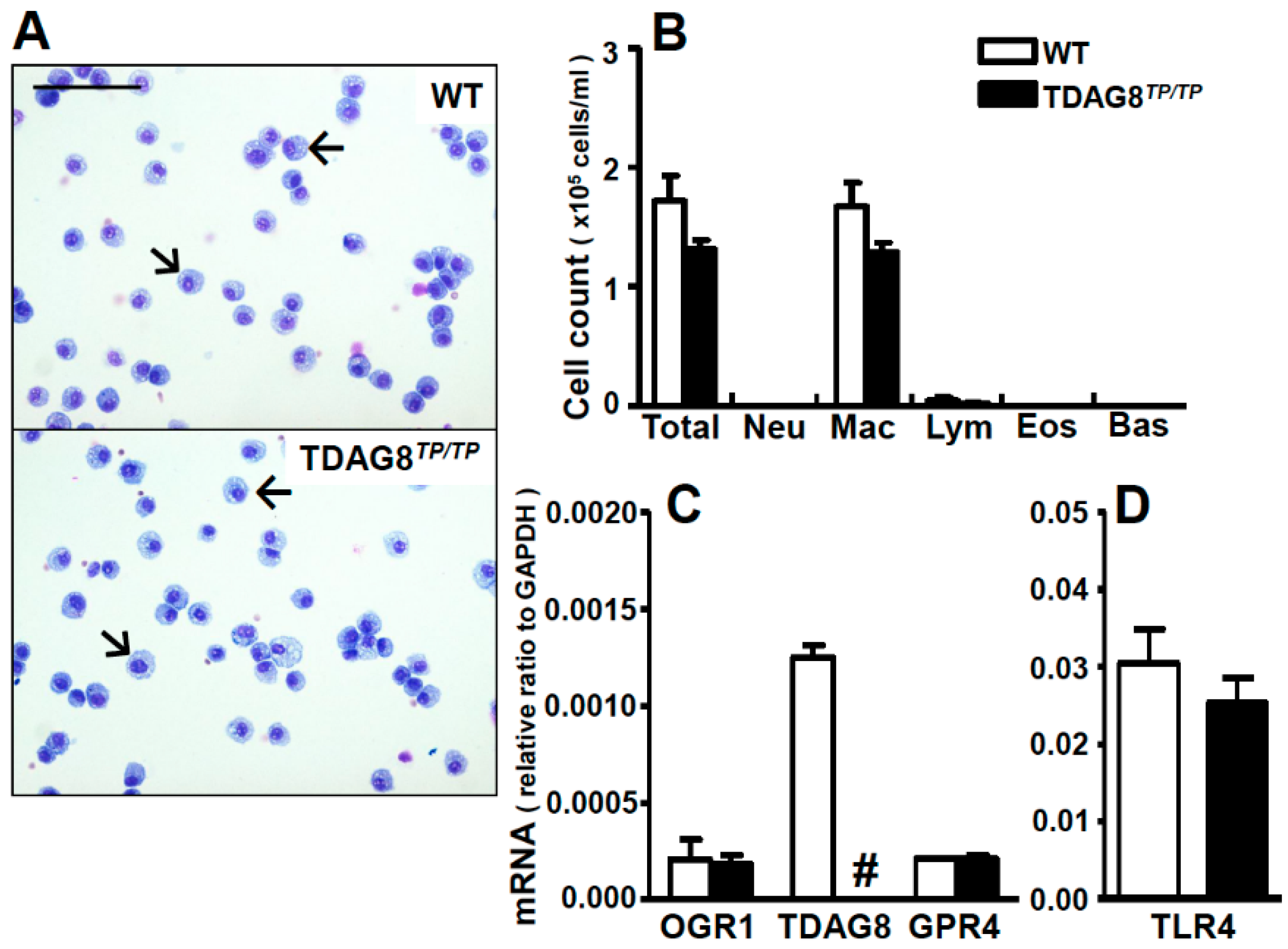
2.1. Expression Profiles of Proton-Sensing GPCRs in Lung Tissues and Bronchoalveolar Lavage (BAL) Fluid Cells


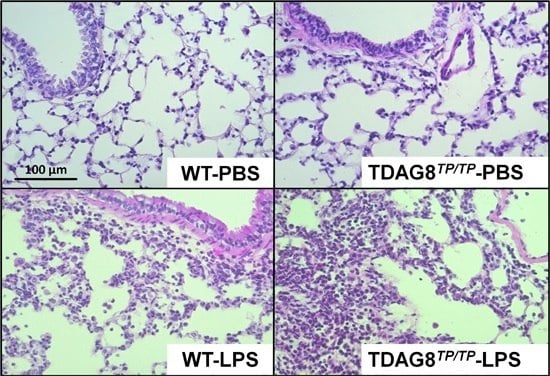
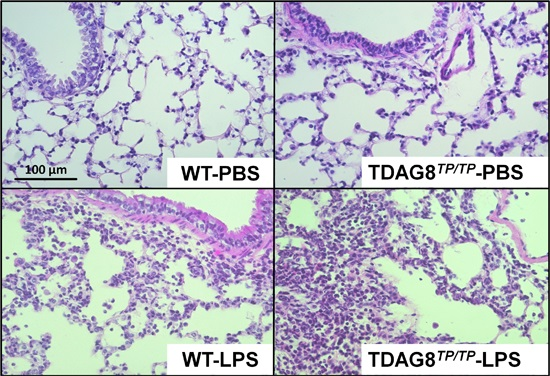
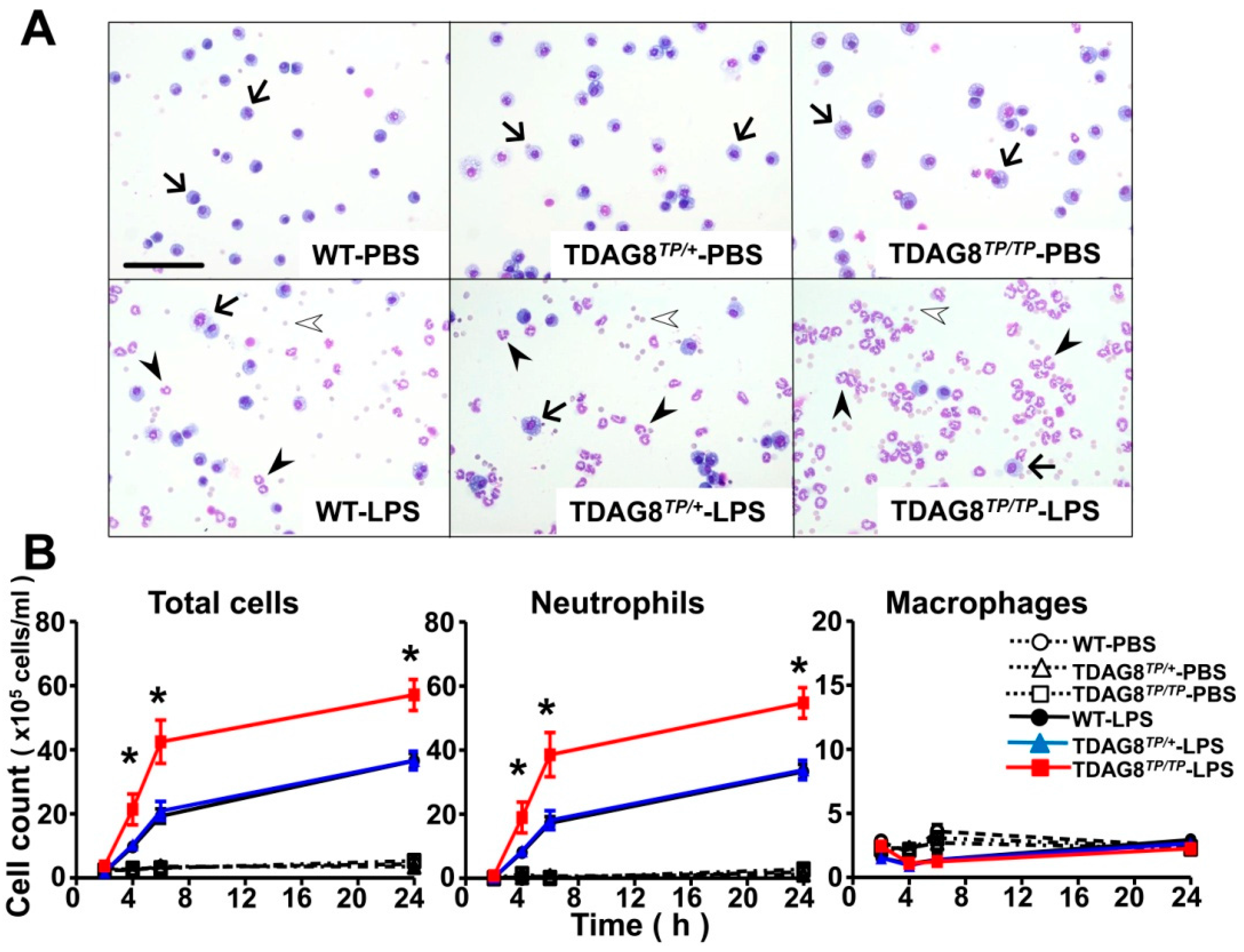
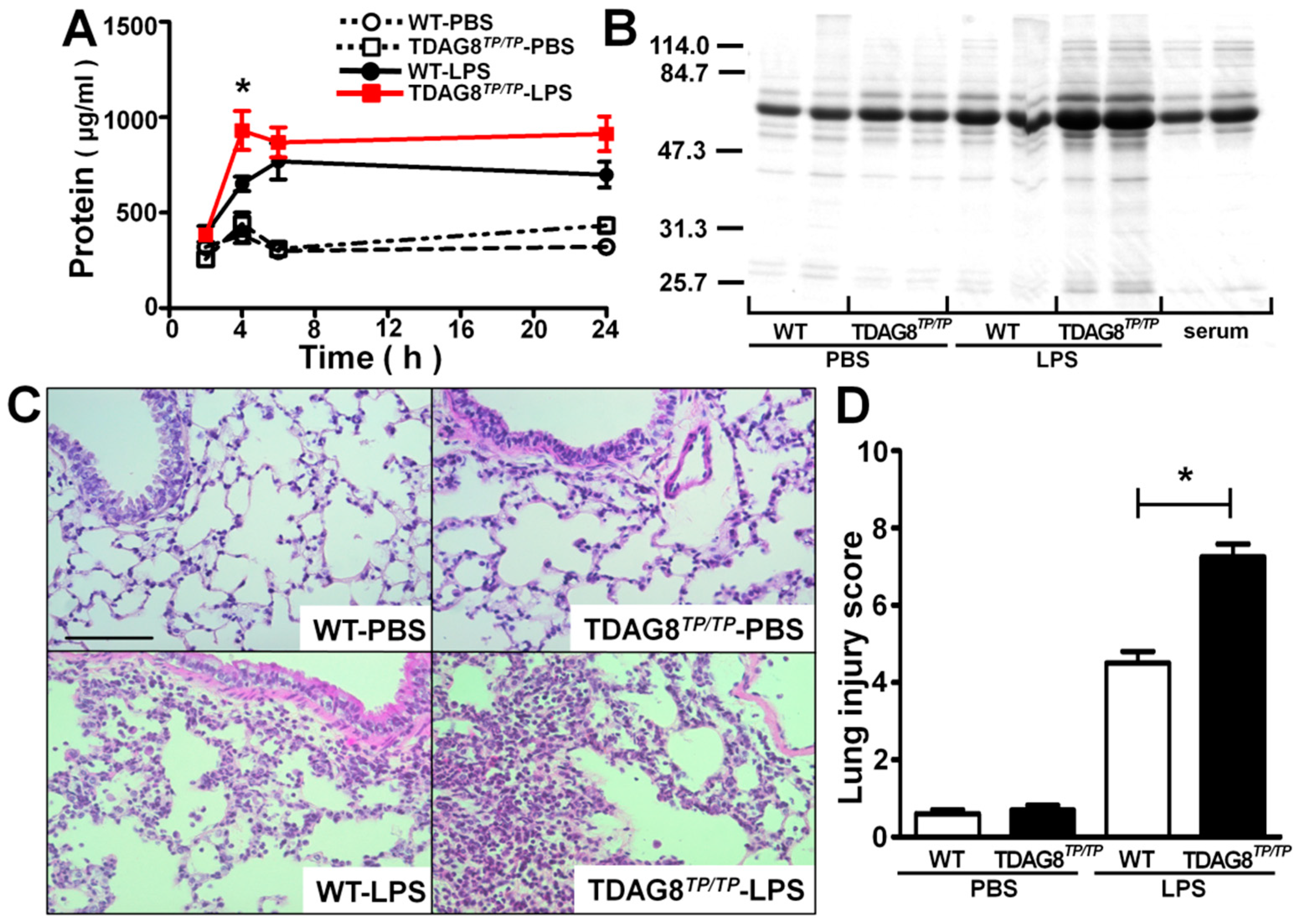
2.2. Enhancement of LPS-Induced Neutrophil Accumulation and Lung Injury by TDAG8 Deficiency


2.3. Involvement of TDAG8 in the Regulation of Cytokine and Chemokine Expression in Lungs

3. Discussion
4. Experimental Section
4.1. Animals
4.2. Murine Model of ALI
4.3. Bronchoalveolar Lavage (BAL)
4.4. Electrophoresis of BAL Fluids
4.5. Measurement of Cytokines and Chemokines
4.6. Histological Studies
4.7. Measurement of mRNAs
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grommes, J.; Soehnlein, O. Contribution of neutrophils to acute lung injury. Mol. Med. 2011, 17, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.E.; Chambers, R.C. The mercurial nature of neutrophils: Still an enigma in ARDS? Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.W.; Bidani, A.; Heming, T.A. Innate host defense of the lung: Effects of lung-lining fluid pH. Lung 2004, 182, 297–317. [Google Scholar] [CrossRef] [PubMed]
- Gessner, C.; Hammerschmidt, S.; Kuhn, H.; Seyfarth, H.J.; Sack, U.; Engelmann, L.; Schauer, J.; Wirtz, H. Exhaled breath condensate acidification in acute lung injury. Respir. Med. 2003, 97, 1188–1194. [Google Scholar] [CrossRef]
- Ludwig, M.G.; Vanek, M.; Guerini, D.; Gasser, J.A.; Jones, C.E.; Junker, U.; Hofstetter, H.; Wolf, R.M.; Seuwen, K. Proton-sensing g-protein-coupled receptors. Nature 2003, 425, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Okajima, F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell Signal. 2013, 25, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Mogi, C.; Okajima, F. Ionotropic and metabotropic proton-sensing receptors involved in airway inflammation in allergic asthma. Mediators Inflamm. 2014, 2014, 712962. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, H.; Zeng, Z.; Su, K.; Fan, P.; Shell, B.K.; Carter, K.C.; Li, Y. Cloning, characterization, and mapping of human homolog of mouse T-cell death-associated gene. DNA Cell. Biol. 1998, 17, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Mogi, C.; Tobo, M.; Tomura, H.; Murata, N.; He, X.D.; Sato, K.; Kimura, T.; Ishizuka, T.; Sasaki, T.; Sato, T.; et al. Involvement of proton-sensing TDAG8 in extracellular acidification-induced inhibition of proinflammatory cytokine production in peritoneal macrophages. J. Immunol. 2009, 182, 3243–3251. [Google Scholar] [CrossRef] [PubMed]
- Murata, N.; Mogi, C.; Tobo, M.; Nakakura, T.; Sato, K.; Tomura, H.; Okajima, F. Inhibition of superoxide anion production by extracellular acidification in neutrophils. Cell. Immunol. 2009, 259, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Kottyan, L.C.; Collier, A.R.; Cao, K.H.; Niese, K.A.; Hedgebeth, M.; Radu, C.G.; Witte, O.N.; Khurana Hershey, G.K.; Rothenberg, M.E.; Zimmermann, N. Eosinophil viability is increased by acidic pH in a cAMP- and GPR65-dependent manner. Blood 2009, 114, 2774–2782. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Mogi, C.; Hisada, T.; Nakakura, T.; Kamide, Y.; Ichimonji, I.; Tomura, H.; Tobo, M.; Sato, K.; Tsurumaki, H.; et al. Proton-sensing ovarian cancer G protein-coupled receptor 1 on dendritic cells is required for airway responses in a murine asthma model. PLoS ONE 2013, 8, e79985. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Frevert, C.W.; Martin, T.R. Animal models of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, 379–399. [Google Scholar] [CrossRef] [PubMed]
- Sender, V.; Stamme, C. Lung cell-specific modulation of LPS-induced TLR4 receptor and adaptor localization. Commun. Integr. Biol. 2014, 7, e29053. [Google Scholar] [CrossRef] [PubMed]
- Raoust, E.; Balloy, V.; Garcia-Verdugo, I.; Touqui, L.; Ramphal, R.; Chignard, M. Pseudomonas aeruginosa LPS or flagellin are sufficient to activate TLR-dependent signaling in murine alveolar macrophages and airway epithelial cells. PLoS ONE 2009, 4, e7259. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.H.; Johnson, M.D. Differential response of primary alveolar type I and type II cells to LPS stimulation. PLoS ONE 2013, 8, e55545. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Nanchal, R.; Husnain, F.; Audi, S.; Konduri, G.G.; Densmore, J.C.; Medhora, M.; Jacobs, E.R. Hypoxia preconditioning increases survival and decreases expression of toll-like receptor 4 in pulmonary artery endothelial cells exposed to lipopolysaccharide. Pulm. Circ. 2013, 3, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, J.W.; Chen, B.J.; Brass, D.M.; Berman, K.; Gunn, M.D.; Cook, D.N.; Schwartz, D.A. The critical role of hematopoietic cells in lipopolysaccharide-induced airway inflammation. Am. J. Respir. Crit. Care Med. 2005, 171, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Andonegui, G.; Bonder, C.S.; Green, F.; Mullaly, S.C.; Zbytnuik, L.; Raharjo, E.; Kubes, P. Endothelium-derived toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J. Clin. Investig. 2003, 111, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.D.; Glidden, D.V.; Eisner, M.D.; Parsons, P.E.; Ware, L.B.; Wheeler, A.; Korpak, A.; Thompson, B.T.; Chertow, G.M.; Matthay, M.A. Predictive and pathogenetic value of plasma biomarkers for acute kidney injury in patients with acute lung injury. Crit. Care Med. 2007, 35, 2755–2761. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, N.; Andres-Hernando, A.; Altmann, C.; Bhargava, R.; Bacalja, J.; Webb, R.G.; He, Z.; Edelstein, C.L.; Faubel, S. Circulating IL-6 mediates lung injury via CXCL1 production after acute kidney injury in mice. Am. J. Physiol. Ren. Physiol. 2012, 303, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, K.C.; Arndt, P.G.; Manos, E.J.; Jones, D.A.; Worthen, G.S. Microarray analysis of lipopolysaccharide-treated human neutrophils. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L663–L670. [Google Scholar] [CrossRef] [PubMed]
- Ichimonji, I.; Tomura, H.; Mogi, C.; Sato, K.; Aoki, H.; Hisada, T.; Dobashi, K.; Ishizuka, T.; Mori, M.; Okajima, F. Extracellular acidification stimulates IL-6 production and Ca2+ mobilization through proton-sensing OGR1 receptors in human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, S.; Ishizuka, T.; Yamada, H.; Kamide, Y.; Hisada, T.; Ichimonji, I.; Aoki, H.; Yatomi, M.; Komachi, M.; Tsurumaki, H.; et al. Extracellular acidification induces connective tissue growth factor production through proton-sensing receptor OGR1 in human airway smooth muscle cells. Biochem. Biophys. Res. Commun. 2011, 413, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Saxena, H.; Deshpande, D.A.; Tiegs, B.C.; Yan, H.; Battafarano, R.J.; Burrows, W.M.; Damera, G.; Panettieri, R.A.; Dubose, T.D., Jr.; An, S.S.; et al. The GPCR OGR1 (GPR68) mediates diverse signalling and contraction of airway smooth muscle in response to small reductions in extracellular pH. Br. J. Pharmacol. 2012, 166, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, Q.; Zhou, X.; Kolosov, V.P.; Perelman, J.M. Regulator of G-protein signaling 2 inhibits acid-induced mucin5AC hypersecretion in human airway epithelial cells. Respir. Physiol. Neurobiol. 2013, 185, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, Z.; Leffler, N.R.; Asch, A.S.; Chi, J.T.; Yang, L.V. Acidosis activation of the proton-sensing GPR4 receptor stimulates vascular endothelial cell inflammatory responses revealed by transcriptome analysis. PLoS ONE 2013, 8, e61991. [Google Scholar] [CrossRef] [PubMed]
- Tobo, A.; Tobo, M.; Nakakura, T.; Ebara, M.; Tomura, H.; Mogi, C.; Im, D.S.; Murata, N.; Kuwabara, A.; Ito, S.; et al. Characterization of imidazopyridine compounds as negative allosteric modulators of proton-sensing GPR4 in extracellular acidification-induced responses. PLoS ONE 2015, 10, e0129334. [Google Scholar] [CrossRef] [PubMed]
- Onozawa, Y.; Komai, T.; Oda, T. Activation of T cell death-associated gene 8 attenuates inflammation by negatively regulating the function of inflammatory cells. Eur. J. Pharmacol. 2011, 654, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Hikiji, H.; Endo, D.; Horie, K.; Harayama, T.; Akahoshi, N.; Igarashi, H.; Kihara, Y.; Yanagida, K.; Takeda, J.; Koji, T.; et al. TDAG8 activation inhibits osteoclastic bone resorption. FASEB J. 2014, 28, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Horie, K.; Yusa, K.; Yae, K.; Odajima, J.; Fischer, S.E.; Keng, V.W.; Hayakawa, T.; Mizuno, S.; Kondoh, G.; Ijiri, T.; et al. Characterization of sleeping beauty transposition and its application to genetic screening in mice. Mol. Cell. Biol. 2003, 23, 9189–9207. [Google Scholar] [CrossRef] [PubMed]
- Sammani, S.; Moreno-Vinasco, L.; Mirzapoiazova, T.; Singleton, P.A.; Chiang, E.T.; Evenoski, C.L.; Wang, T.; Mathew, B.; Husain, A.; Moitra, J.; et al. Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. Am. J. Respir. Cell Mol. Biol. 2010, 43, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Rittirsch, D.; Flierl, M.A.; Day, D.E.; Nadeau, B.A.; McGuire, S.R.; Hoesel, L.M.; Ipaktchi, K.; Zetoune, F.S.; Sarma, J.V.; Leng, L.; et al. Acute lung injury induced by lipopolysaccharide is independent of complement activation. J. Immunol. 2008, 180, 7664–7672. [Google Scholar] [CrossRef] [PubMed]
- Hisada, T.; Hellewell, P.G.; Teixeira, M.M.; Malm, M.G.; Salmon, M.; Huang, T.J.; Chung, K.F. Alpha4 integrin-dependent eotaxin induction of bronchial hyperresponsiveness and eosinophil migration in interleukin-5 transgenic mice. Am. J. Respir. Cell Mol. Biol. 1999, 20, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Hassoun, P.M.; Sammani, S.; McVerry, B.J.; Burne, M.J.; Rabb, H.; Pearse, D.; Tuder, R.M.; Garcia, J.G. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am. J. Respir. Crit. Care Med. 2004, 169, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, J.; Fukumoto, I.; Parthasarathy, P.T.; Cox, R.; Huynh, B.; Ramanathan, G.K.; Venugopal, R.B.; Allen-Gipson, D.S.; Lockey, R.F.; Kolliputi, N. NLRP3 deletion protects from hyperoxia-induced acute lung injury. Am. J. Physiol. Cell Physiol. 2013, 305, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Sue, R.D.; Belperio, J.A.; Burdick, M.D.; Murray, L.A.; Xue, Y.Y.; Dy, M.C.; Kwon, J.J.; Keane, M.P.; Strieter, R.M. CXCR2 is critical to hypoxia-indced lung injury. J. Immunol. 2004, 172, 3860–3868. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Kon, J.; Mogi, C.; Tobo, M.; Damirin, A.; Sato, K.; Komachi, M.; Malchinkhuu, E.; Murata, N.; Kimura, T.; et al. TDAG8 is a proton-sensing and psychosine-sensitive G-protein-coupled receptor. J. Biol. Chem. 2004, 279, 45626–45633. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Sato, K.; Tobo, A.; Mogi, C.; Tobo, M.; Murata, N.; Ishii, S.; Im, D.S.; Okajima, F. Inhibition of interleukin-1β production by extracellular acidification through the TDAG8/cAMP pathway in mouse microglia. J. Neurochem. 2014, 129, 683–695. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsurumaki, H.; Mogi, C.; Aoki-Saito, H.; Tobo, M.; Kamide, Y.; Yatomi, M.; Sato, K.; Dobashi, K.; Ishizuka, T.; Hisada, T.; et al. Protective Role of Proton-Sensing TDAG8 in Lipopolysaccharide-Induced Acute Lung Injury. Int. J. Mol. Sci. 2015, 16, 28931-28942. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226145
Tsurumaki H, Mogi C, Aoki-Saito H, Tobo M, Kamide Y, Yatomi M, Sato K, Dobashi K, Ishizuka T, Hisada T, et al. Protective Role of Proton-Sensing TDAG8 in Lipopolysaccharide-Induced Acute Lung Injury. International Journal of Molecular Sciences. 2015; 16(12):28931-28942. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226145
Chicago/Turabian StyleTsurumaki, Hiroaki, Chihiro Mogi, Haruka Aoki-Saito, Masayuki Tobo, Yosuke Kamide, Masakiyo Yatomi, Koichi Sato, Kunio Dobashi, Tamotsu Ishizuka, Takeshi Hisada, and et al. 2015. "Protective Role of Proton-Sensing TDAG8 in Lipopolysaccharide-Induced Acute Lung Injury" International Journal of Molecular Sciences 16, no. 12: 28931-28942. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226145