
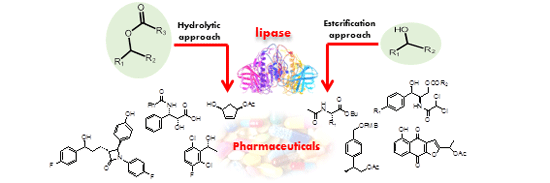
Recent Advances in Lipase-Mediated Preparation of Pharmaceuticals and Their Intermediates


 ,
,
Abstract
:
1. Introduction
2. Hydrolytic Approach
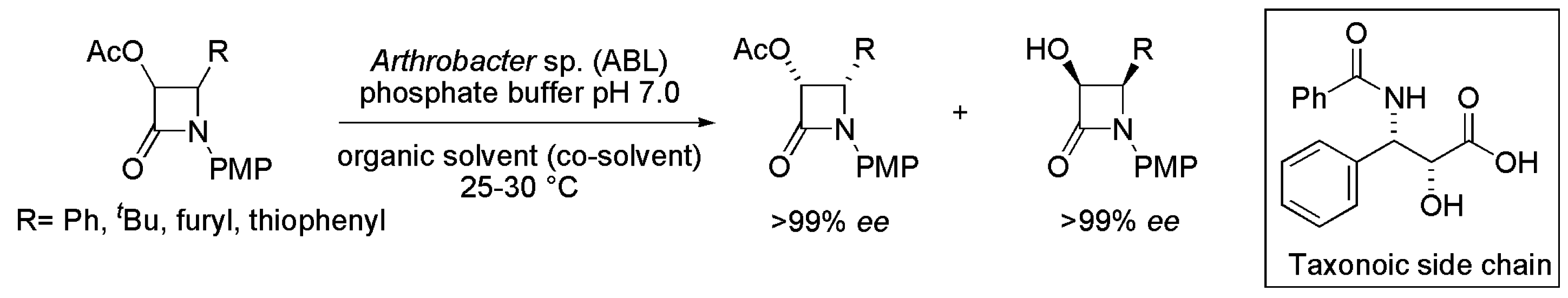
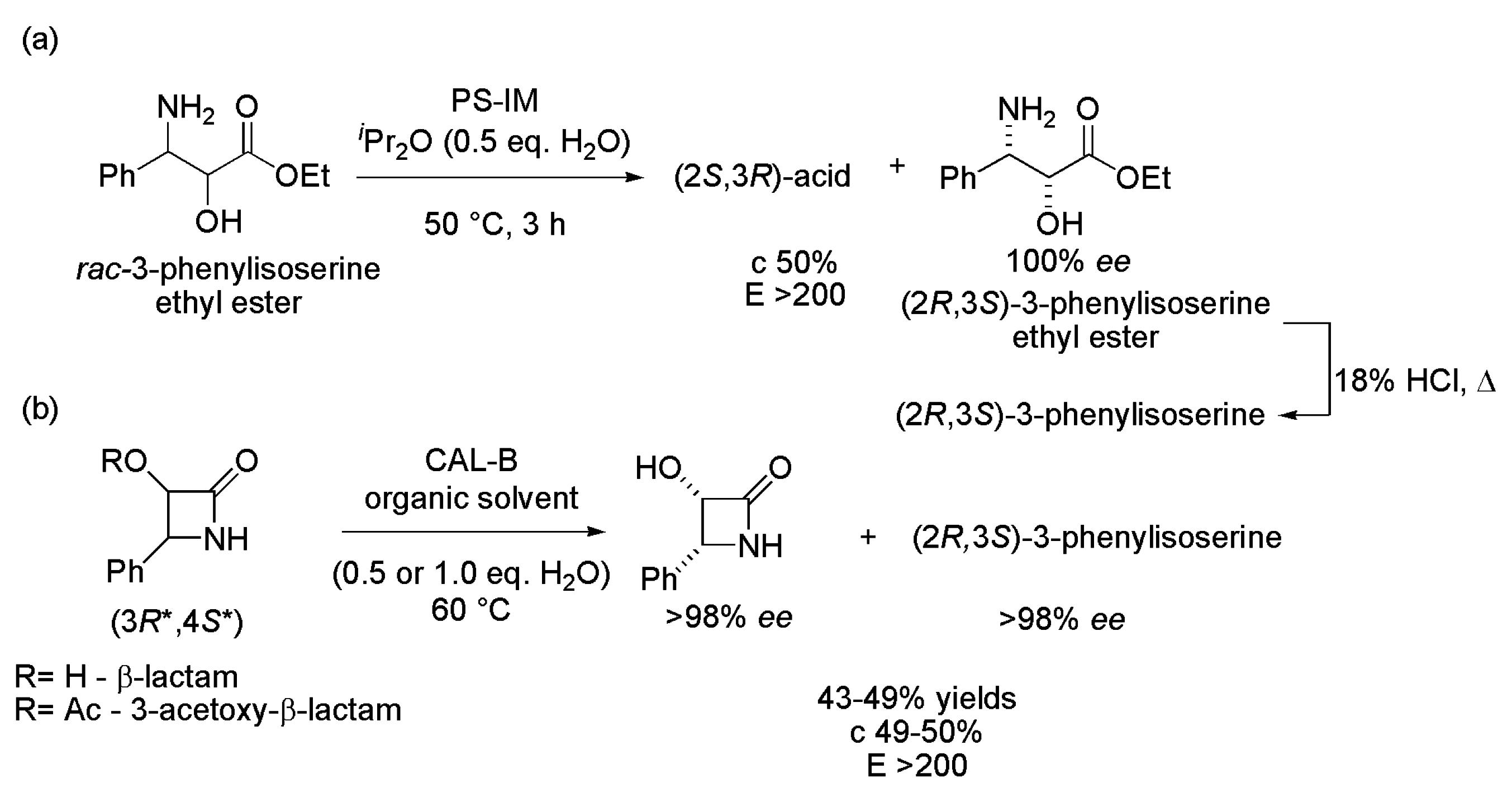
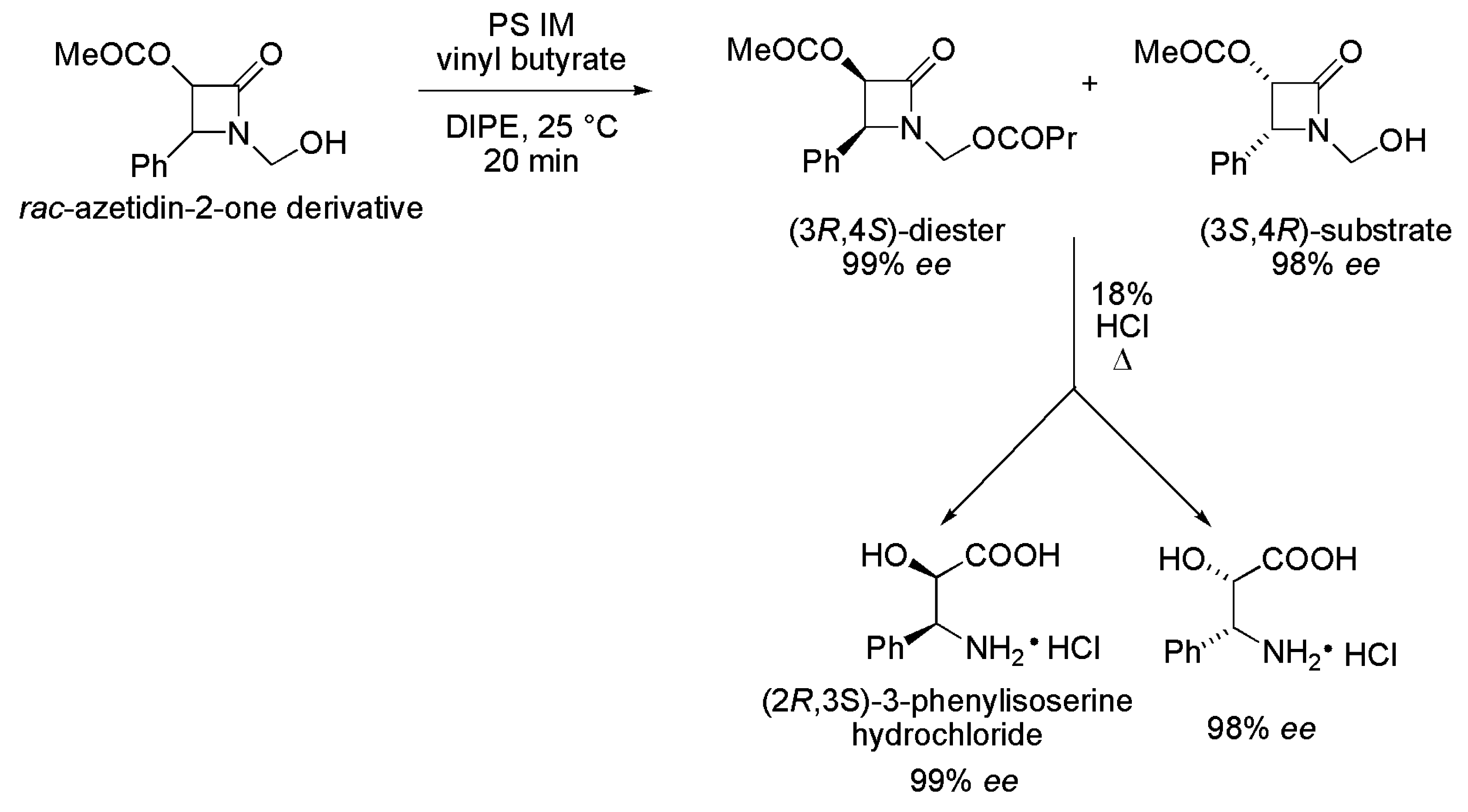
2.1. Key Intermediates of Paclitaxel Side Chain


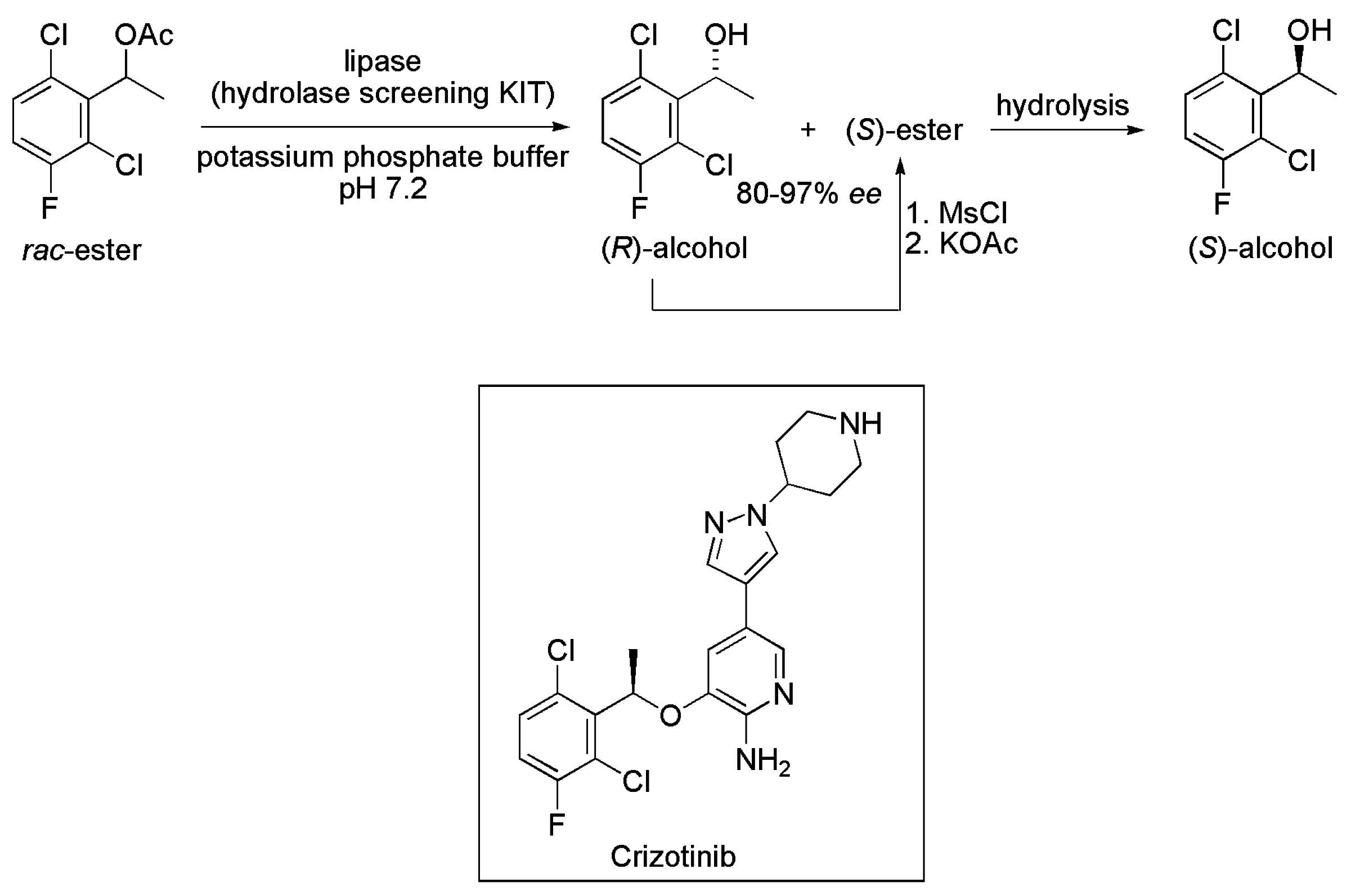
2.2. Key Intermediate of Crizotinib

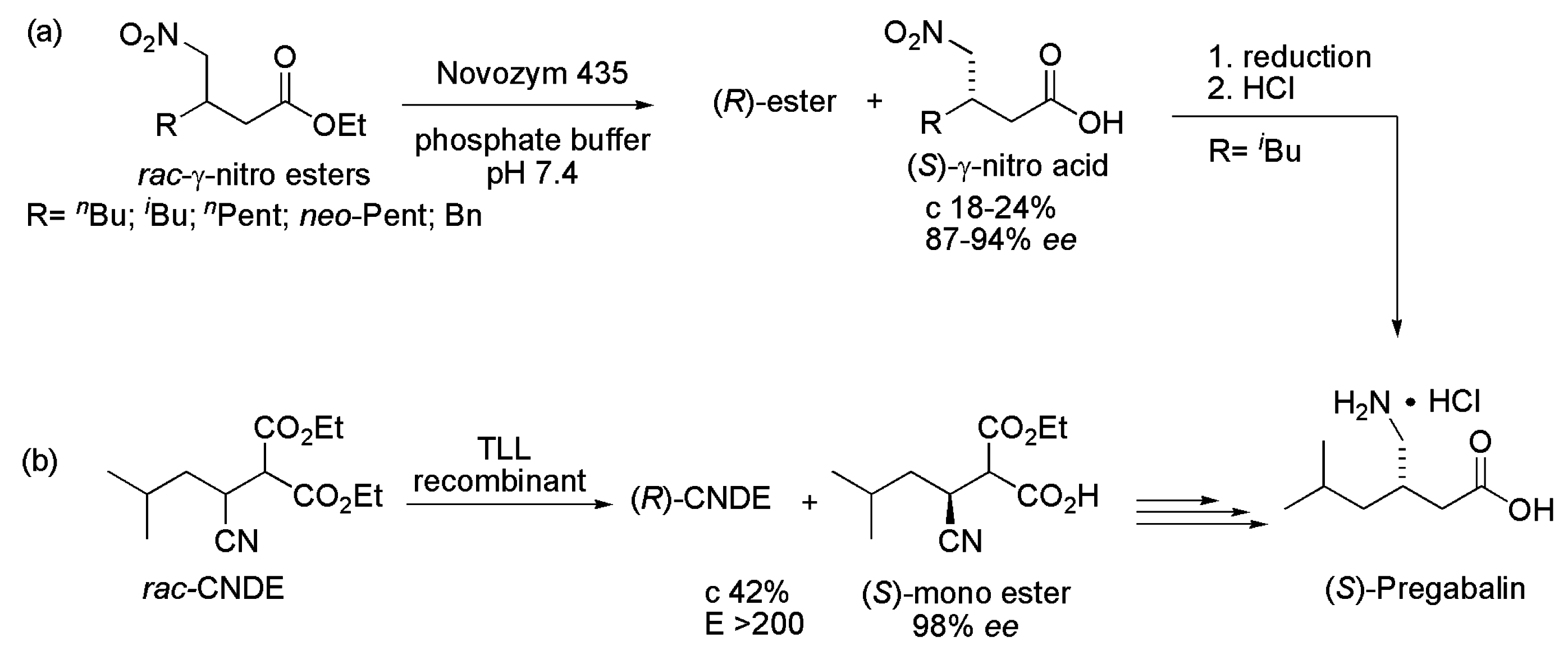
2.3. Pregabalin and Analogues


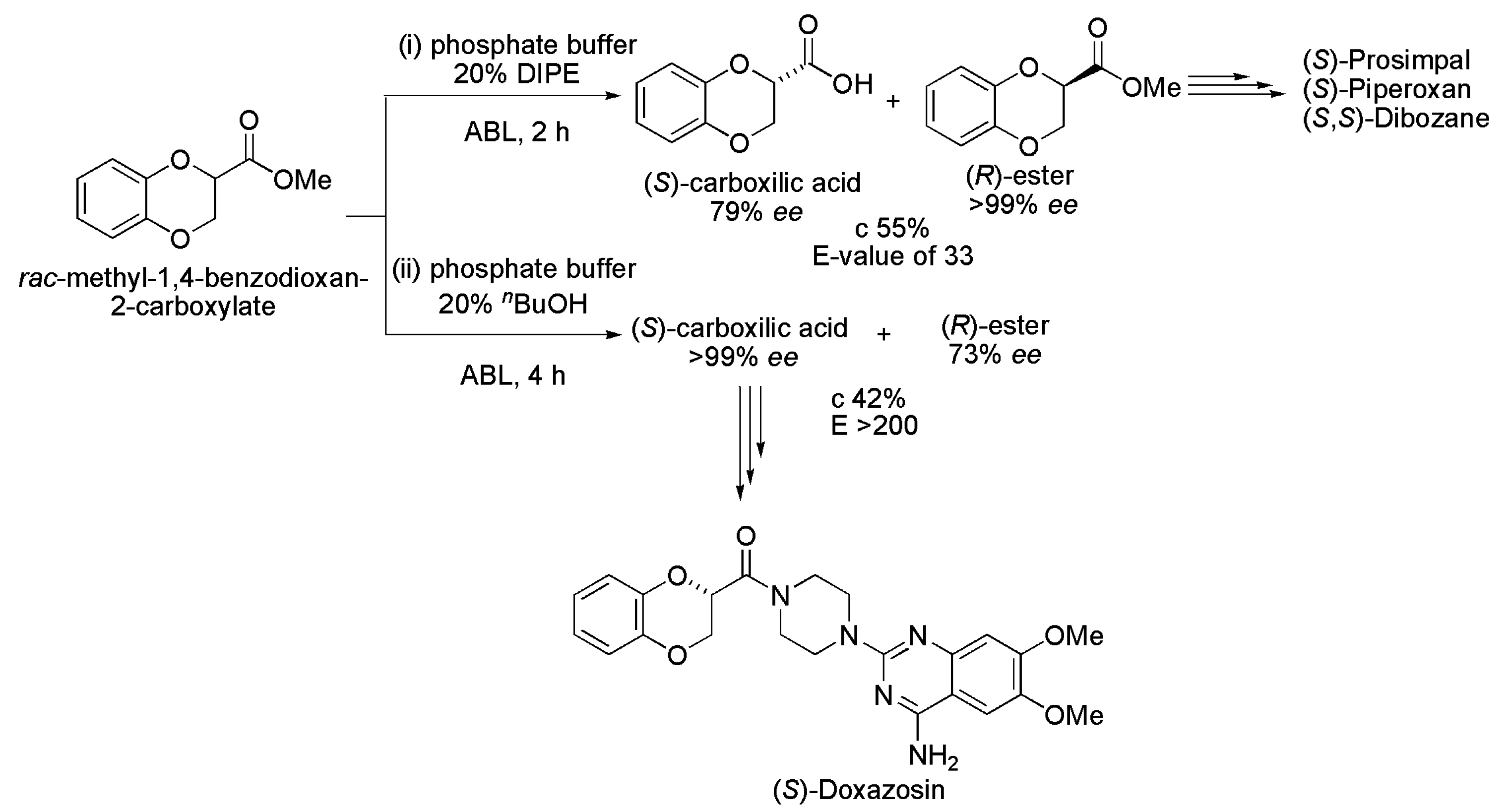
2.4. Prosimpal, Piperoxan, Dibozane and Doxazosin
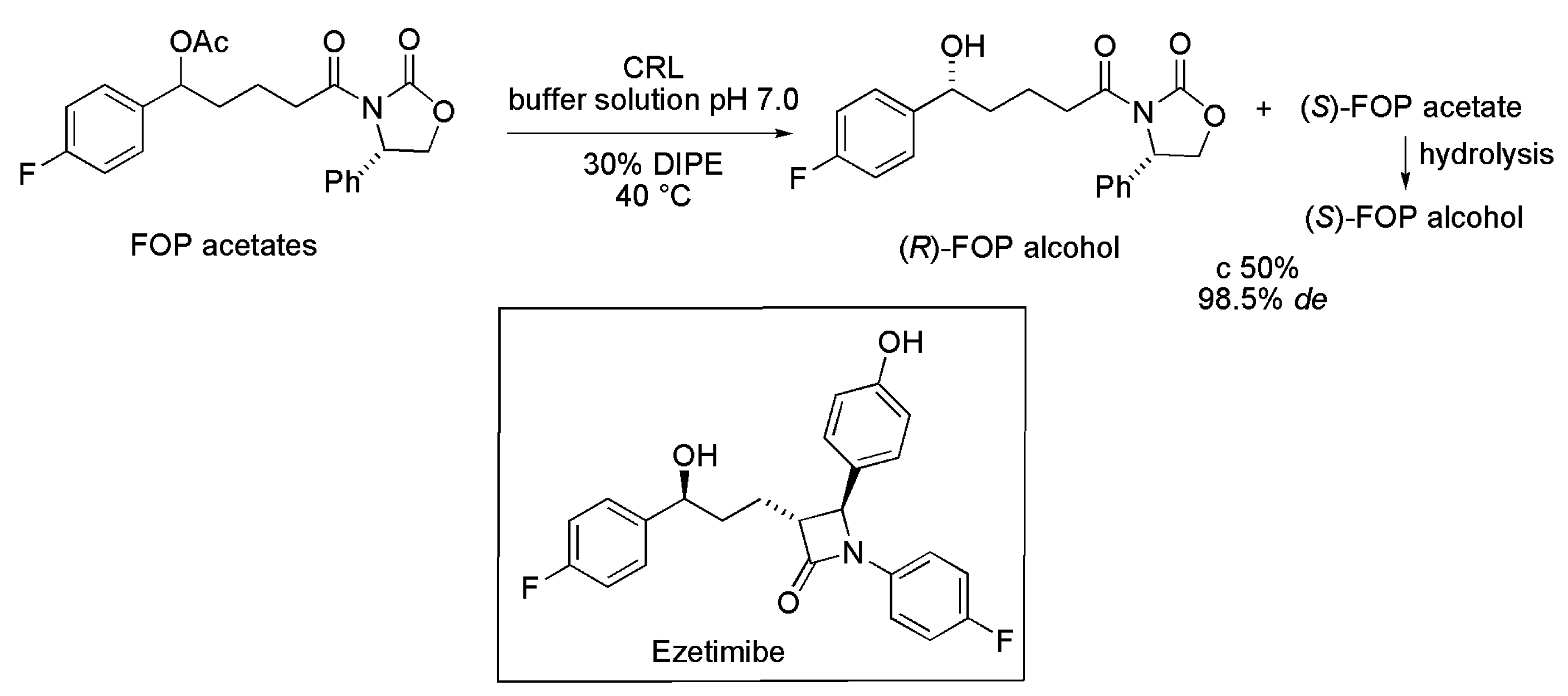
2.5. Key Intermediate of Ezetimibe

2.6. Key Intermediate of Hepatitis C Virus Protease Inhibitor

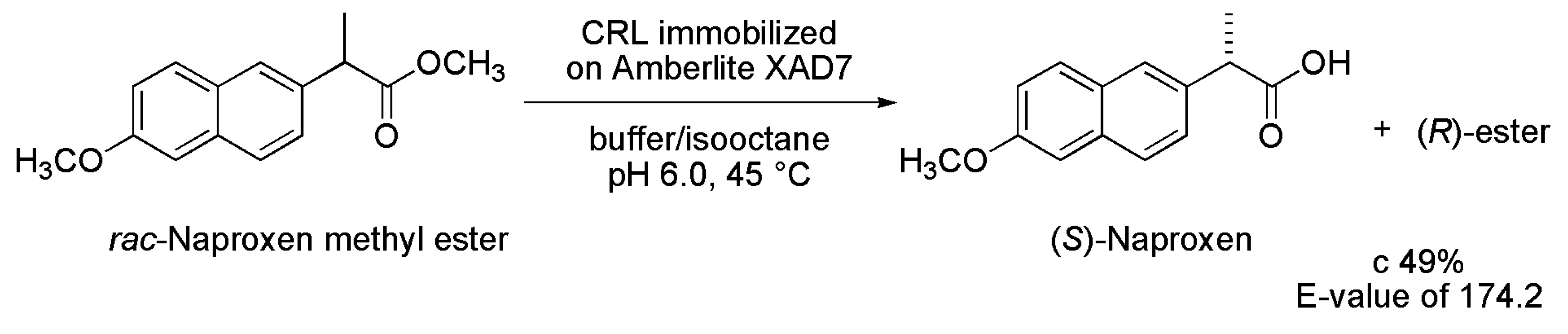
2.7. Naproxen

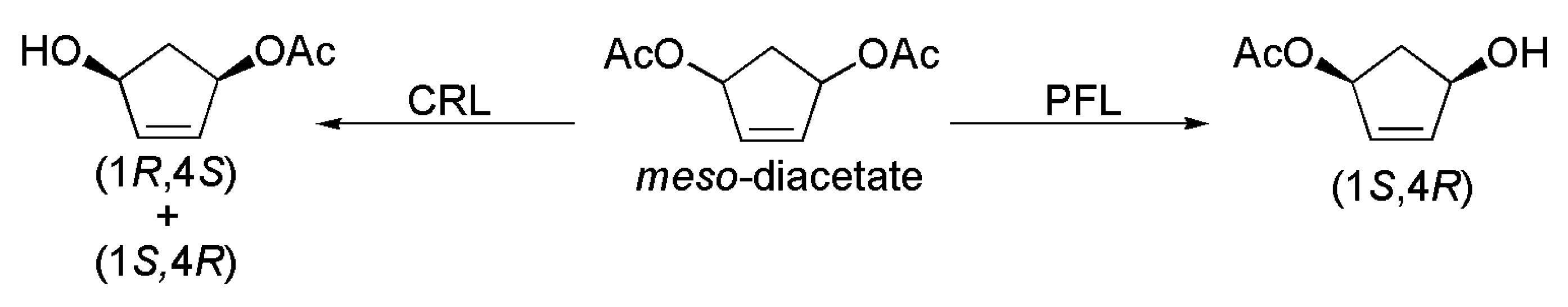
2.8. Key Intermediate of Prostaglandins, Prostacyclins and Thromboxane

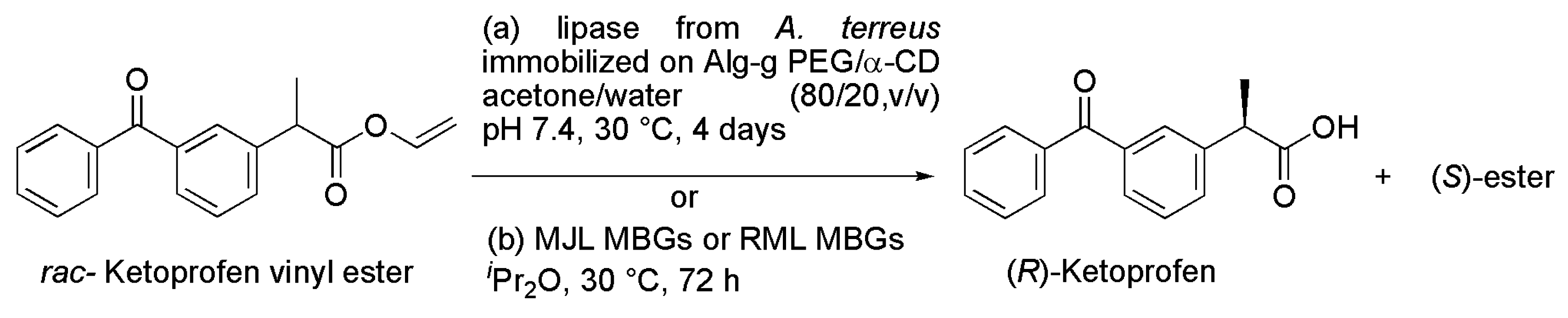
2.9. Ketoprofen


| Method | Enzyme | Conditions | Reference |
|---|---|---|---|
| a | lipase from A. terreus immobilized on Alg-g-PEG/α-CD, hollow spheres | acetone/water (80/20, v/v), pH 7.4, 30 °C, 4 days | [21] |
| b | MJL MBGs or RML MBGs | DIPE, 30 °C, 72 h | [22] |
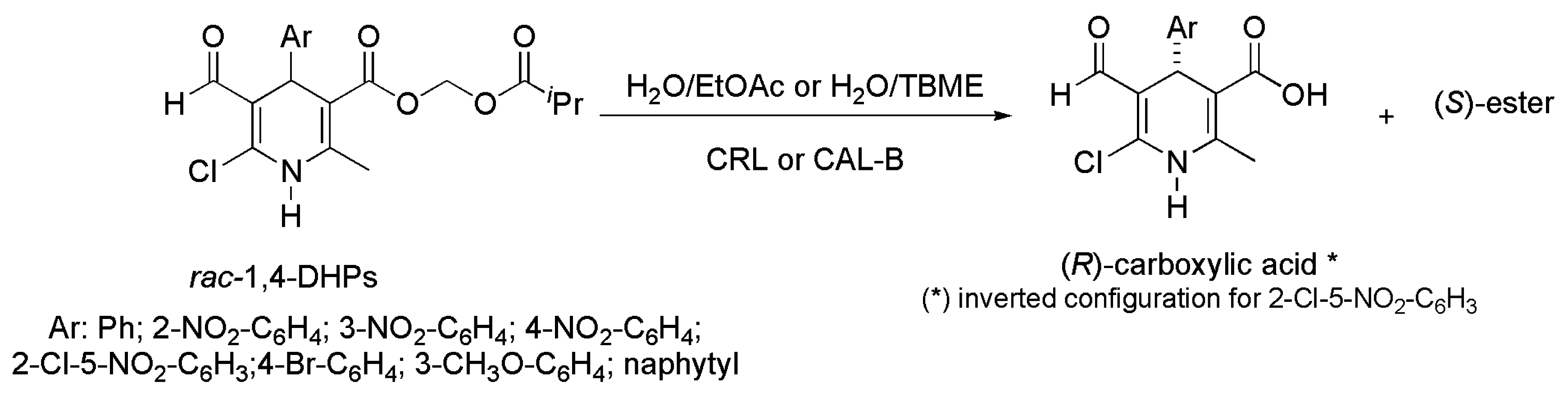
2.10. Dihydropyridine Derivatives

3. Esterification Approach
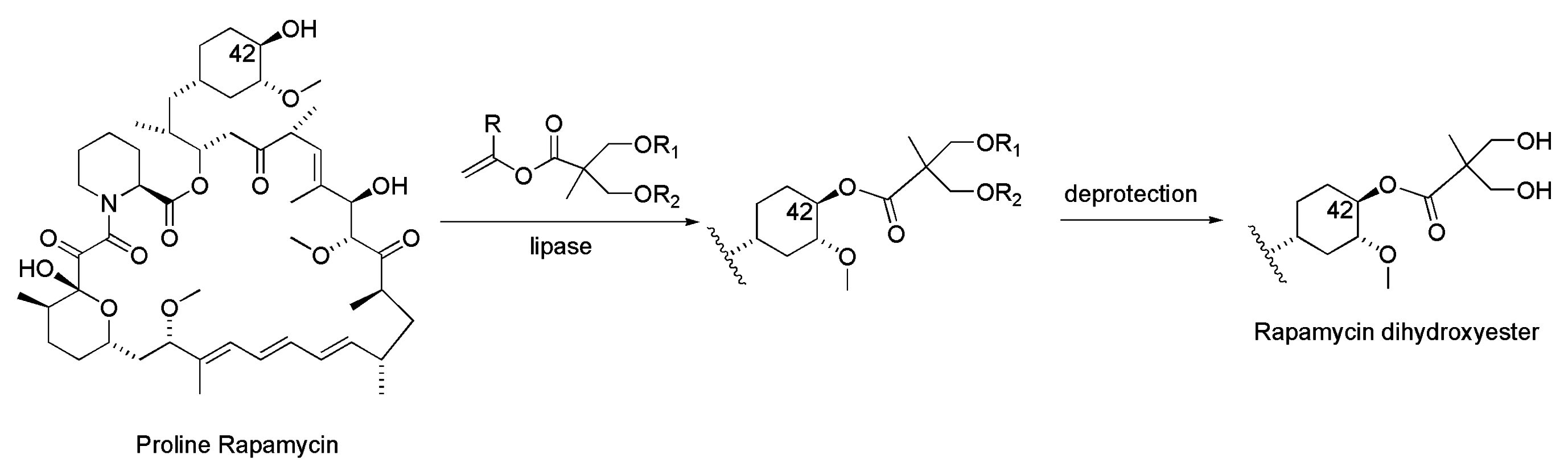
3.1. Rapamycin

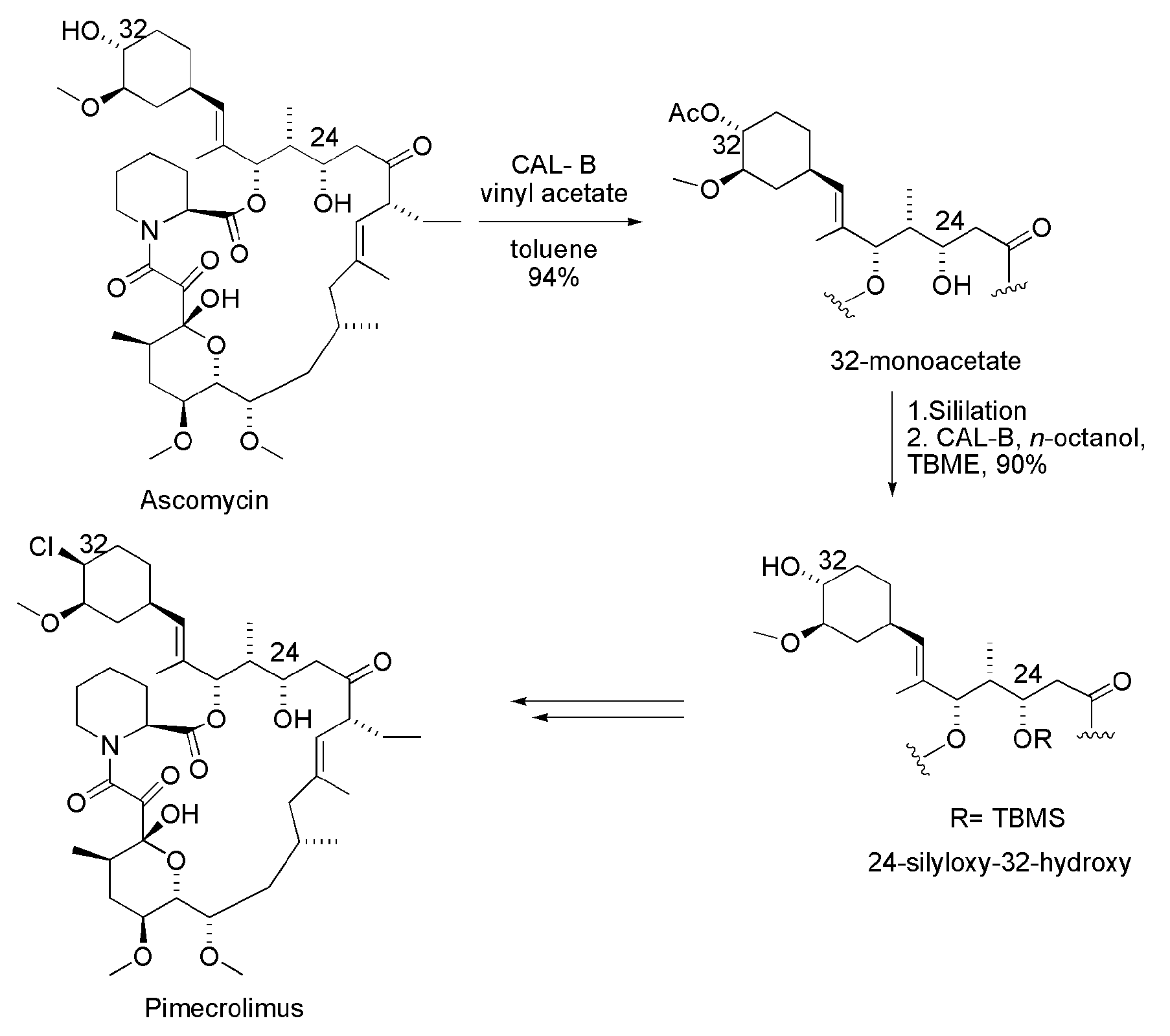
3.2. Pimecrolimus

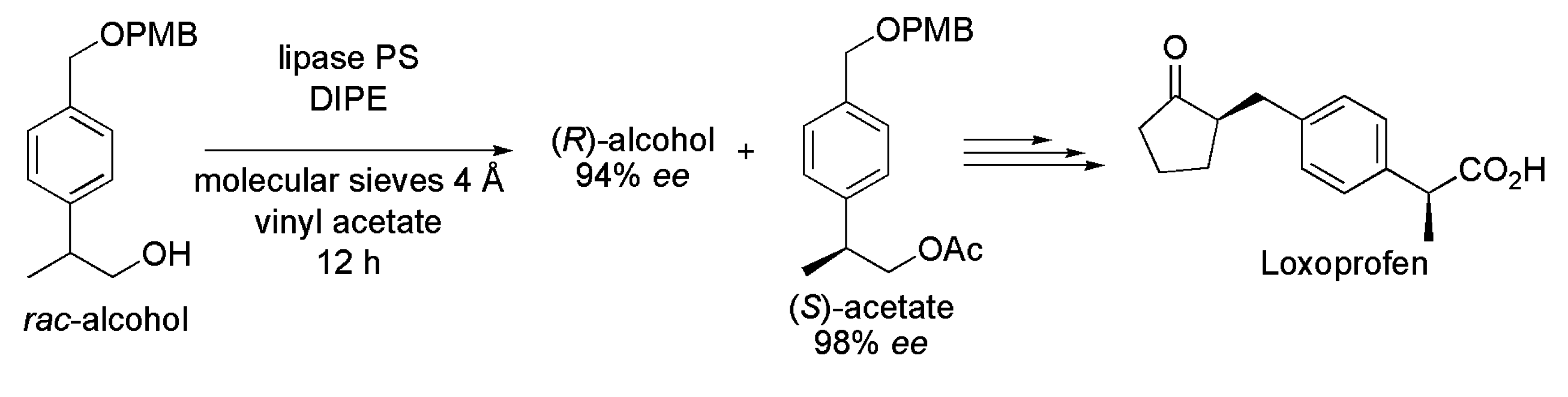
3.3. Loxoprofen

3.4. Flurbiprofen

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
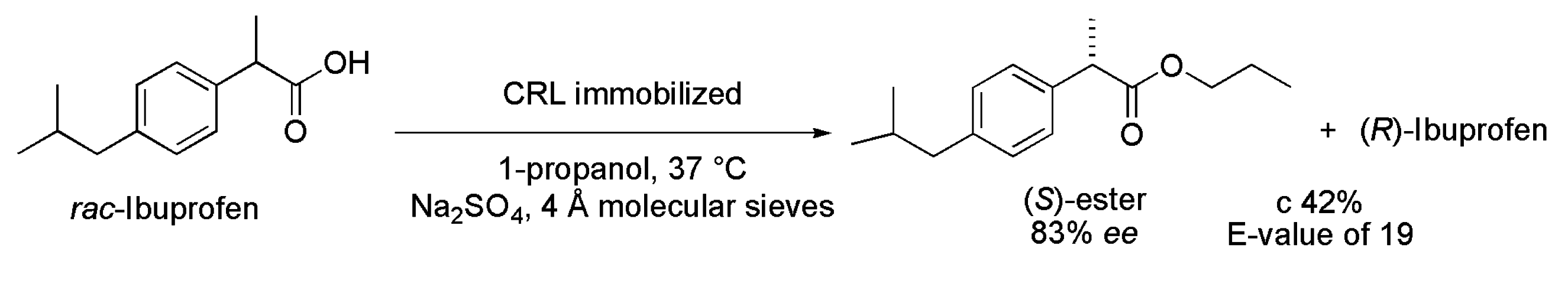
3.5. Ibuprofen

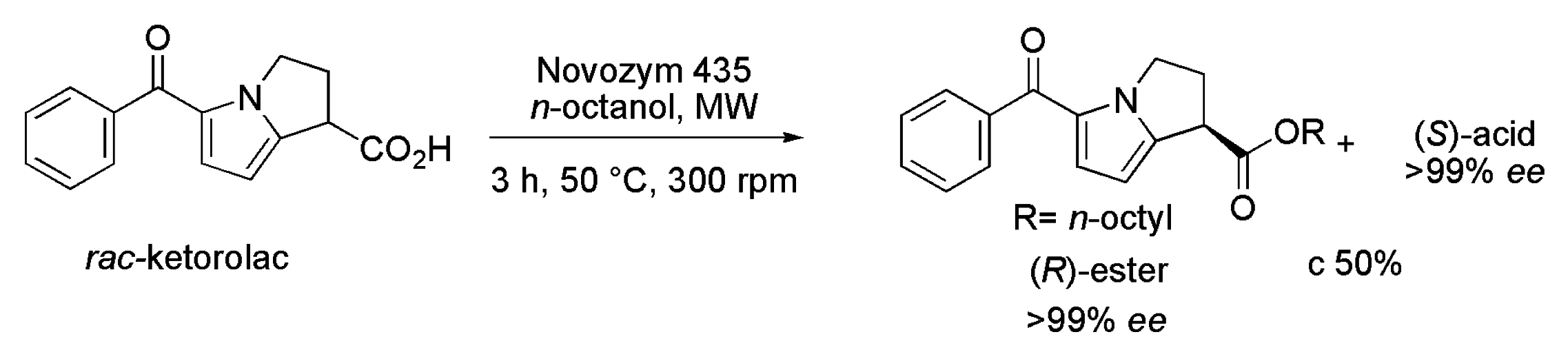
3.6. Ketorolac

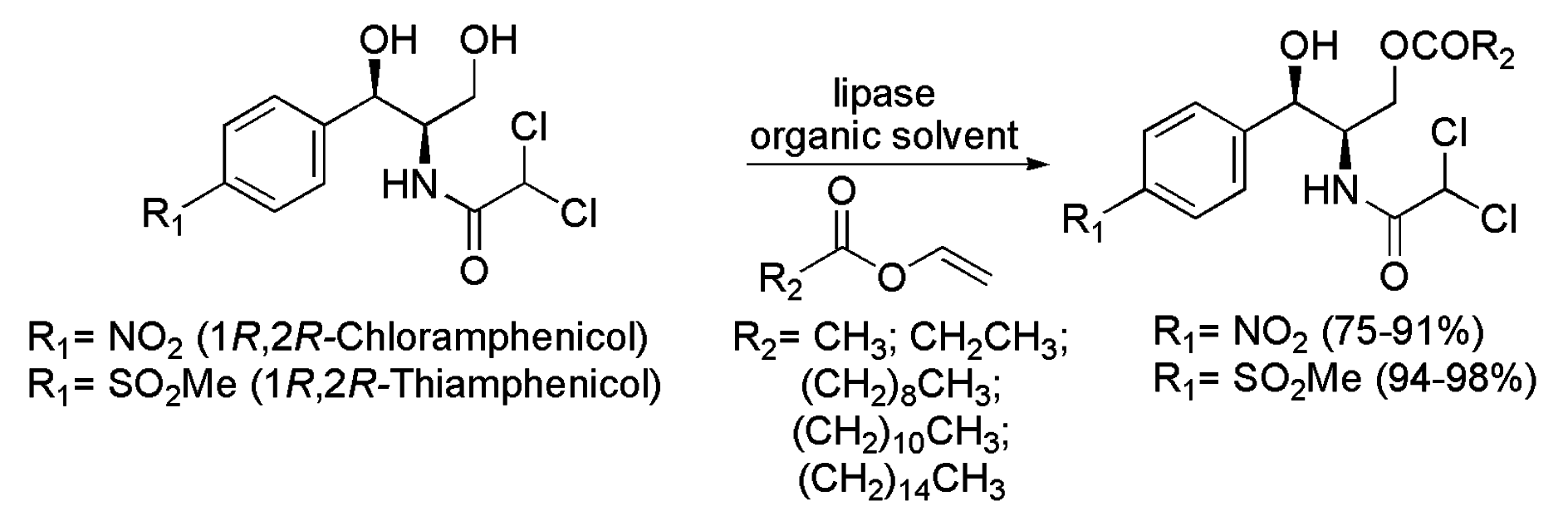
3.7. Chloramphenicol and Thiamphenicol

3.8. Key Intermediates of Modified Cephalosporins

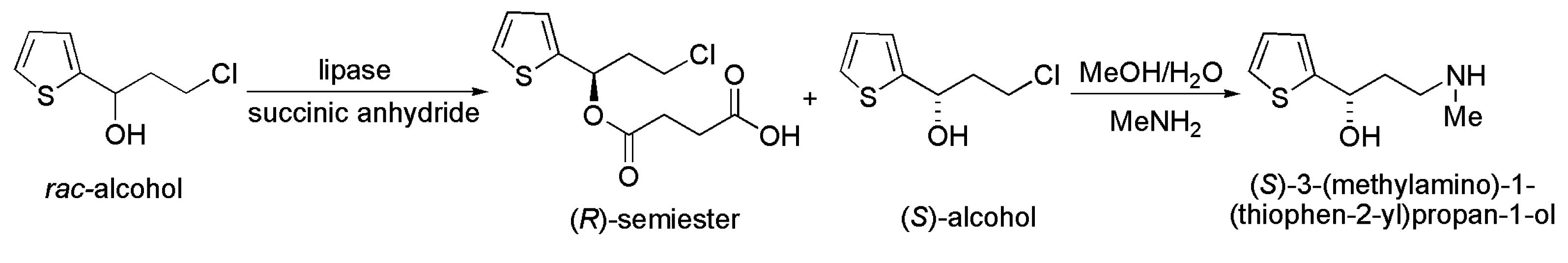
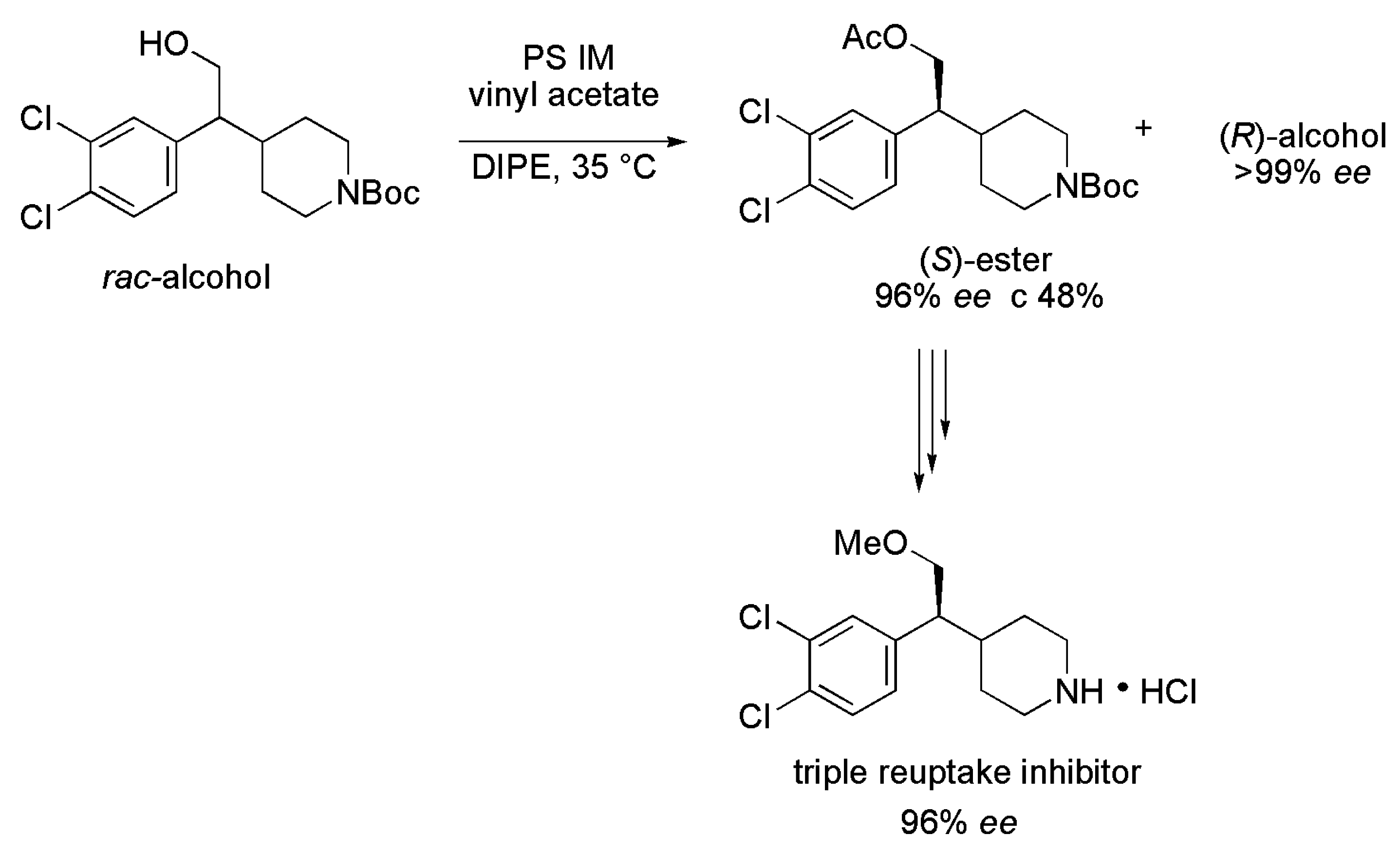
3.9. Key Intermediate of Duloxetin

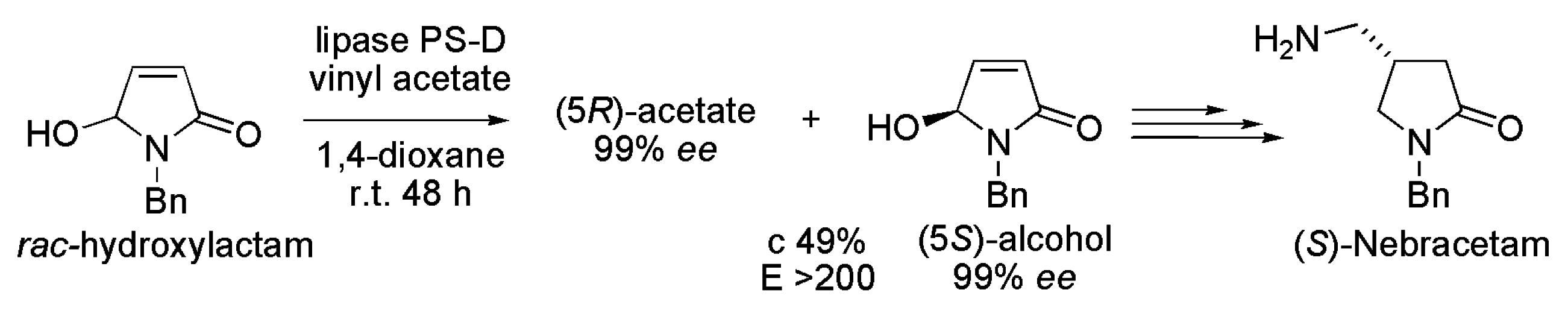
3.10. Nebracetam

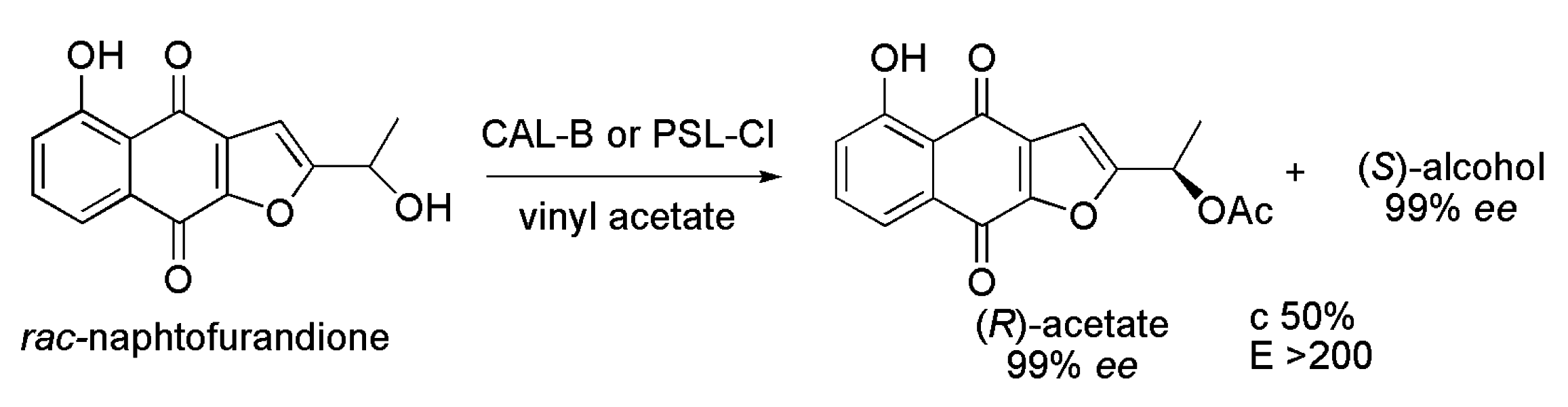
3.11. Naphthofurandione Derivative

3.12. GABOB and Carnitine

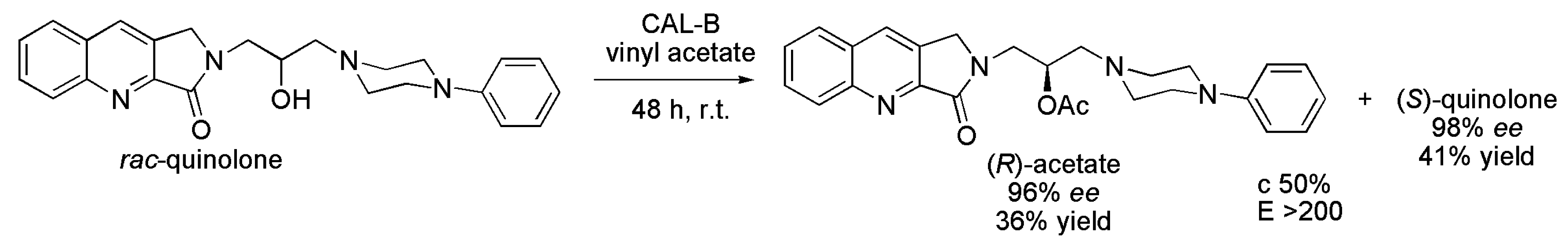
3.13. Quinolone Derivatives

3.14. Key Intermediate of Mevinic Acid Analogues

3.15. Key Intermediate of Tetrahydrofuran Derivatives

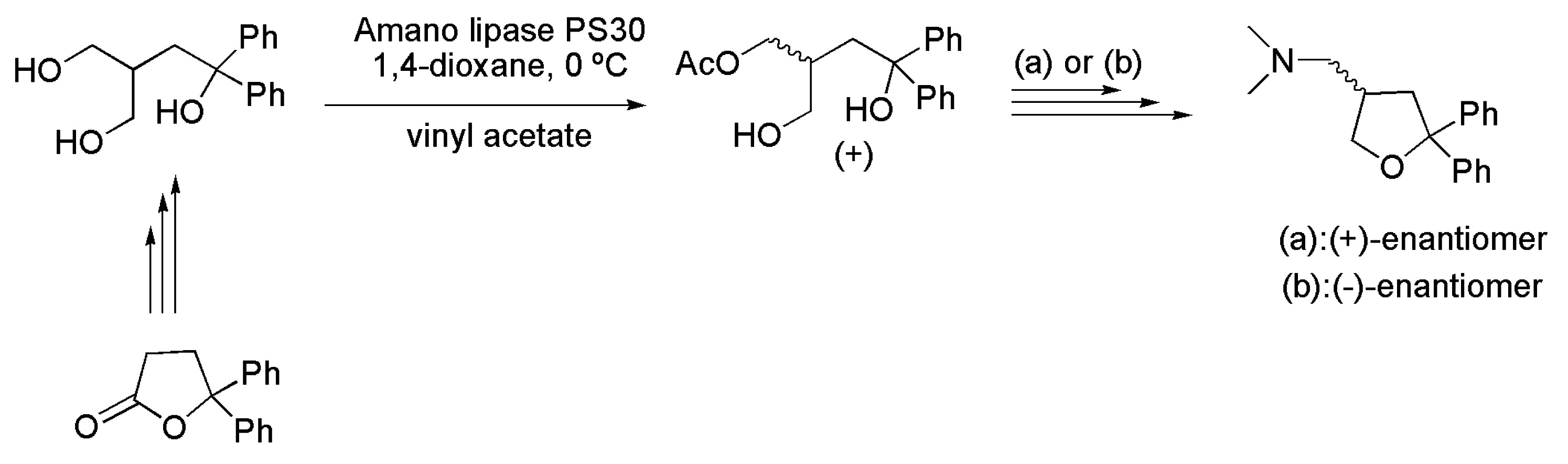
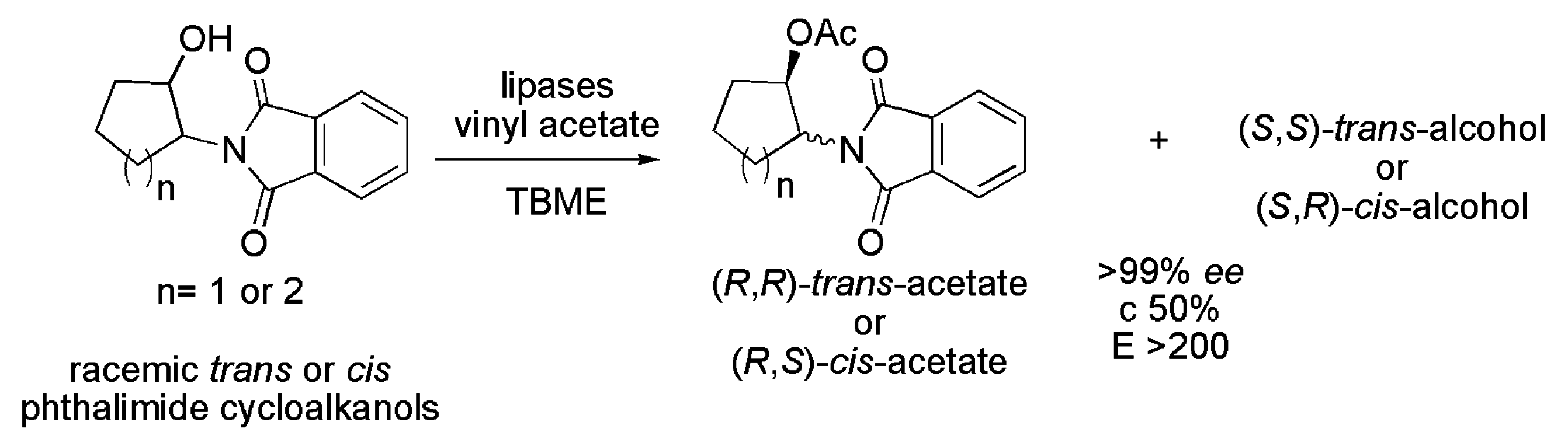
3.16. Key Intermediates of β-Amino Alcohols

| Compound | Lipase |
|---|---|
| trans-2-phthalimidocyclopentanol | P. cepacia (PS IM) |
| cis-2-phthalimidocyclopentanol | C. antarctica (CAL-A), R. miehei (RM IM), P. fluorescens (AK) and P. cepacia (PS IM) |
| trans-2-phthalimidocyclohexanol | P. cepacia (PS IM) |
| cis-2-phthalimidocyclohexanol | P. cepacia (PS IM) |
3.17. Key Intermediates of Iminocyclitols

3.18. N-Acetyl Phenylalanine and Analogues

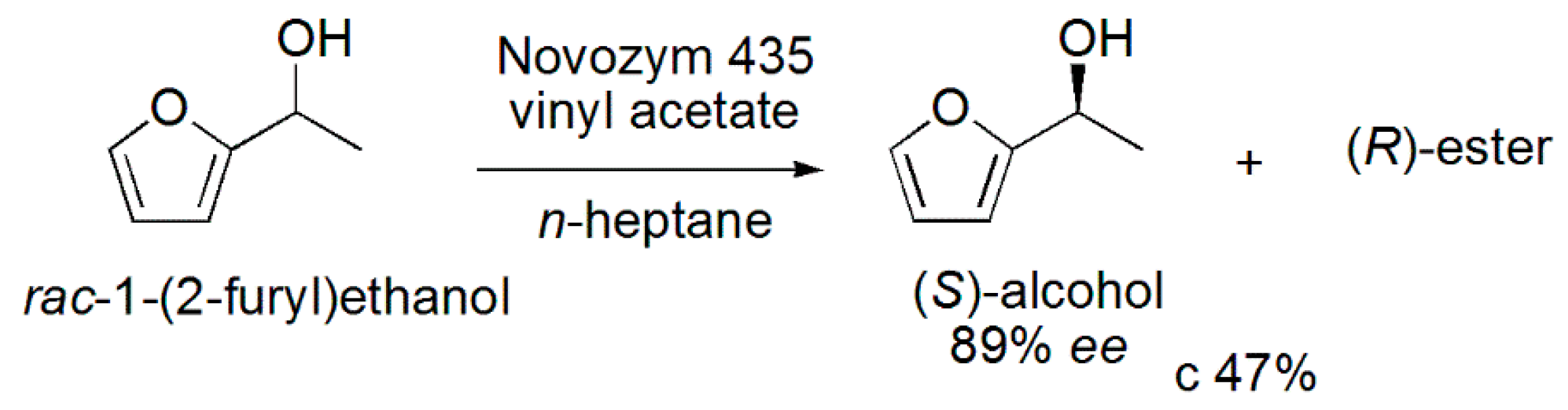
3.19. (S)-1-(2-Furyl)ethanol

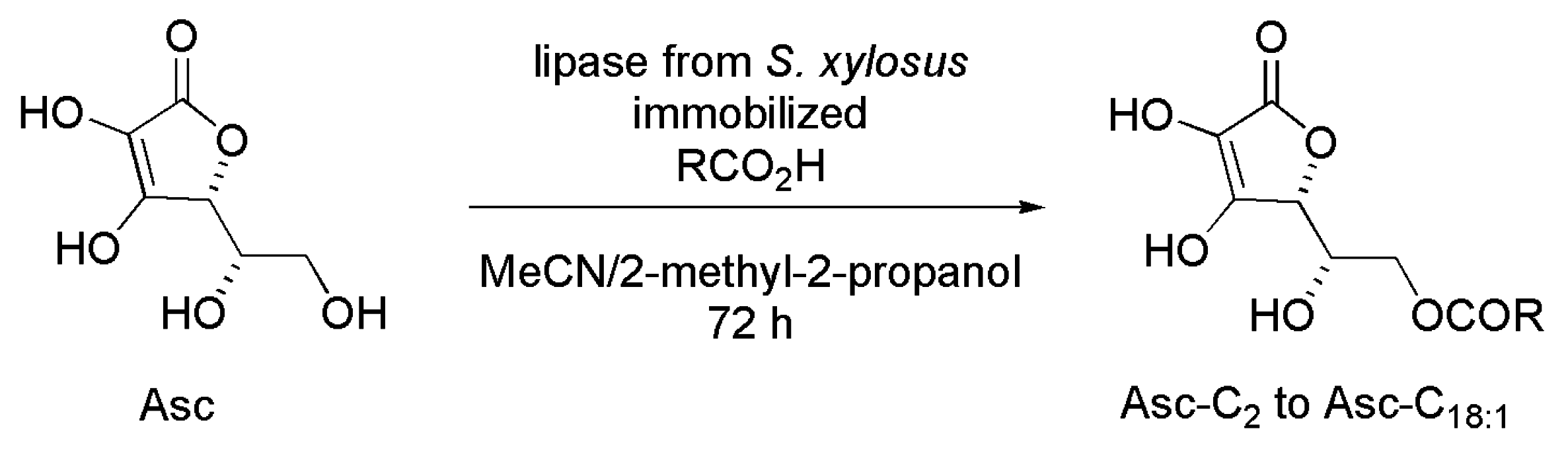
3.20. Ascorbyl Ester Derivatives

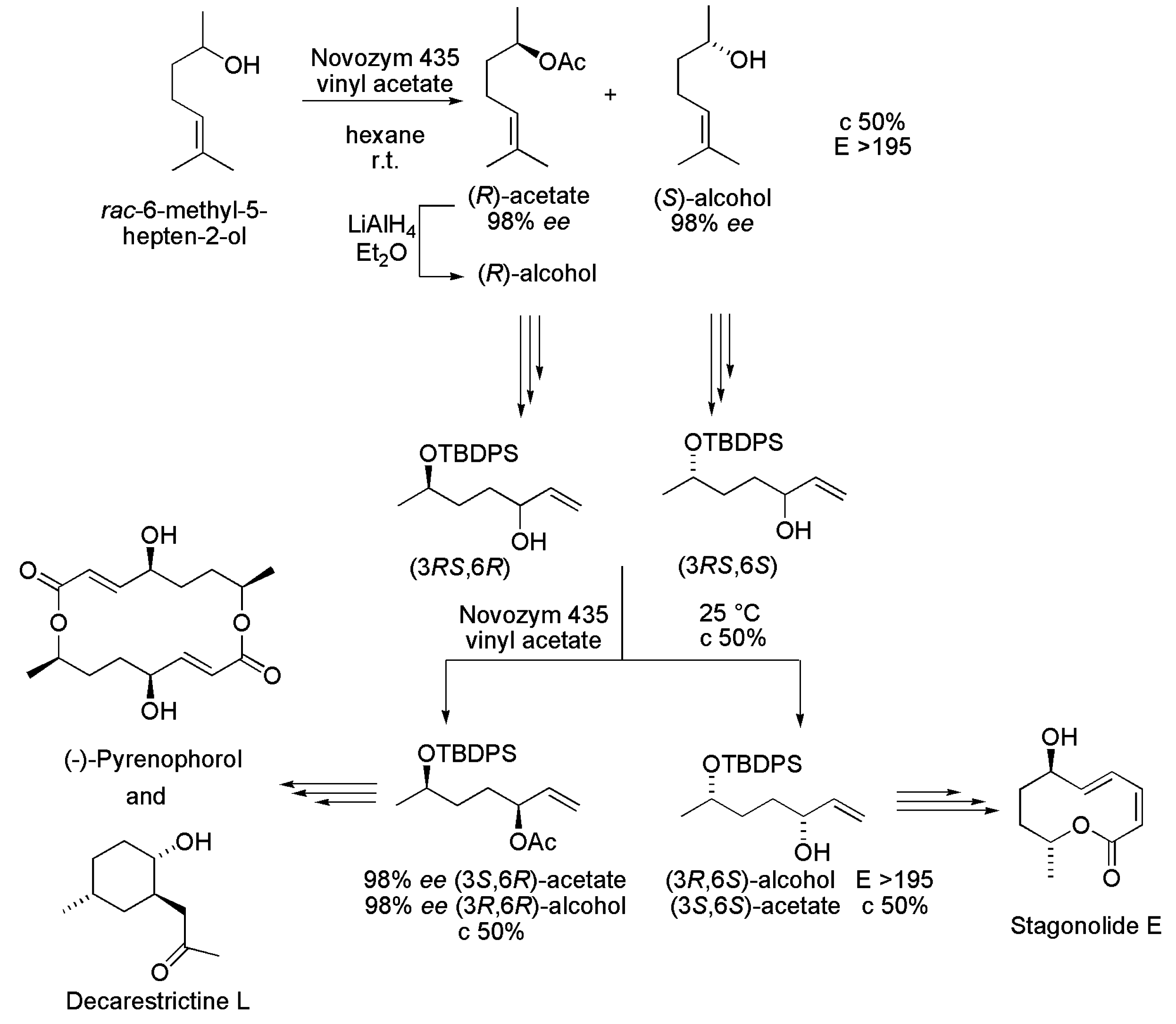
3.21. Key Intermediates of Stagonolide E, Pyrenophorol and Decarestrictine L

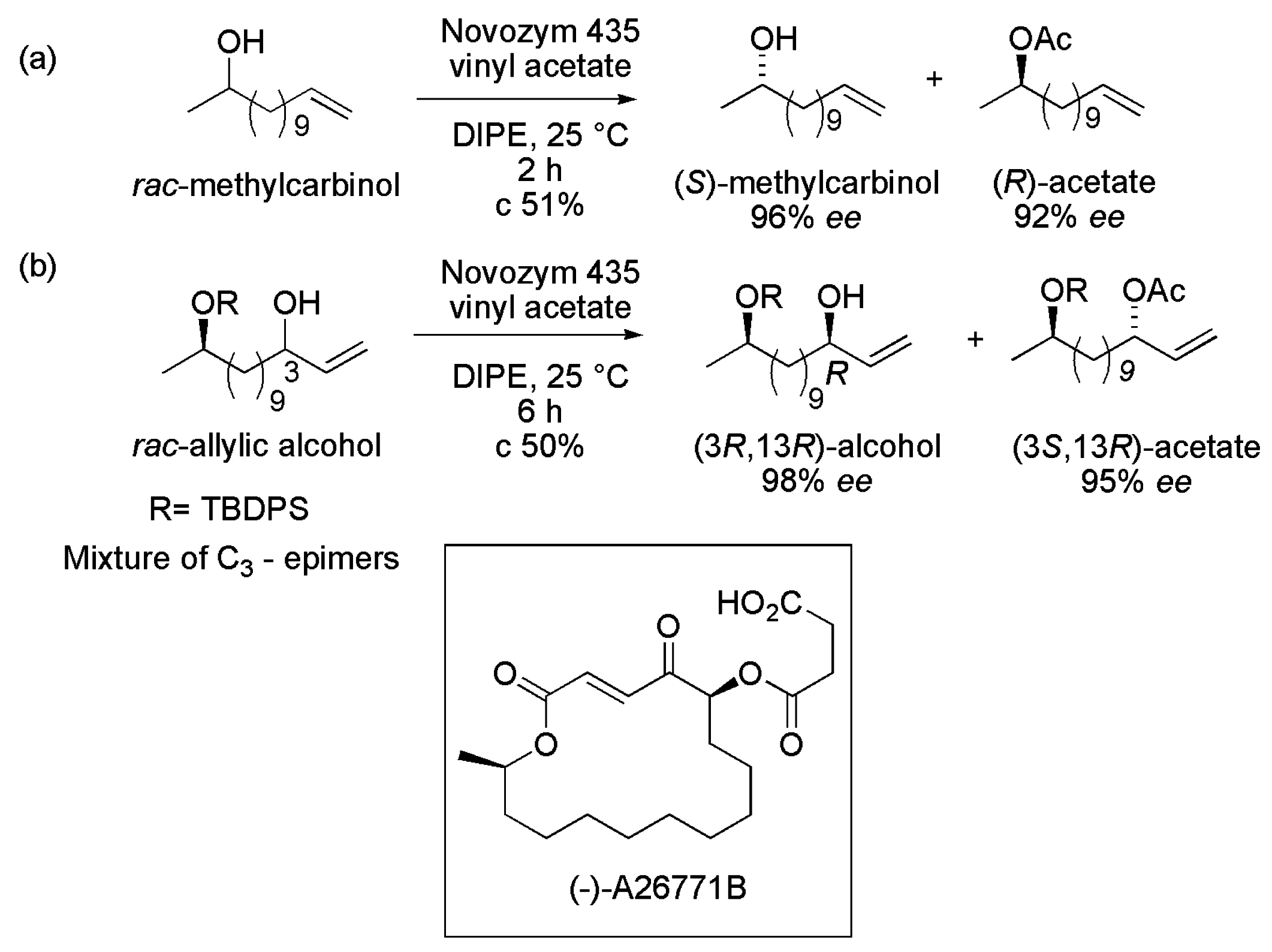
3.22. Key Intermediates of Macrolide Antibiotic (−)-A26771B

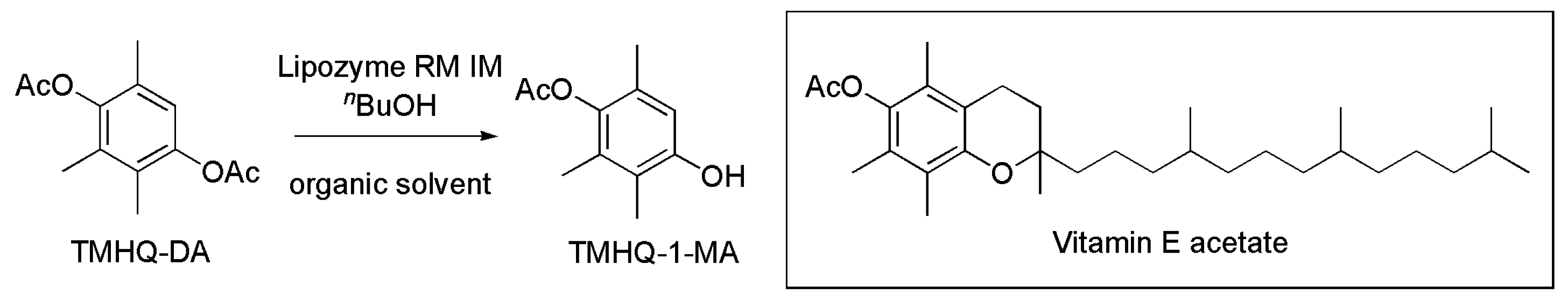
3.23. Key Intermediate of Vitamin E Acetate

3.24. Rasagiline Mesylate


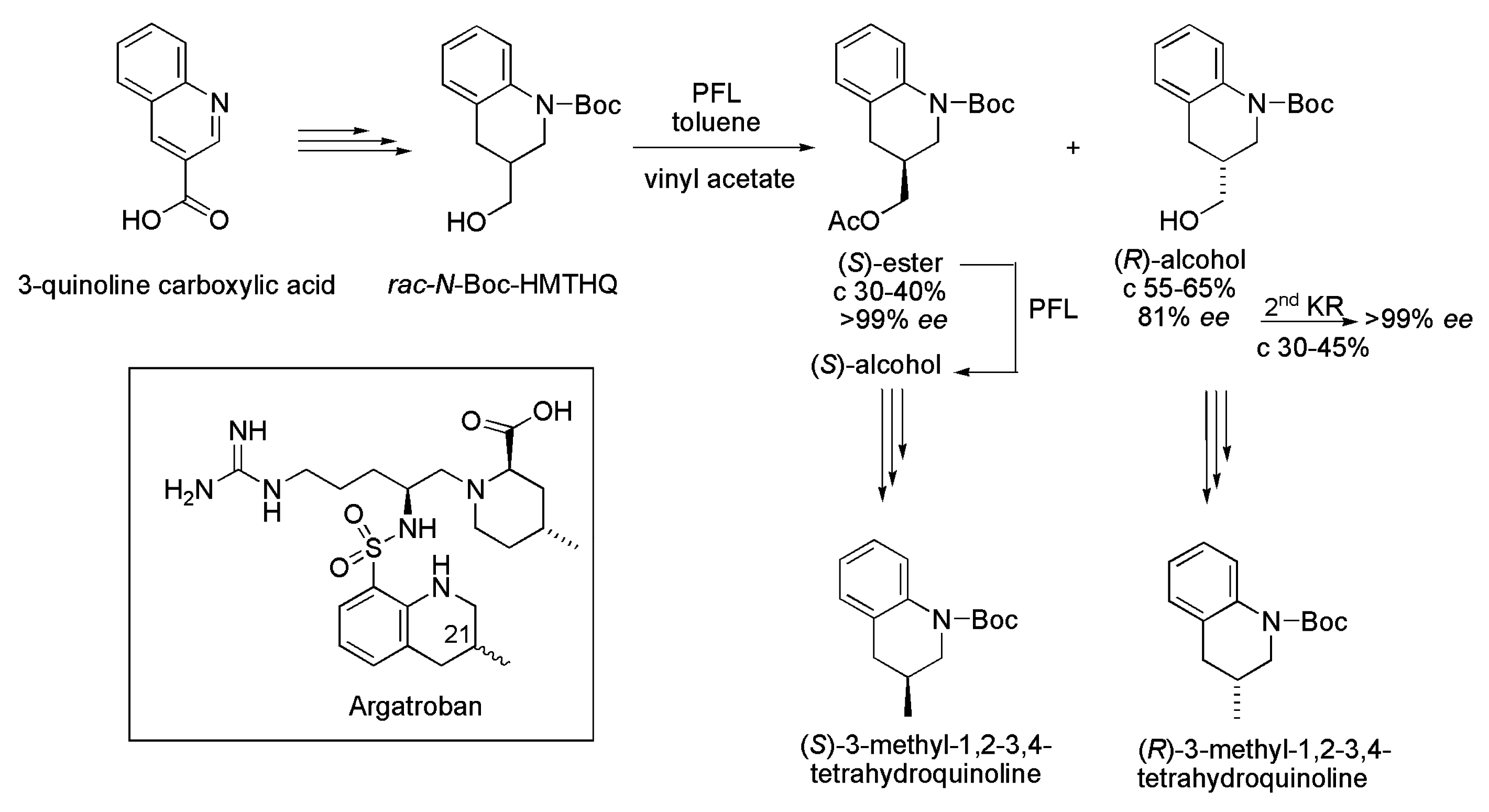
3.25. Argatroban
3.26. Key Intermediate of the Paclitaxel Side Chain

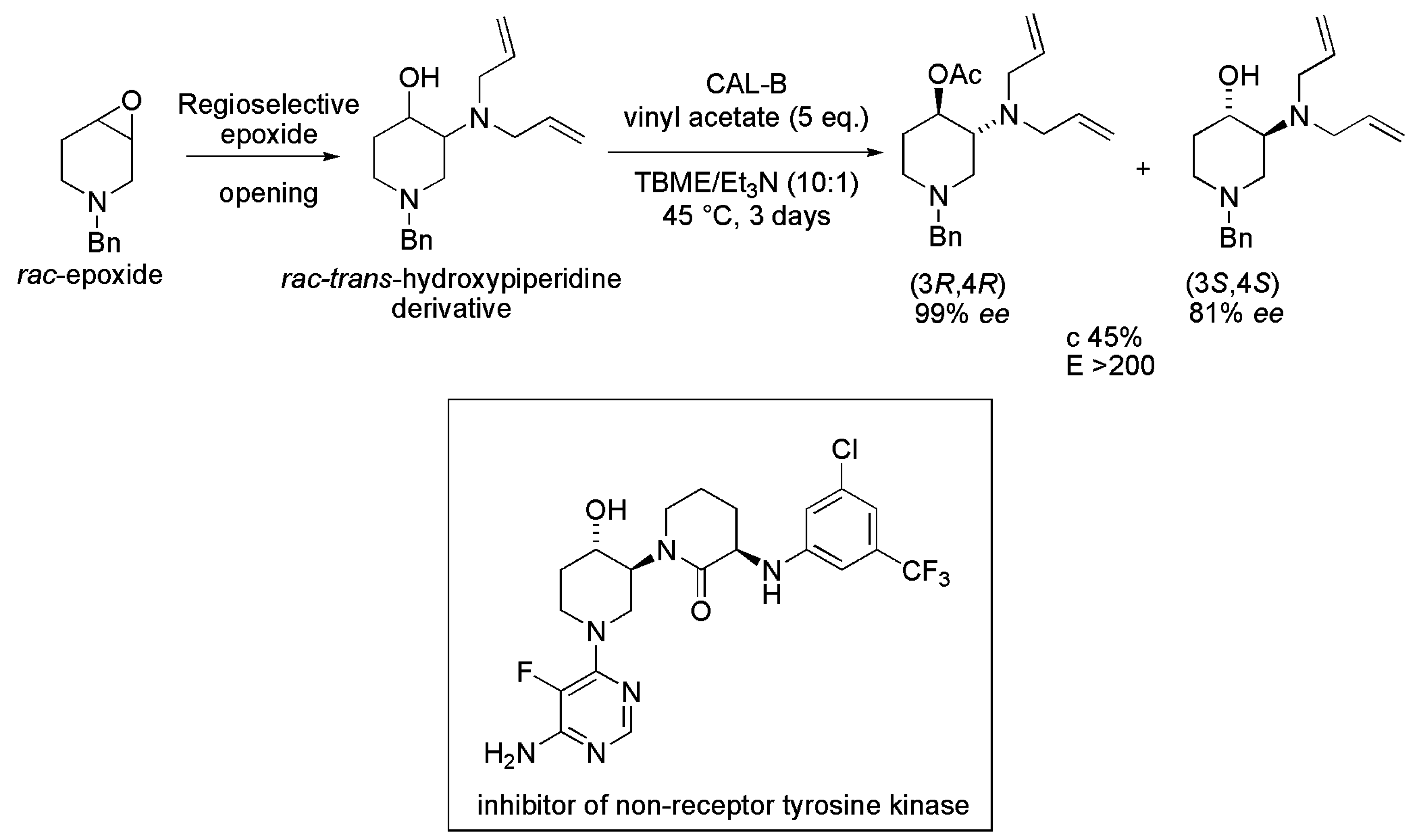
3.27. Piperidine Derivative

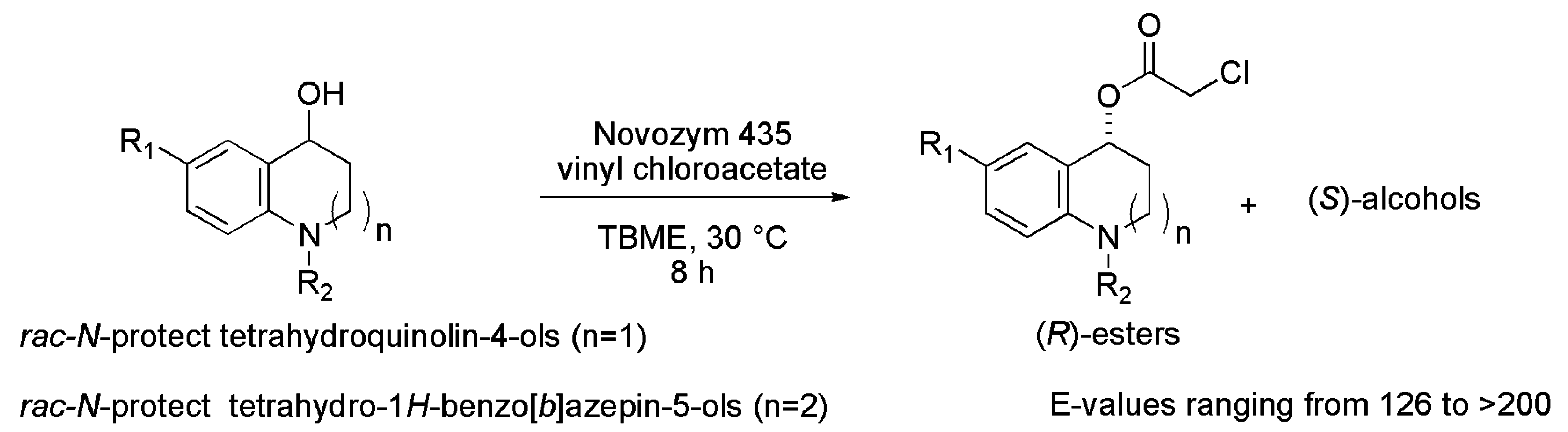
3.28. Tetrahydroquinolinol and Tetrahydrobenzoazepinol Derivatives

| Substrates | n | R1 | R2 |
|---|---|---|---|
| rac-N-protect tetrahydroquinolin-4-ols | 1 | H | Boc |
| Cl | Boc | ||
| Br | Boc | ||
| OMe | Ac | ||
| H | CO2Ph | ||
| H | Cbz | ||
| rac-N-protect tetrahydro-1H-benzo[b]azepin-5-ols | 2 | H | Bz |
| H | 2-furoyl | ||
| H | Cbz |
3.29. Aminohydroxypiperidine Derivatives

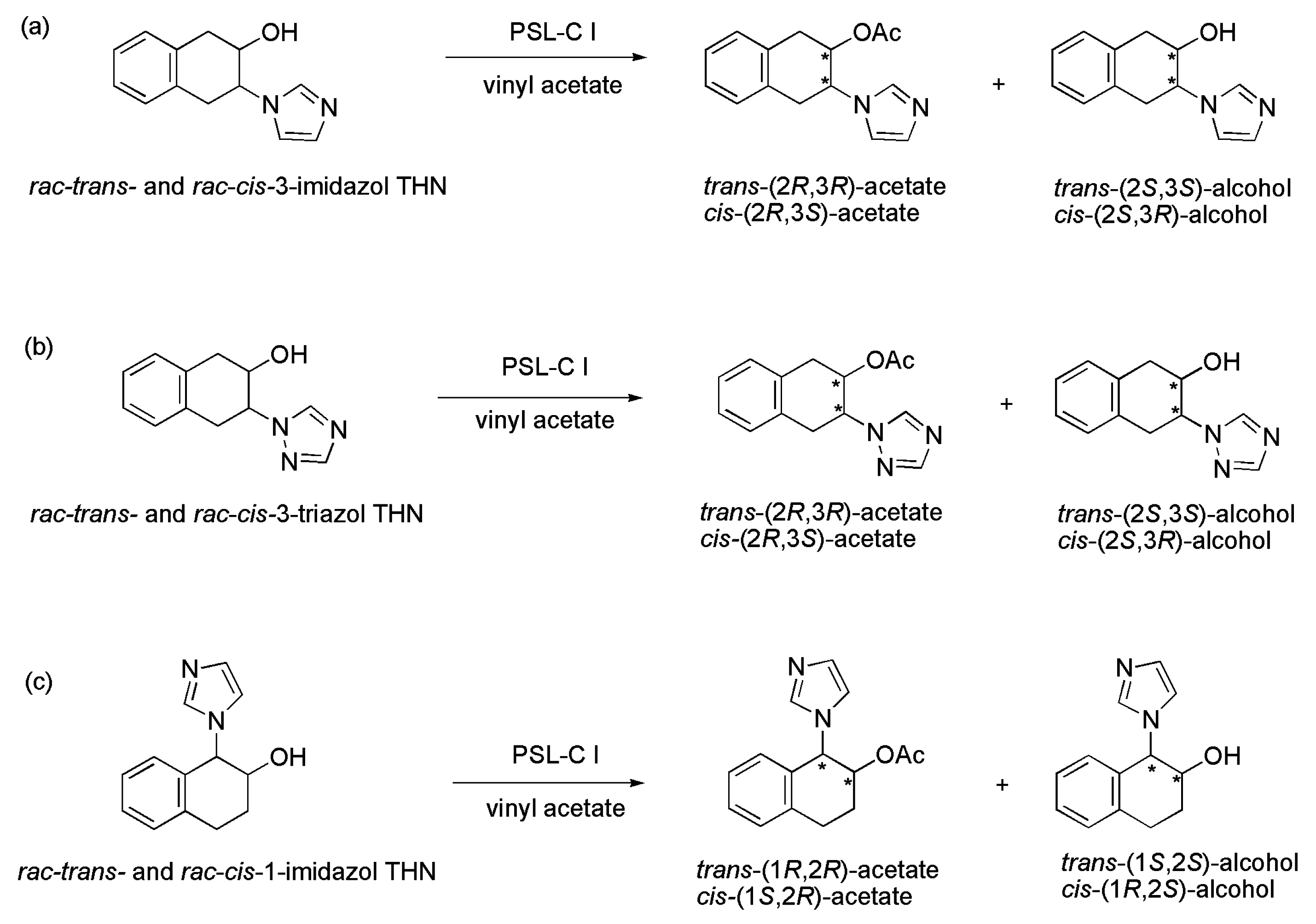
3.30. Azole Derivatives

| Scheme 41 | Substrate | c (%) | eeP (%) Acetate | eeS (%) Alcohol | E |
|---|---|---|---|---|---|
| a | trans-3-imidazol THN | 46 | 95 | 83 | 102 |
| rac-cis-3-imidazol THN | 43 | 95 | 71 | 83 | |
| b | rac-trans-3-triazol THN | 48 | 99 | 93 | >200 |
| rac-cis-3-triazol THN | 23 | >99 | 29 | >200 | |
| c | rac-trans-1-imidazol THN | 43 | 96 | 85 | 133 |
| rac-cis-1-imidazol THN | 50 | 98 | 98 | >200 |
3.31. Benzoin Derivative

4. Complementary Approaches: Hydrolysis and Esterification
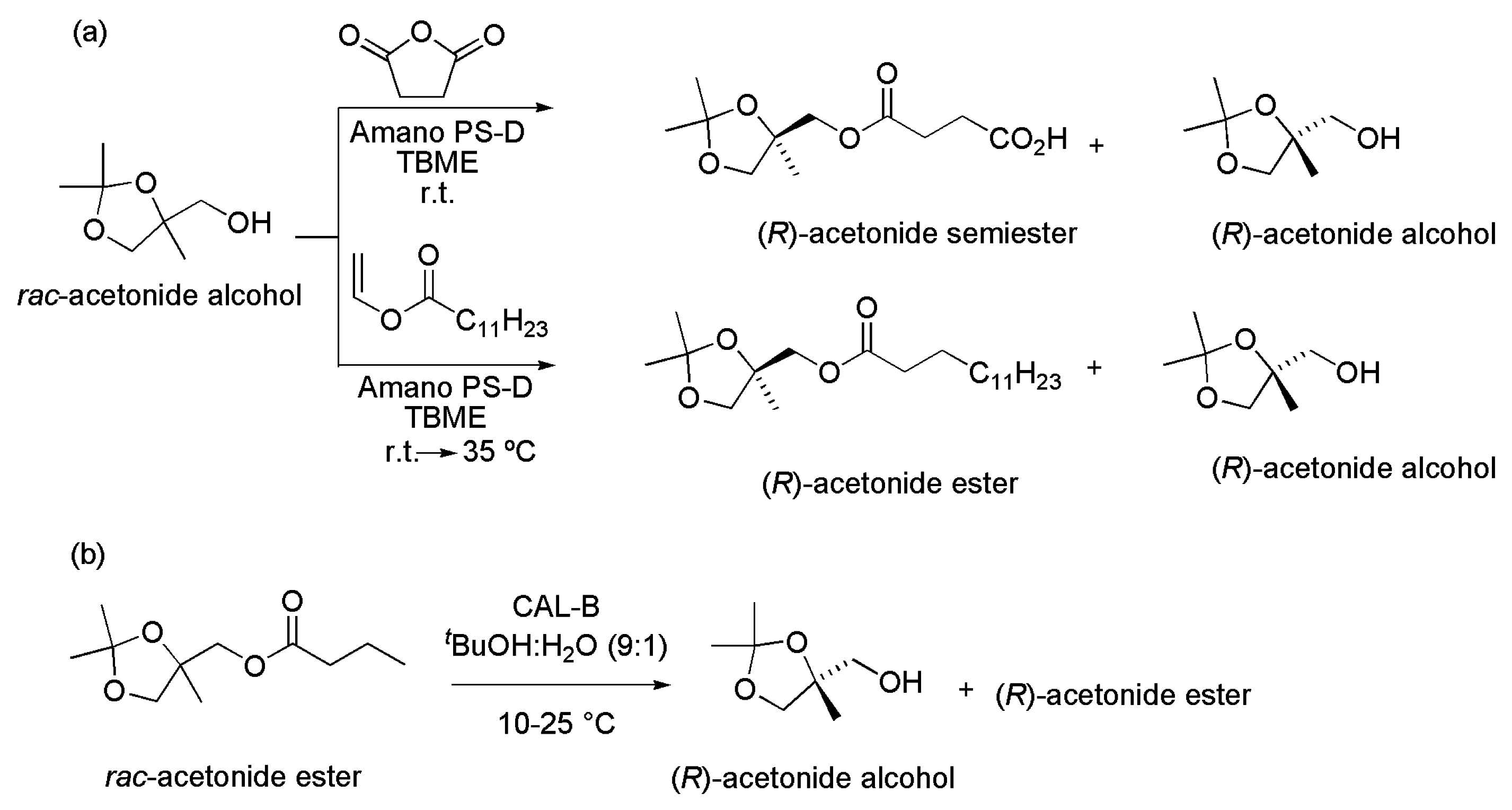
4.1. Acetonide Derivatives

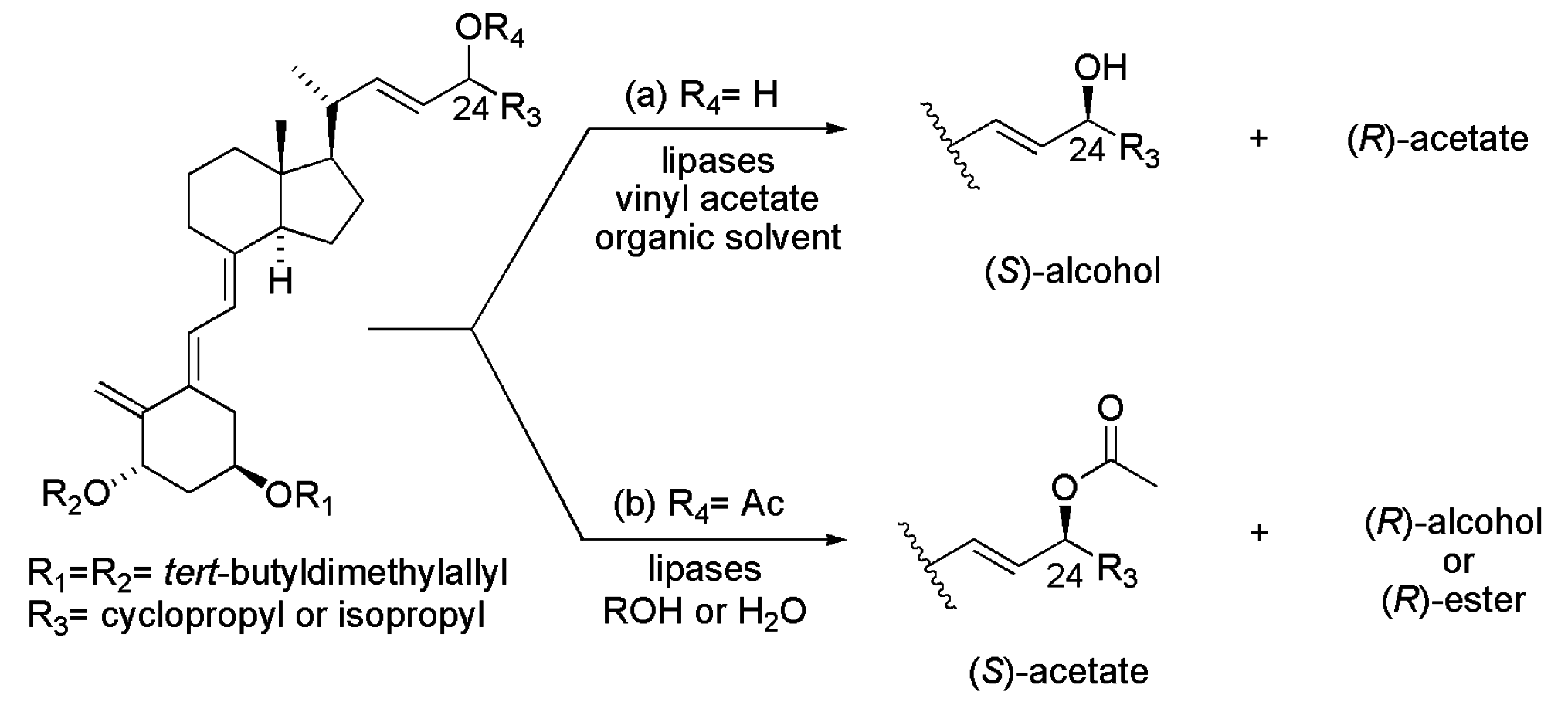
4.2. Vitamin D Analogues

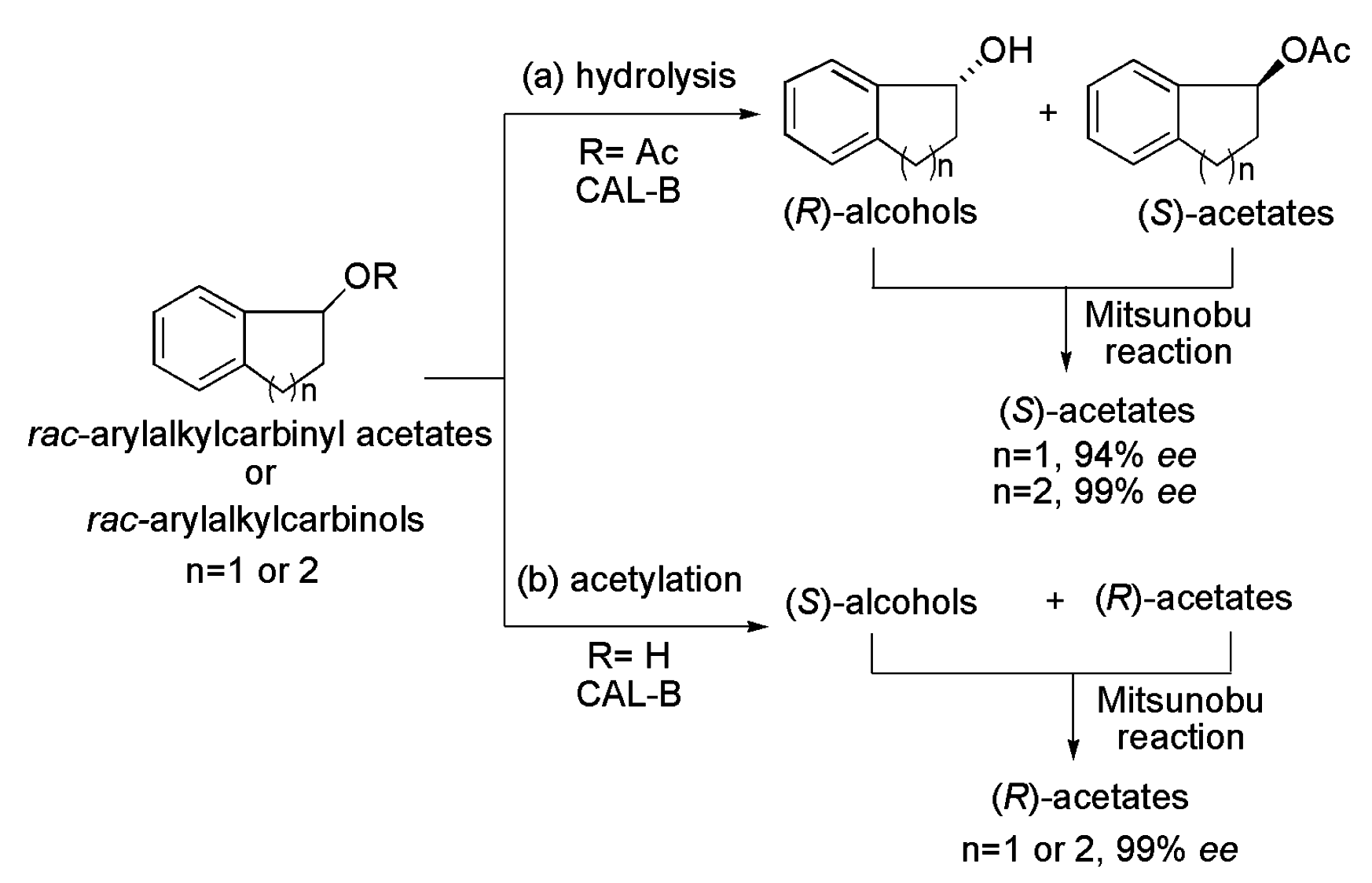
4.3. Arylalkylcarbinol Derivatives

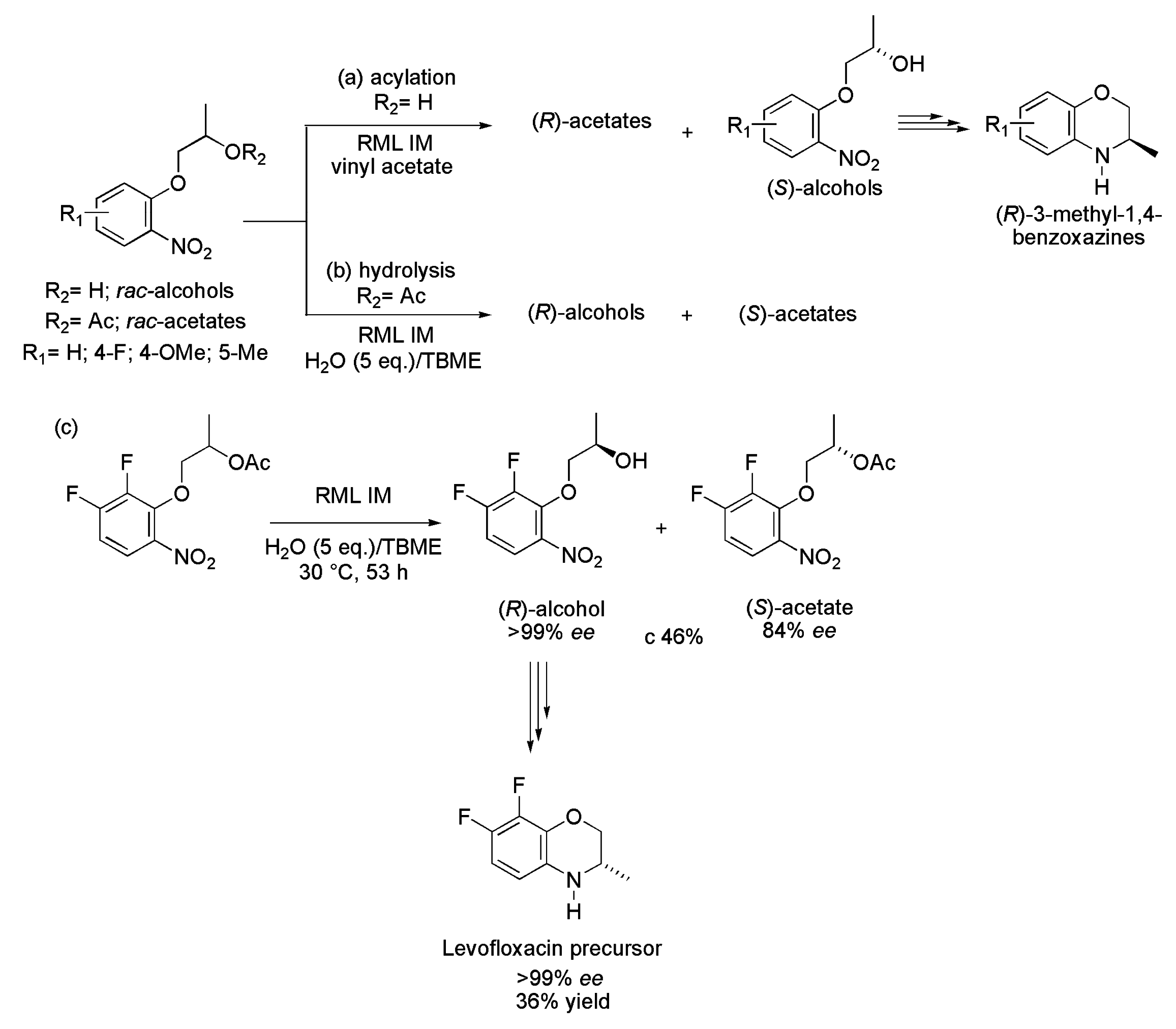
4.4. Key Intermediate of Levofloxacin

| Scheme 46 | Acetates | Alcohols | c (%) | E | ||
|---|---|---|---|---|---|---|
| ee (%) | Yield (%) | ee (%) | Yield (%) | |||
| a | 93–95 | 45–47 | 89–94 | 44–48 | 48–50 | 103–>200 |
| b | 91–97 | 41–48 | 96–>99 | 44–47 | 48–50 | >200 |
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gotor-Fernandez, V.; Brieva, R.; Gotor, V. Lipases: Useful biocatalysts for the preparation of pharmaceuticals. J. Mol. Catal. B. 2006, 40, 111–120. [Google Scholar] [CrossRef]
- Tao, J.; Zhao, L.; Ran, N. Recent advances in developing chemoenzymatic processes for active pharmaceutical ingredients. Org. Process Res. Dev. 2007, 11, 259–267. [Google Scholar]
- Pollard, D.J.; Woodley, J.M. Biocatalysis for pharmaceutical intermediates: The future is now. Trends Biotechnol. 2007, 25, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.N. Synthesis of chiral pharmaceutical intermediates by Biocatalysis. Coord. Chem. Rev. 2008, 252, 659–701. [Google Scholar] [CrossRef]
- Tao, J.; Xu, J.-H. Biocatalysis in development of green pharmaceutical processes. Curr. Opin. Chem. Biol. 2009, 13, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.N. Biocatalysis: Synthesis of key intermediates for development of pharmaceuticals. ACS Catal. 2011, 1, 1056–1074. [Google Scholar] [CrossRef]
- Anand, N.; Kapoor, M.; Ahmad, K.; Koul, S.; Parshad, R.; Manhas, K.S.; Sharma, L.R.; Qazi, G.N.; Taneja, S.C. Arthrobacter sp.: A lipase of choice for the kinetic resolution of racemic arylazetidinone precursors of taxanoid side chains. Tetrahedron 2007, 18, 1059–1069. [Google Scholar] [CrossRef]
- Rimoldi, M.; Pellizzoni, G.; Facchetti, F.E.; Molinari, F.; Zerla, D.S.; Gandolfi, R. Chemo- and biocatalytic strategies to obtain phenylisoserine, lateral chain of taxol by asymmetric reduction. Tetrahedron 2011, 22, 2110–2116. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. A new enzymatic strategy for the preparation of (2R,3S)-3-phenylisoserine: A key intermediate for the Taxol side chain. Tetrahedron 2010, 21, 637–639. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. New enzymatic two-step cascade reaction for the preparation of a key intermediate for the taxol side-chain. Eur. J. Org. Chem. 2010, 16, 3074–3079. [Google Scholar] [CrossRef]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef] [PubMed]
- Kung, P.-P.; Martinez, C.; Tao, J. Enantioselective Biotransformation for Preparation of Protein Tyrosine Kinase Inhibitor Intermediates. US 7465842 B2, 16 December 2008. [Google Scholar]
- Felluga, F.; Pitacco, G.; Valentin, E.; Venneri, C.D. A facile chemoenzymatic approach to chiral non-racemic β-alkyl-γ-amino acids and 2-alkylsuccinic acids. A concise synthesis of (S)-(+)-Pregabalin. Tetrahedron 2008, 19, 945–955. [Google Scholar] [CrossRef]
- Li, X.-J.; Zheng, R.-C.; Ma, H.-Y.; Zheng, Y.-G. Engineering of Thermomyces lanuginosus lipase Lip: Creation of novel biocatalyst for efficient biosynthesis of chiral intermediate of Pregablin. Appl. Microbiol. Biotechnol. 2014, 98, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Rouf, A.; Gupta, P.; Aga, M.A.; Kumar, B.; Chaubey, A.; Parshad, R.; Taneja, S.C. Chemoenzymatic synthesis of piperoxan, prosympal, dibozane, and doxazosin. Tetrahedron 2012, 23, 1615–1623. [Google Scholar] [CrossRef]
- Singh, A.; Goel, Y.; Rai, A.K.; Banerjee, U.C. Lipase catalyzed kinetic resolution for the production of (S)-3-[5-(4-fluoro-phenyl)-5-hydroxy-pentanoyl]-4-phenyl-oxazolidin-2-one: An intermediate for the synthesis of ezetimibe. J. Mol. Catal. B 2013, 85–86, 99–104. [Google Scholar] [CrossRef]
- Goswami, A.; Guo, Z.; Qiu, Y. Process for Resolving cyclopropyl diesters. EP2817412 A1, 31 December 2014. [Google Scholar]
- Takaç, S.; Bakkal, M. Impressive effect of immobilization conditions on the catalytic activity and enantioselectivity of Candida rugosa lipase toward S-Naproxen production. Pro. Biochem. 2007, 42, 1021–1027. [Google Scholar] [CrossRef]
- Sayin, S.; Akoz, E.; Yilmaz, M. Enhanced catalysis and enantioselective resolution of racemic naproxen methyl ester by lipase encapsulated within iron oxide nanoparticles coated with calix[8]arene valeric acid complexes. Org. Biomol. Chem. 2014, 12, 6634–6642. [Google Scholar] [CrossRef] [PubMed]
- Sharifabad, M.E.; Hodgson, B.; Jellite, M.; Mercer, T.; Sen, T. Enzyme immobilised novel core–shell superparamagnetic nanocomposites for enantioselective formation of 4-(R)-hydroxycyclopent-2-en-1-(S)-acetate. Chem. Commun. 2014, 50, 11185–11187. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Wang, N.; Zhang, W.; Zhang, S.; Meng, Y.; Yu, X. Immobilization of Aspergillus terreus lipase in self-assembled hollow nanospheres for enantioselective hydrolysis of ketoprofen vinyl ester. J. Biotechnol. 2015, 194, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-W.; Jia, J.-Q.; Wang, N.; Hu, C.-L.; Yang, S.-Y.; Yu, X.-Q. Improved activity of lipase immobilized in microemulsion-based organogels for (R,S)-ketoprofen ester resolution: Long-term stability and reusability. Biotechnol. Rep. 2015, 7, 1–8. [Google Scholar] [CrossRef]
- Reis, P.; Holmberg, K.; Watzke, H.; Leser, M.E.; Miller, R. Lipases at interfaces: A review. Adv. Colloid Interface Sci. 2009, 147–148, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.Y.; Verdecia, Y.; Rebolledo, F. Chemoenzymatic approach to optically active 1,4-dihydropyridine derivatives. Tetrahedron 2015, 71, 3976–3984. [Google Scholar] [CrossRef]
- Gu, J.; Rupeen, M.E.; Raveendranath, P.; Chew, W.; Shaw, C.C. Proline cci-779 (Proline-Rapamycin 42-Ester with 2,2-Bis(hydroxymethyl) Propionic Acid) and Two-Step Enzymatic Synthesis of Proline cci-779 and cci-779 Using Microbial Lipase. EP1751168A1, 14 February 2007. [Google Scholar]
- Ferraboschi, P.; Colombo, D.; de Mieri, M.; Grisenti, P. First chemoenzymatic synthesis of immunomodulating macrolactam pimecrolimus. Tetrahedron Lett. 2009, 50, 4384–4388. [Google Scholar] [CrossRef]
- Grisenti, P.; Reza, E.S.; Verza, E. A Chemo-Enzymatic Approach to the Synthesis of Primecrolimus. EP2432791B1, 8 July 2015. [Google Scholar]
- Bhuniya, R.; Nanda, S. Asymmetric synthesis of the active form of loxoprofen and its analogue. Tetrahedron 2011, 22, 1125–1132. [Google Scholar] [CrossRef]
- Spizzo, P.; Basso, A.; Ebert, C.; Gardossi, L.; Ferrario, V.; Romano, D.; Molinari, F. Resolution of (R,S)-flurbiprofen catalysed by dry mycelia in organic solvente. Tetrahedron 2007, 63, 11005–11010. [Google Scholar] [CrossRef]
- Tamborini, L.; Romano, D.; Pinto, A.; Bertolani, A.; Molinari, F.; Conti, P. An efficient method for the lipase-catalysed resolution and in-line purification of racemic flurbiprofen in a continuous-flow reactor. J. Mol. Catal. B 2012, 84, 78–82. [Google Scholar] [CrossRef]
- Tamborini, L.; Romano, D.; Pinto, A.; Contente, M.; Iannuzzi, M.C.; Conti, P.; Molinari, F. Biotransformation with whole microbial systems in a continuous flow reactor: Resolution of (RS)-flurbiprofen using Aspergillus oryzae by direct esterification with ethanol in organic solvent. Tetrahedron Lett. 2013, 54, 6090–6093. [Google Scholar] [CrossRef]
- Pastre, J.C.; Browne, D.L.; Ley, S.V. Flow chemistry syntheses of natural products. Chem. Soc. Rev. 2013, 42, 8849–8869. [Google Scholar] [CrossRef] [PubMed]
- Itabaiana, I.; Miranda, L.S.M.; Souza, R.O.M.A. Towards a continuous flow environment for lipase-catalyzed reactions. J. Mol. Catal. B 2013, 85–86, 1–9. [Google Scholar] [CrossRef]
- Marszałł, M.P.; Siódmiak, T. Immobilization of Candida rugosa lipase onto magnetic beads for kinetic resolution of (R,S)-ibuprofen. Catal. Commun. 2012, 24, 80–84. [Google Scholar] [CrossRef]
- Shinde, S.D.; Yadav, G.D. Insight into microwave assisted immobilized Candida Antarctica lipase B catalyzed kinetic resolution of R,S-(±)-ketorolac. Process Biochem. 2015, 50, 230–236. [Google Scholar] [CrossRef]
- Bizerra, A.M.C.; Montenegro, T.G.C.; Lemos, T.L.G.; de Oliveira, M.C.F.; de Mattos, M.C.; Lavandera, I.; Gotor-Fernandez, V.; Gonzalo, G.; Gotor, V. Enzymatic regioselective production of chloramphenicol esters. Tetrahedron 2011, 67, 2858–2862. [Google Scholar] [CrossRef]
- Da Silva, M.R.; Montenegro, T.G.C.; de Mattos, M.C.; de Oliveira, M.C.F.; de Lemos, T.L.G.; de Gonzalo, G.; Lavandera, I.; Gotor-Fernandez, V.; Gotor, V. Regioselective preparation of thiamphenicol esters through lipase-catalyzed processes. J. Braz. Chem. Soc. 2014, 25, 987–994. [Google Scholar] [CrossRef]
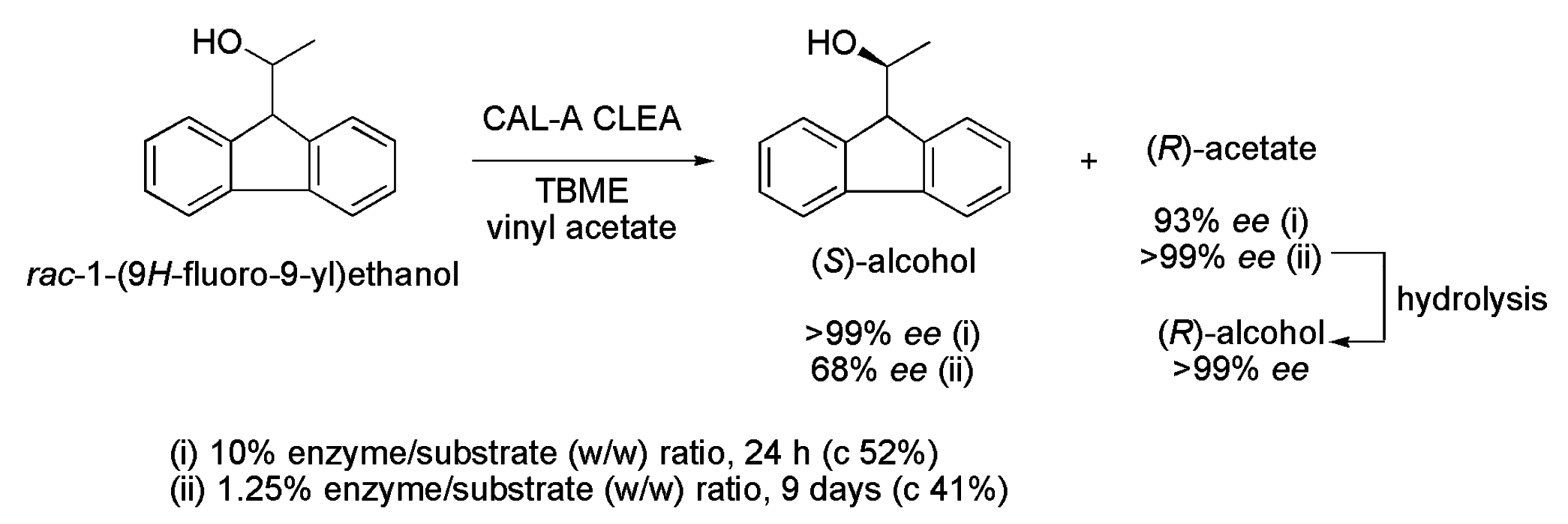
- Borowiecki, P.; Balter, S.; Justyniak, I.; Ochal, Z. First chemoenzymatic synthesis of (R)- and (S)-1-(9H-fluoren-9-yl)ethanol. Tetrahedron 2013, 24, 1120–1126. [Google Scholar] [CrossRef]
- Stürmer, R. Method for producing enantiomer-pure aminoalcohols. US 7,435,835 B2, 14 October 2008. [Google Scholar]
- Yamashita, S.; Mase, N.; Takabe, K. Chemoenzymatic total synthesis and determination of the absolute configuration of (S)-nebracetam. Tetrahedron 2008, 19, 2115–2118. [Google Scholar] [CrossRef]
- Araujo, D.M.F.; Vieira, G.A.B.; Mattos, M.C.; Lemos, T.L.G.; Oliveira, M.C.F.; Melo, V.M.M.; Gonzalo, G.; Gotor-Fernandez, V.; Gotor, V. Chemoenzymatic preparation of a biologically active naphthoquinone from Tabebuia impetiginosa using lipases or alcohol dehydrogenases. J. Mol. Catal. B 2009, 61, 279–283. [Google Scholar] [CrossRef]
- Kamal, A.; Khanna, G.; Krishnaji, T.; Ramu, R. Chemoenzymatic Process for the Stereoselective Preparation of (R)-γ-Amino-β-hydroxybutyric Acid or (R)-Carnitine from 3,4-Dihydroxybutanenitrile. US 7816119B2, 19 October 2010. [Google Scholar]
- Nagarapu, L.; Gaikwad, H.K.; Bantu, R.; Manikonda, S.R. Chemoenzymatic synthesis with lipase catalyzed resolution and evaluation of antitumor activity of (R/S)-2-[2-hydroxy-3-(4-phenylpiperazin-1-yl)propyl]-1H-pyrrolo[3,4-b]quinolin-3(2H)-one. Eur. J. Med. Chem. 2011, 46, 2152–2156. [Google Scholar] [CrossRef] [PubMed]
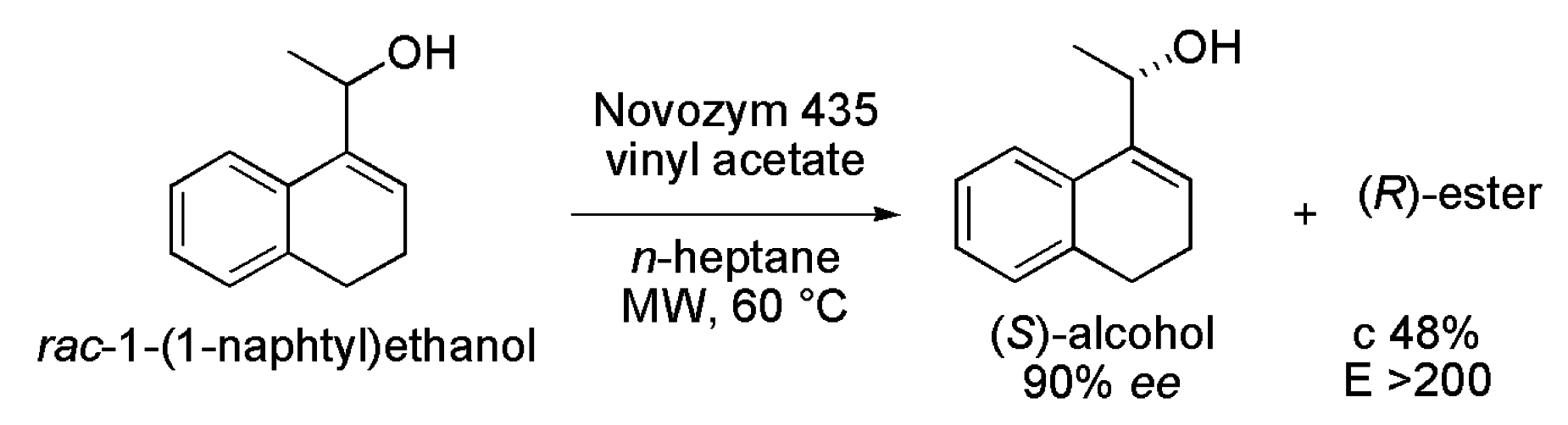
- Yadav, G.D.; Devendran, S. Lipase catalyzed kinetic resolution of (±)-1-(1-naphthyl) ethanol under microwave irradiation. J. Mol. Catal. B 2012, 81, 58–65. [Google Scholar] [CrossRef]
- Wamvakides, A.; Moutsos, V.; Schmitt, M. Synthesis of (+) and (ࢤ) 1-(5,5-Diphenyltetrahydrofuran-3-yl)-N,N-dimethylmethanamine, (+) and (ࢤ) 1-(2,2-Diphenyltetrahydrofuran-3-yl)-N,N-dimethylmethanamine and (+) and (ࢤ) 1-(2,2-Diphenyltetrahydrofuran-3-yl)-N-methylmethanamine. WO2013008044 A1, 17 January 2013. [Google Scholar]
- González-Sabín, J.; Ríos-Lombardía, N.; Gotor, V.; Morís, F. Enzymatic transesterification of pharmacologically interesting β-aminocycloalkanol precursors. Tetrahedron 2013, 24, 1421–1425. [Google Scholar] [CrossRef]
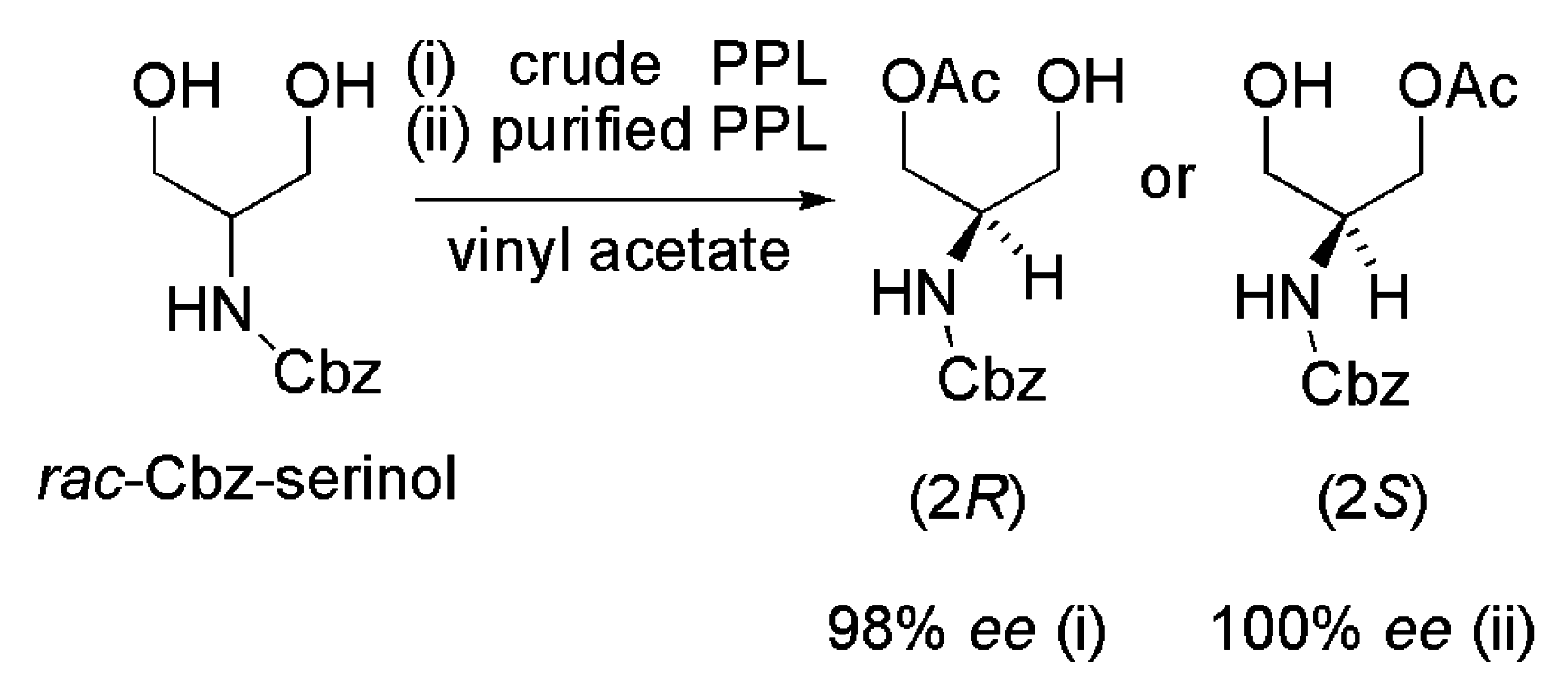
- Jacobsen, E.E.; Lie, A.; Frigstad, M.M.H.; el-Behairy, M.F.; Ljones, T.; Wohlgemuth, R.; Anthonsen, T. Desymmetrization of cbz-serinol catalyzed by crude pig pancreatic lipase reveals action of lipases with opposite enantioselectivity. J. Mol. Catal. B 2013, 85–86, 134–139. [Google Scholar] [CrossRef]
- Da Silva, M.R.; de Mattos, M.C.; de Oliveira, M.C.F.; de Lemos, T.L.G.; Ricardo, N.M.P.S.; de Gonzalo, G.; Lavandera, I.; Gotor-Fernández, V.; Gotor, V. Asymmetric chemoenzymatic synthesis of N-acetyl-α-amino esters based on lipase-catalyzed kinetic resolutions through interesterification reactions. Tetrahedron 2014, 70, 2264–2271. [Google Scholar] [CrossRef]
- Devendran, S.; Yadav, G.D. Lipase-catalyzed kinetic resolution of (±)-1-(2-furyl) ethanol in nonaqueous media. Chirality 2014, 26, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Karmee, S.K. The synthesis, properties, and applications of ascorbyl esters. Lipid. Technol. 2011, 23, 227–229. [Google Scholar] [CrossRef]
- Kharrat, N.; Aissa, I.; Sghaier, M.; Bouaziz, M.; Sellami, M.; Laouini, D.; Gargouri, Y. Lipophilization of ascorbic acid: A monolayer study and biological and antileishmanial activities. J. Agric. Food Chem. 2014, 62, 9118–9127. [Google Scholar]
- Chatterjee, S.; Ghadigaonkar, S.; Sur, P.; Sharma, A.; Chattopadhyay, S. A chemoenzymatic synthesis of hept-6-ene-2,5-diol stereomers: Application to asymmetric synthesis of decarestrictine L, pyrenophorol, and stagonolide E. J. Org. Chem. 2014, 79, 8067–8076. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Sharma, A.; Chattopadhyay, S. Chemoenzymatic synthesis of macrolide antibiotic (−)-A26771B. RSC Adv. 2014, 4, 42697–42705. [Google Scholar] [CrossRef]
- Wei, C.; Fu, X.-F.; Wang, Z.; Yu, X.-J.; Zhang, Y.-J.; Zheng, J.-Y. Efficient synthesis of vitamin E intermediate by lipase-catalyzed regioselective transesterification. J. Mol. Catal. B 2014, 106, 90–94. [Google Scholar] [CrossRef]
- Fonseca, T.S.; Silva, M.R.; Oliveira, M.C.F.; Lemos, T.L.G.; Marques, R.A.; Mattos, M.C. Chemoenzymatic synthesis of rasagiline mesylate using lipases. Appl. Catal. A 2015, 492, 76–82. [Google Scholar] [CrossRef]
- Carvalho, A.C.L.M.; Araujo, D.M.F.; Gonçalves, L.R.B.; Mattos, M.C.; Silva, M.R.; Oliveira, M.C.F.; Marques, R.A.; Lemos, T.L.G.; Fonseca, T.S.; Oliveira, U.M.F. Desenvolvimento de um Processo Biocatalítico para a Produção do (S)-Indanol, Precursor do Fármaco Mesilato de Rasagilina. BR102013024675–1A2, 15 September 2015. [Google Scholar]
- Grisenti, P. Biocatalyzed Synthesis of the Optically Pure (R) and (S) 3-Methyl-1,2,3,4-tetrahydroquinoline and Their Use as Chiral Synthons for the Preparation of the Antithrombotic (21R)- and (21S)-Argatroban. WO2015004015 A1, 15 January 2015. [Google Scholar]
- Forró, E.; Galla, Z.; Nádasdia, Z.; Árva, J.; Fülöp, F. Novel chemo-enzymatic route to a key intermediate for the taxol side-chain through enantioselective O-acylation. Unexpected acyl migration. J. Mol. Catal. B 2015, 116, 101–105. [Google Scholar] [CrossRef]
- Yamashita, M.; Taya, N.; Nishitani, M.; Oda, K.; Kawamoto, T.; Kimura, E.; Ishichi, Y.; Terauchi, J.; Yamano, T. Preparation of (S)-4-(1-(3,4-dichlorophenyl)-2-methoxyethyl)piperidine. Tetrahedron 2015, 26, 935–942. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, D.; Cui, B.; Han, W.; Chen, Y. Novozym 435 lipase mediated enantioselective kinetic resolution: A facile method for the synthesis of chiral tetrahydroquinolin-4-ol and tetrahydro1H-benzo[b]azepin-5-ol derivatives. Tetrahedron 2015, 71, 4738–4744. [Google Scholar] [CrossRef]
- Villa-Barro, A.; Gotor, V.; Brieva, R. Highly selective chemoenzymatic synthesis of enantiopure orthogonally protected trans-3-amino-4-hydroxypiperidines. Tetrahedron 2015, 71, 6907–6912. [Google Scholar] [CrossRef]
- Méndez-Sánchez, D.; Ríos-Lombardía, N.; Gotor, V.; Gotor-Fernández, V. Asymmetric synthesis of azolium-based 1,2,3,4-tetrahydronaphthalen-2-ols through lipase-catalyzed resolutions. Tetrahedron 2015, 26, 760–767. [Google Scholar] [CrossRef]
- Petrenz, A.; de Maria, P.D.; Ramanathan, A.; Hanefeld, U.; Ansorge-Schumacher, M.B.; Kara, S. Medium and reaction engineering for the establishment of a chemo-enzymatic dynamic kinetic resolution of rac-benzoin in batch and continuous mode. J. Mol. Catal. B 2015, 114, 42–49. [Google Scholar] [CrossRef]
- Hoyos, P.; Sansottera, G.; Fernández, M.; Molinari, F.; Sinisterra, J.V.; Alcántara, A.R. Enantioselective monoreduction of different 1,2-diaryl-1,2-diketones catalysed by lyophilised whole cells from Pichia glucozyma. Tetrahedron 2008, 64, 7929–7936. [Google Scholar] [CrossRef]
- Fragnelli, M.C.; Hoyos, P.; Romano, D.; Gandolfi, R.; Alcántara, A.R.; Molinari, F. Enantioselective reduction and deracemisation using the non-conventional yeast Pichia glucozyma in water-organic solvent biphasic systems: Preparation of (S)-1,2-diaryl-2-hydroxyethanones (benzoins). Tetrahedron 2012, 68, 523–528. [Google Scholar] [CrossRef]
- De Miranda, A.S.; Miranda, L.S.M.; de Souza, R.O.M.A. Lipases: Valuable catalysts for dynamic kinetic resolutions. Biotechnol. Adv. 2015, 33, 372–393. [Google Scholar] [CrossRef] [PubMed]
- Ainge, D.; Gnad, F.; Sinclair, R.; Vaz, L.M.; Wells, A. Use of Intermediates (R)-2,2,4-Trimethyl-l,3-dioxolane-4-yl) methanol (a), 3-Fluoro-4-nitro-phenol (b) and 1-(4-Chloro-benzyl)-piperidin-4-ylamine (c). WO2009035407 A1, 19 March 2009. [Google Scholar]
- Shapiro, E.; Fishman, A.; Effenberger, R.; Maymon, A.; Schwartz, A. Selective Enzymatic Esterification and Solvolysis of Epimeric Vitamin D Analog and Separation of the Epimers. US 8129173 B2, 6 March 2012. [Google Scholar]
- Bouzemi, N.; Grib, I.; Houiene, Z.; Aribi-Zouioueche, L. Enantiocomplementary preparation of (S)- and (R)-arylalkylcarbinols by lipase-catalysed resolution and Mitsunobu inversion: Impact of lipase amount. Catalysts 2014, 4, 215–225. [Google Scholar] [CrossRef]
- López-Iglesias, M.; Busto, E.; Gotor, V.; Gotor-Fernández, V. Chemoenzymatic asymmetric synthesis of 1,4-benzoxazine derivatives: Application in the synthesis of a levofloxacin precursor. J. Org. Chem. 2015, 80, 3815–3824. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carvalho, A.C. L.d.M.; Fonseca, T.D.S.; Mattos, M.C.d.; Oliveira, M.D.C.F.d.; Lemos, T.L.G.d.; Molinari, F.; Romano, D.; Serra, I. Recent Advances in Lipase-Mediated Preparation of Pharmaceuticals and Their Intermediates. Int. J. Mol. Sci. 2015, 16, 29682-29716. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226191
Carvalho ACLdM, Fonseca TDS, Mattos MCd, Oliveira MDCFd, Lemos TLGd, Molinari F, Romano D, Serra I. Recent Advances in Lipase-Mediated Preparation of Pharmaceuticals and Their Intermediates. International Journal of Molecular Sciences. 2015; 16(12):29682-29716. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226191
Chicago/Turabian StyleCarvalho, Ana Caroline Lustosa de Melo, Thiago De Sousa Fonseca, Marcos Carlos de Mattos, Maria Da Conceição Ferreira de Oliveira, Telma Leda Gomes de Lemos, Francesco Molinari, Diego Romano, and Immacolata Serra. 2015. "Recent Advances in Lipase-Mediated Preparation of Pharmaceuticals and Their Intermediates" International Journal of Molecular Sciences 16, no. 12: 29682-29716. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226191