
The Complete Mitochondrial Genome of Mindarus keteleerifoliae (Insecta: Hemiptera: Aphididae) and Comparison with Other Aphididae Insects

Abstract
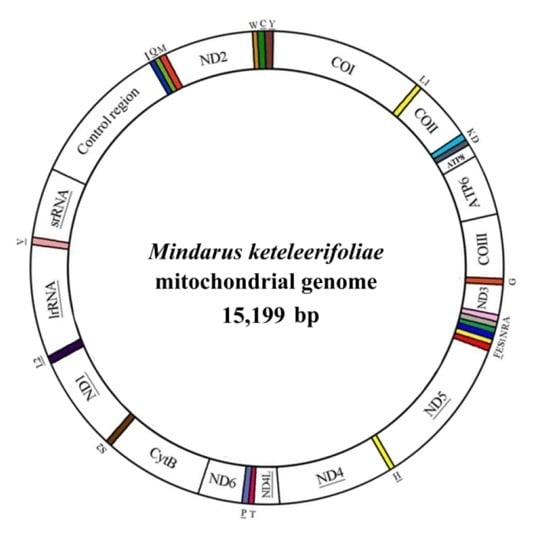
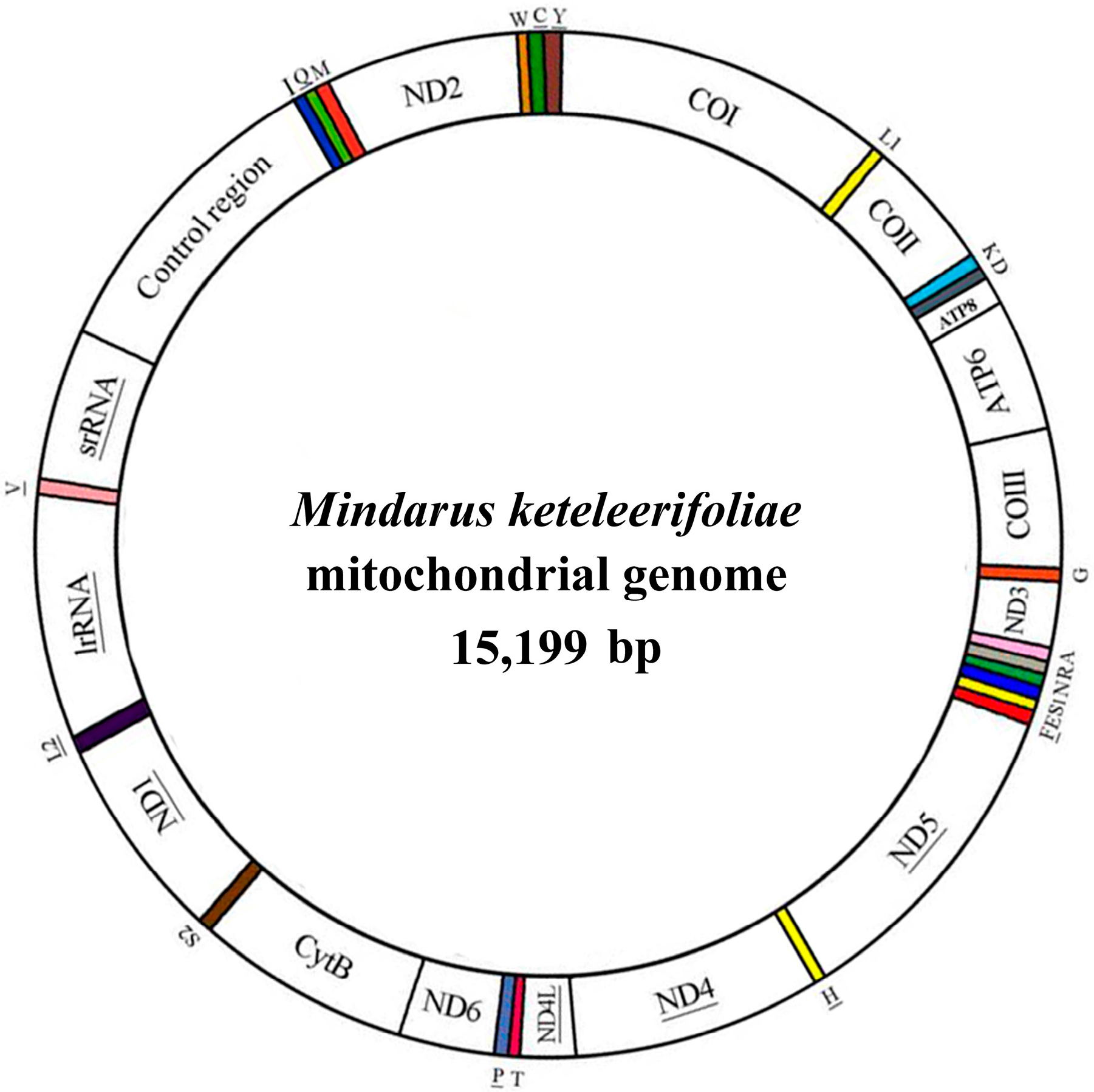
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Species | Length (bp) | GenBank No. | Reference | |
|---|---|---|---|---|---|
| Aphididae | Aphidinae | Schizaphis graminum | 15,721 | NC_006158 | Thao et al., 2004 [13] |
| Acyrthosiphon pisum | 16,971 | NC_011594 | IAGC, 2010 [14] | ||
| Diuraphis noxia | 15,784 | NC_022727 | Zhang et al., 2014 [15] | ||
| Sitobion avenae | 15,180 | NC_024683 | Zhang et al., 2014 [16] | ||
| Cavariella salicicola | 16,317 | NC_022682 | Wang et al., 2013 [17] | ||
| Aphis gossypii | 15,869 | NC_024581 | Zhang et al., 2014 [18] | ||
| Greenideinae | Cervaphis quercus | 15,272 | NC_024926 | Wang et al., 2014 [12] | |
| Mindarinae | Mindarus keteleerifoliae | 15,199 | KP722576 | This study | |
| Phylloxeridae | Viteus vitifoliae * | 12,349 | DQ021446 | Direct submission | |
2. Results and Discussion
2.1. Genome Organization and Composition

| Gene | Strand | Position | Anticodon | Size (bp) | Start Codon | Stop Codon | Intergenic Nucleotides * |
|---|---|---|---|---|---|---|---|
| cox1 | J | 1–1531 | – | 1531 | ATA | T | −1 |
| tRNA-Leu | J | 1531–1596 | TAA | 66 | – | – | 3 |
| cox2 | J | 1600–2271 | – | 672 | ATA | TAA | – |
| tRNA-Lys | J | 2272–2344 | CTT | 73 | – | – | 7 |
| tRNA-Asp | J | 2352–2419 | GTC | 68 | – | – | 17 |
| atp8 | J | 2437–2586 | – | 150 | ATA | TAA | −20 |
| atp6 | J | 2567–3220 | – | 654 | ATT | TAA | |
| cox3 | J | 3221–4006 | – | 786 | ATG | TAA | −1 |
| tRNA-Gly | J | 4007–4068 | TCC | 62 | – | – | −3 |
| nad3 | J | 4066–4422 | – | 357 | ATA | TAA | – |
| tRNA-Ala | J | 4423–4487 | TGC | 65 | – | – | −1 |
| tRNA-Arg | J | 4487–4555 | TCG | 69 | – | – | −1 |
| tRNA-Asn | J | 4555–4623 | GTT | 69 | – | – | −1 |
| tRNA-Ser | J | 4623–4684 | TCT | 62 | – | – | 6 |
| tRNA-Glu | J | 4691–4764 | TTC | 74 | – | – | 3 |
| tRNA-Phe | N | 4768–4837 | GAA | 70 | – | – | – |
| nad5 | N | 4838–6508 | – | 1671 | ATT | TAA | – |
| tRNA-His | N | 6509–6574 | GTG | 66 | – | – | – |
| nad4 | N | 6575–7883 | – | 1309 | ATA | T | 5 |
| nad4L | N | 7889–8179 | – | 291 | ATA | TAA | – |
| tRNA-Thr | J | 8180–8245 | TGT | 66 | – | – | – |
| tRNA-Pro | N | 8246–8316 | TGG | 71 | – | – | 1 |
| nad6 | J | 8318–8812 | – | 495 | ATT | TAA | −1 |
| cob | J | 8812–9927 | – | 1116 | ATG | TAA | −2 |
| tRNA-Ser | J | 9926–9990 | TGA | 65 | 10 | ||
| nad1 | N | 10001–10936 | – | 936 | ATT | TAA | – |
| tRNA-Leu | N | 10937–11001 | TAG | 65 | – | – | – |
| rrnL | N | 11002–12267 | – | 1266 | – | – | 1 |
| tRNA-Val | N | 12269–12330 | TAC | 62 | – | – | 3 |
| rrnS | N | 12334–13098 | – | 765 | – | – | – |
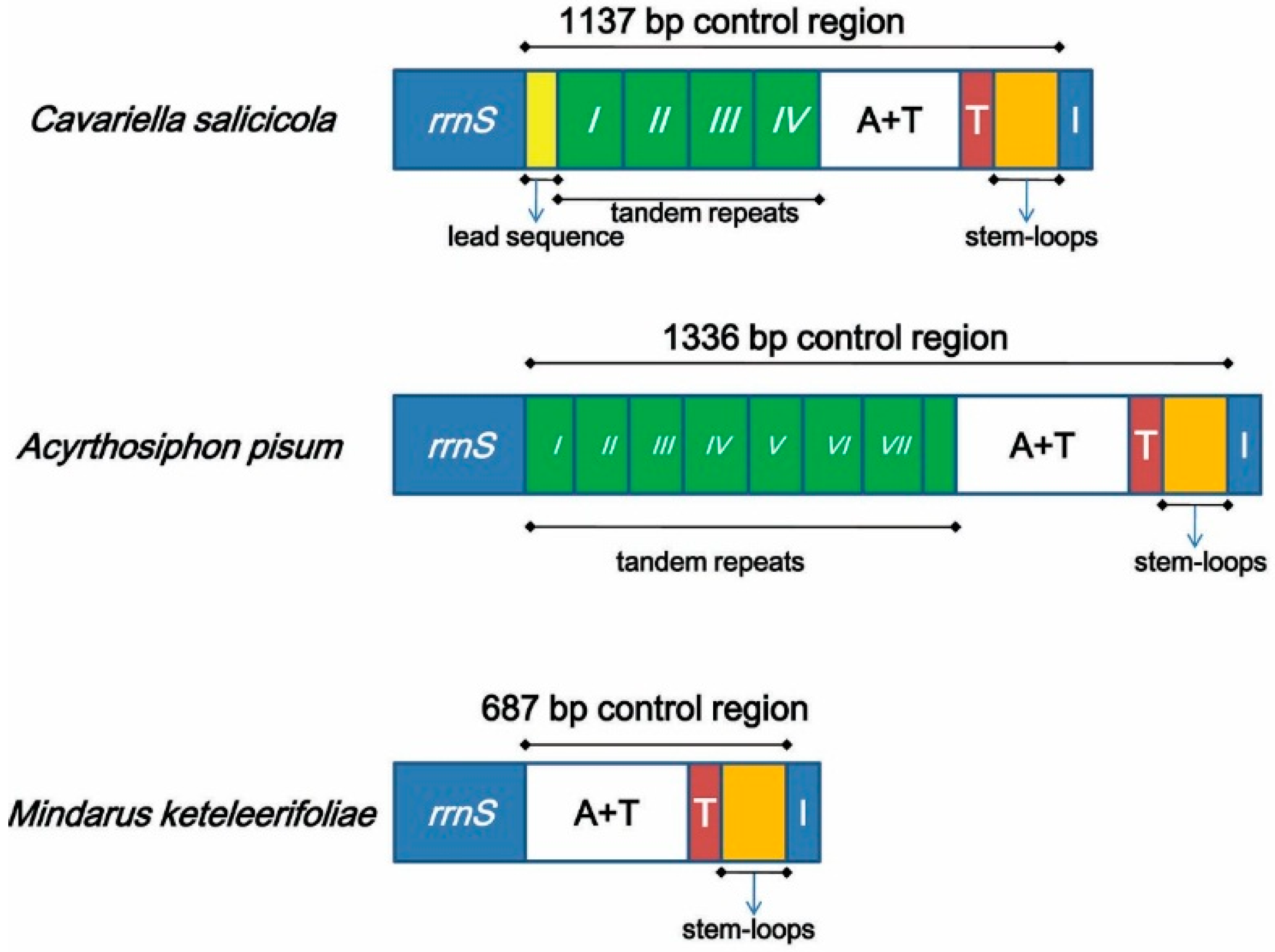
| control region | – | 13099–13785 | – | 687 | – | – | – |
| tRNA-Ile | J | 13786–13851 | GAT | 66 | – | – | 3 |
| tRNA-Gln | N | 13855–13920 | TTG | 66 | – | – | 29 |
| tRNA-Met | J | 13950–14017 | CAT | 68 | – | – | – |
| nad2 | J | 14018–14995 | – | 978 | ATA | TAA | – |
| tRNA-Trp | J | 14996–15063 | TCA | 68 | – | – | −2 |
| tRNA-Cys | N | 15062–15127 | GCA | 66 | – | – | 4 |
| tRNA-Tyr | N | 15132–15198 | GTA | 67 | – | – | 1 |
2.2. Protein-Coding Genes
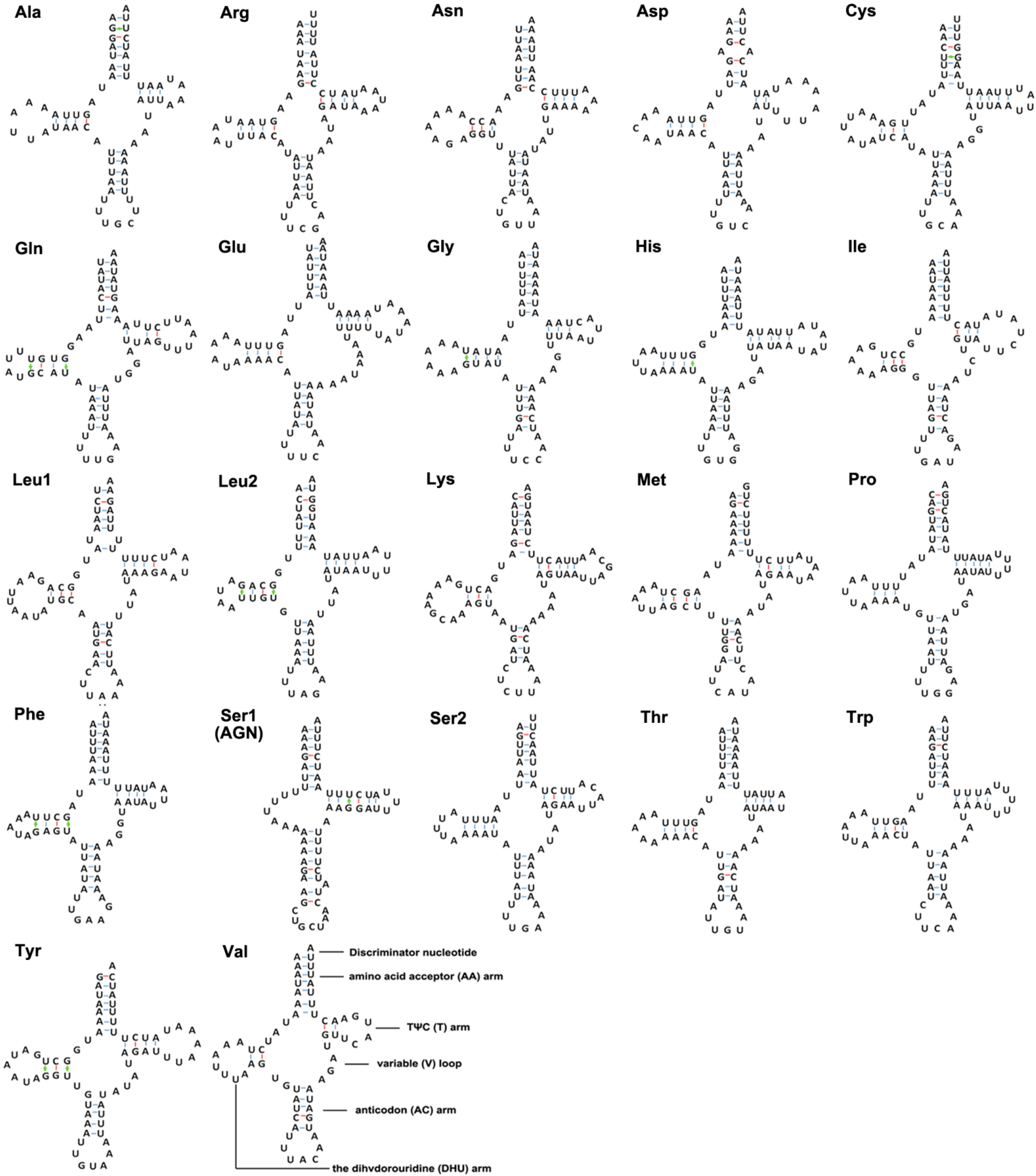
2.3. tRNA and rRNA

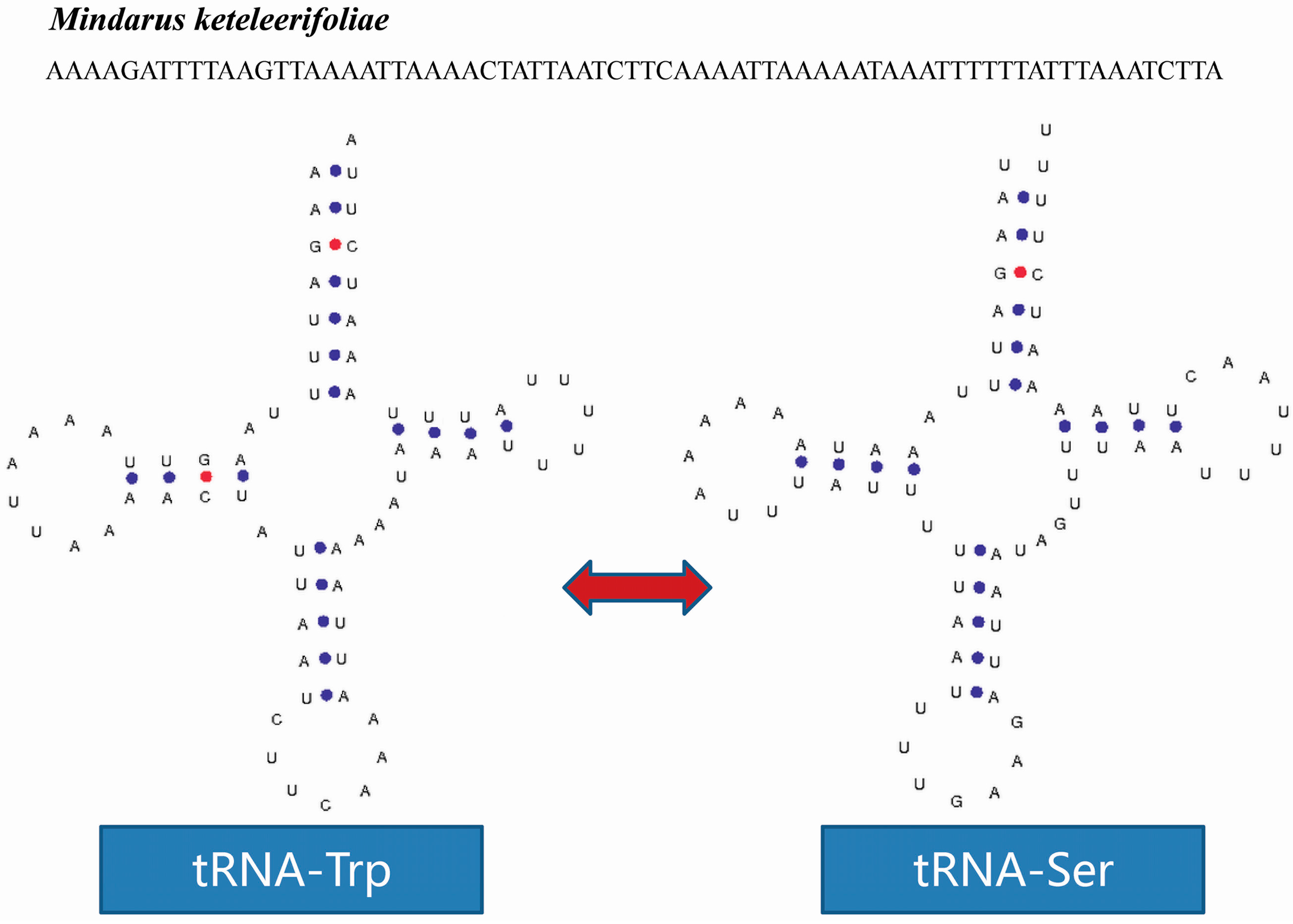
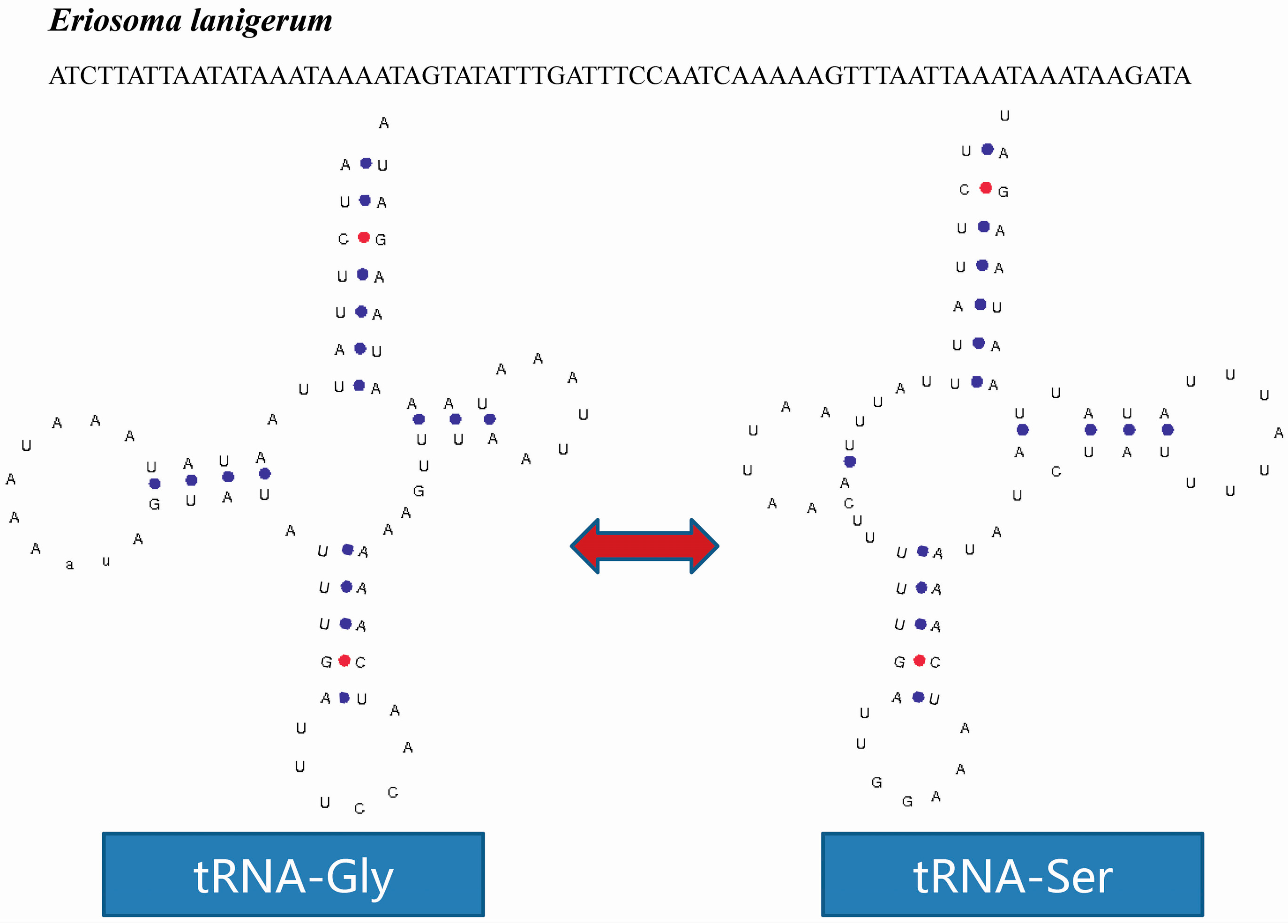
2.4. tRNA Isomerism

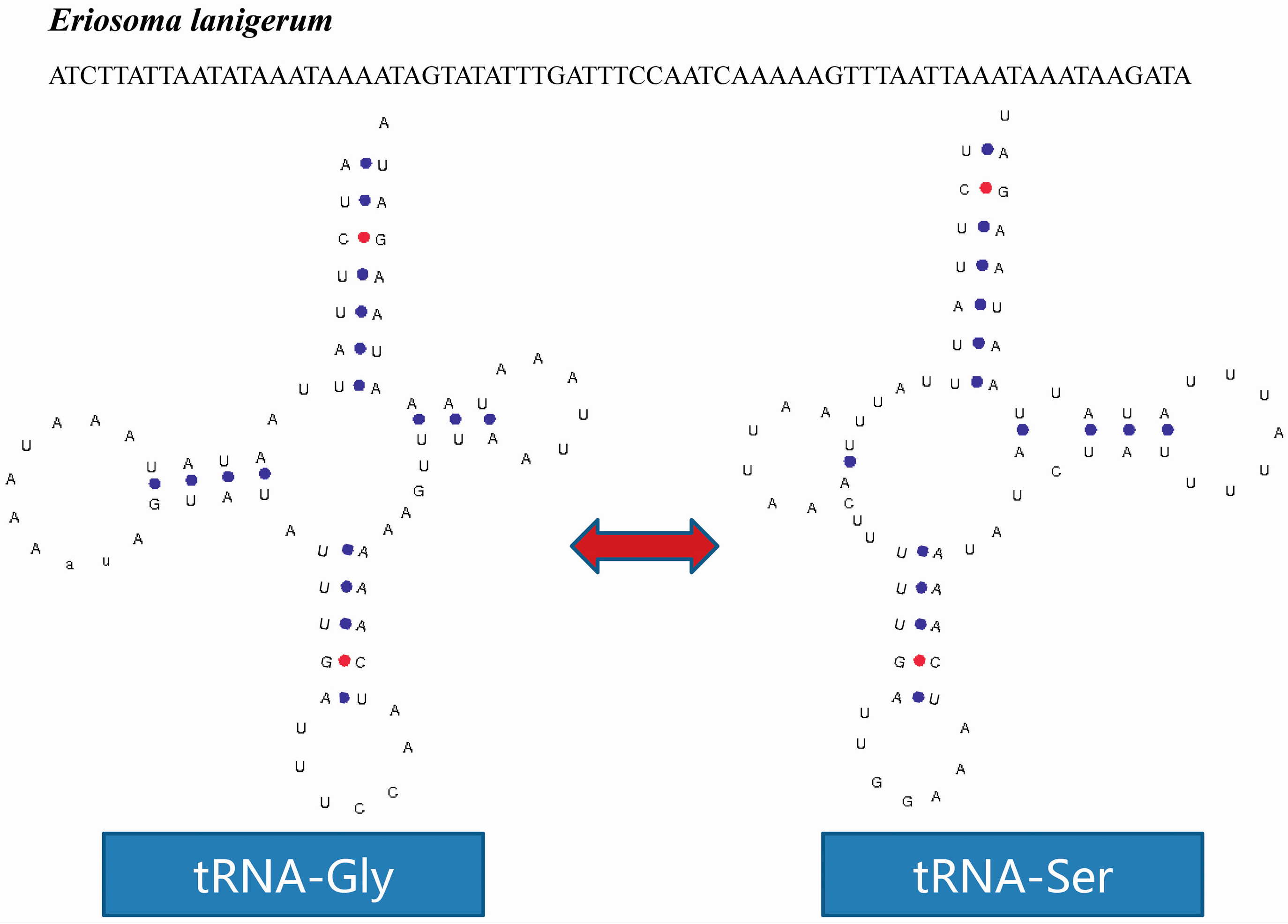
2.5. Non-Coding Regions

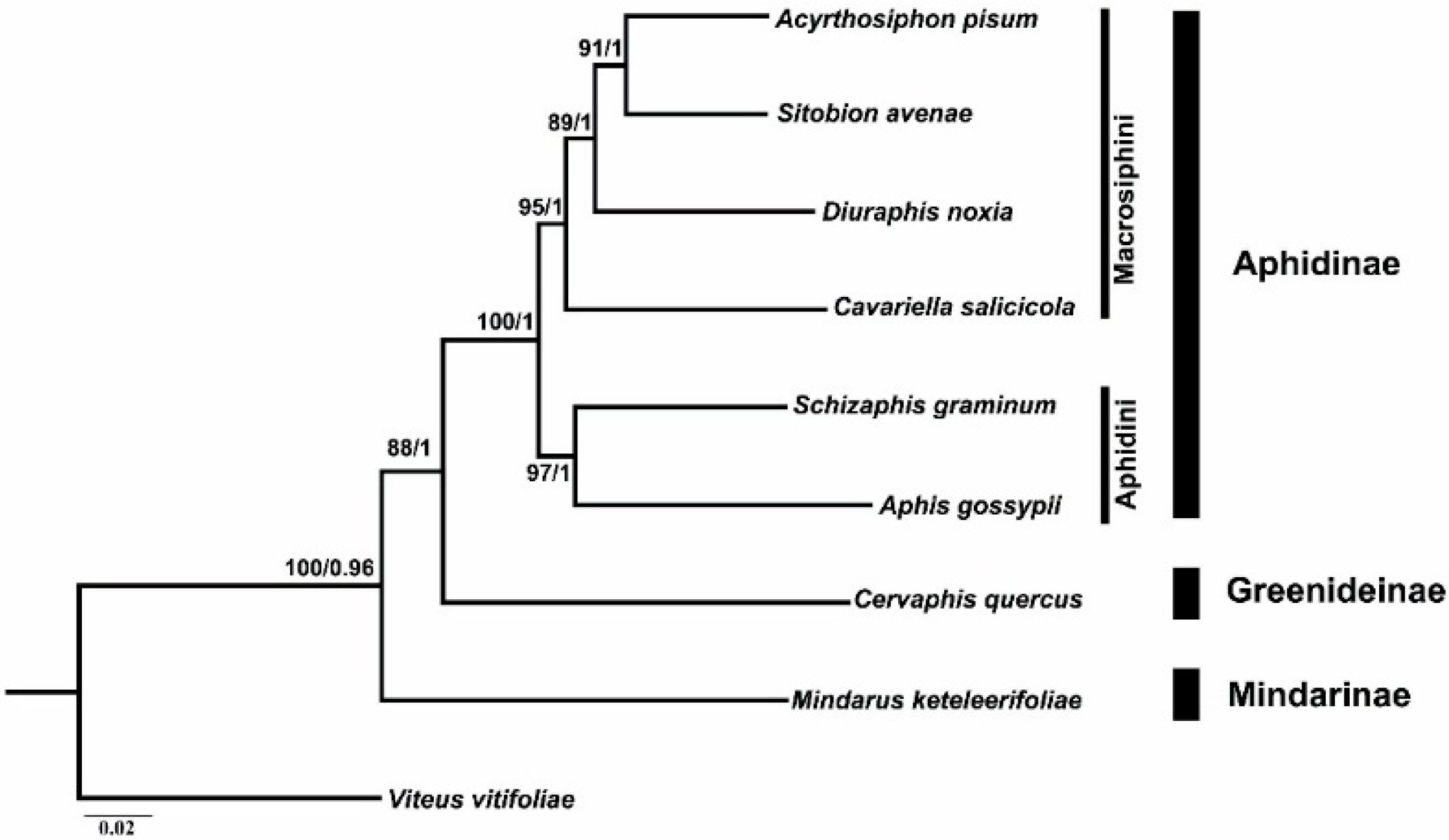
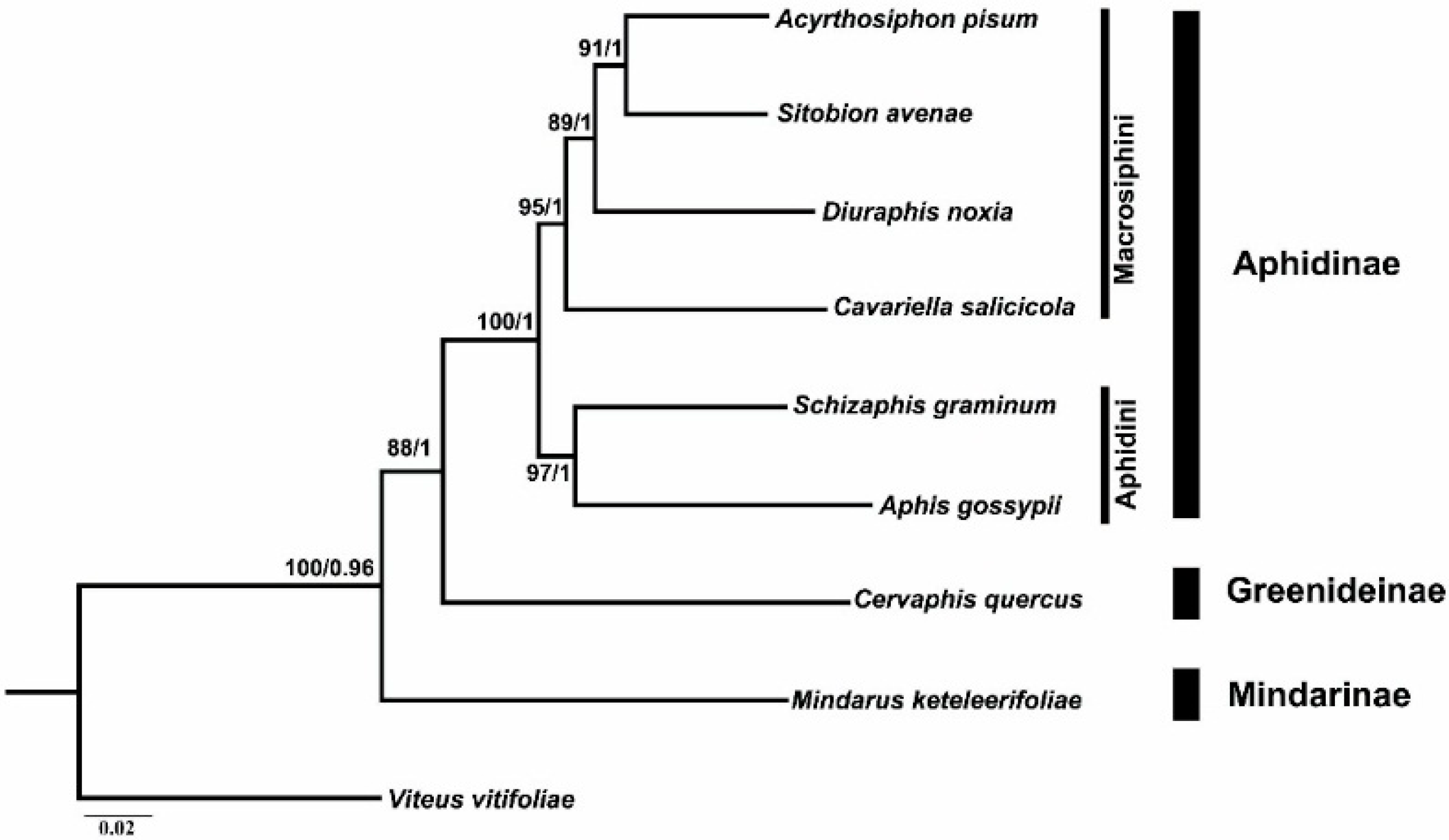
2.6. Phylogenetic Analyses
3. Materials and Methods
3.1. Experimental Sample
3.2. DNA Extraction, Amplification and Sequencing
3.3. Mitogenome Annotation and Analysis
3.4. Phylogenetic Analysis
4. Conclusion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [PubMed]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, J.M.; Li, M.; Dong, P.Z.; Cui, Y.; Xie, Q.; Bu, W.J. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insecta: Hemiptera: Heteroptera). BMC Genom. 2008, 9, 610. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, H.Y.; Song, F.; Shi, A.M.; Zhou, X.G.; Cai, W.Z. Comparative mitogenomic analysis of damsel bugs representing three tribes in the family Nabidae (Insecta: Hemiptera). PLoS ONE 2012, 7, e45925. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Yang, P.C.; Jiang, F.; Shali, Y.; Chapuis, M.P.; Sword, G.; Kang, L. Mitochondrial genomes reveal the global phylogeography and dispersal routes of the migratory locust. Mol. Ecol. 2012, 21, 4344–4358. [Google Scholar] [CrossRef] [PubMed]
- Remaudière, G.; Remaudière, M. Catalogue des Aphididae du Monde; Institut National de la Recherche Agronomique: Paris, France, 1997. [Google Scholar]
- Version 5.0/5.0. Available online: http://Aphid.SpeciesFile.org (accessed on 8 October 2015).
- Blackman, R.L.; Eastop, V.F. Aphids on the World’s Crops: An Identification and Information Guide, 2nd ed.; John Wiley & Sons: Chichester, UK, 2000. [Google Scholar]
- Huang, X.L.; Qiao, G.X. Aphids as models for ecological and evolutionary studies. Insect. Sci. 2014, 21, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, X.L.; Qiao, G.X. The complete mitochondrial genome of Cervaphis quercus (Insecta: Hemiptera: Aphididae: Greenideinae). Insect. Sci. 2014, 21, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Thao, M.L.; Baumann, L.; Baumann, P. Organization of the mitochondrial genomes of whiteflies, aphids, and psyllids (Hemiptera, Sternorrhyncha). BMC Evol. Biol. 2004, 4, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The International Aphid Genomics Consortium. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 2010, 8, e1000313. [Google Scholar]
- Zhang, B.; Ma, C.; Edwards, O.; Fuller, S.; Kang, L. The mitochondrial genome of the Russian wheat aphid Diuraphis noxia: Large repetitive sequences between trnE and trnF in aphids. Gene 2014, 533, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zheng, J.; Liang, L.; Fuller, S.; Ma, C.S. The complete mitochondrial genome of Sitobion avenae (Hemiptera: Aphididae). Mitochondrial DNA 2014. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, X.L.; Qiao, G.X. Comparative analysis of mitochondrial genomes of five aphid species (Hemiptera: Aphididae) and phylogenetic implications. PLoS ONE 2013, 8, e77511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Luo, J.; Wang, C.; Lv, L.; Li, C.; Jiang, W.; Cui, J.; Rajput, L.B. Complete mitochondrial genome of Aphis gossypii Glover (Hemiptera: Aphididae). Mitochondrial DNA 2014. [Google Scholar] [CrossRef]
- Heie, O.E. Paleontology and phylogeny. In Aphids: Their Biology, Natural Enemies and Control; Elsevier: Amsterdam, The Netherlands, 1987. [Google Scholar]
- Quednau, F.W. Atlas of the Drepanosiphine Aphids of the World Part III; The American Entomological Institute: Gainesville, FL, USA, 2010. [Google Scholar]
- Zhang, G.X.; Qiao, G.X.; Zhong, T.S.; Zhang, W.Y. Homoptera: Mindaridae and Pemphigidae; Science Press: Beijing, China, 1999. [Google Scholar]
- Clary, D.O.; Wolstenholme, D.R. The mitochondrial DNA molecular of Drosophila yakuba: Nucleotide sequence, gene organization and genetic code. J. Mol. Evol. 1985, 22, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Boore, J.L.; Brown, W.M. The complete mitochondrial DNA sequence of the horseshoe crab Limulus polyphemus. Mol. Biol. Evol. 2000, 17, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, J.J.; Johnston, J.S.; Cannone, J.J.; Gutell, R.R. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): Structure, organization and retrotransposable elements. Insect Mol. Biol. 2006, 15, 657–686. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.J.; Shi, M.; Sharkey, M.J.; van Achterberg, C.; Chen, X.X. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to holometabolous insects. BMC Genom. 2010, 11, 371. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Jiang, L.Y.; Qiao, G.X. Hemipteran mitochondrial genomes: Features, structures and implications for phylogeny. Int. J. Mol. Sci. 2015, 16, 12382–12404. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Peccoud, J.; Ollivier, A.; Plantegenest, M.; Simon, J.C. A continuum of genetic divergence from sympatric host races to species in the pea aphid complex. Proc. Natl. Acad. Sci. USA 2009, 106, 7495–7500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Edwards, O.R.; Kang, L.; Fuller, S.J. Russian wheat aphids (Diuraphis noxia) in China: Native range expansion or recent introduction? Mol. Ecol. 2012, 21, 2130–2144. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Kang, J.; Jung, C.; Hoelmer, K.; Lee, S.; Lee, S. Complete mitochondrial genome of brown marmorated stink bug Halyomorpha halys (Hemiptera: Pentatomidae), and phylogenetic relationships of hemipteran suborders. Mol. Cell 2009, 28, 155. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gao, J.; Liu, H.; Liang, A.; Zhou, X.; Cai, W.Z. The architecture and complete sequence of mitochondrial genome of an assassin bug Agriosphodrus dohrni (Hemiptera: Reduviidae). Int. J. Biol. Sci. 2011, 7, 792. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Watanabe, Y.; Watanabe, K.; Ohtsuki, T. A protein extension to shorten RNA: Elongated elongation factor-Tu recognizes the D-arm of T-armless tRNAs in nematode mitochondria. Biochem. J. 2006, 399, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Von Dohlen, C.D.; Moran, N.A. Molecular data support a rapid radiation of aphids in the Cretaceous and multiple origins of host alternation. Biol. J. Linn. Soc. 2000, 71, 689–717. [Google Scholar] [CrossRef]
- Heie, O.E.; Wegierek, P. A classification of the Aphidomorpha (Hemiptera: Sternorrhyncha) under consideration of the fossil taxa. Redia 2009, 92, 69–77. [Google Scholar]
- Heie, O.E. The aphidoidea (hemiptera) of fennoscandia and Denmark. IV. In Family Aphididae: Part 1 of Tribe Macrosiphini of Subfamily Aphidinae; E.J. Brill/Scandinavian Science Press Ltd.: Leiden, The Netherlands, 1992. [Google Scholar]
- Ortiz-Rivas, B.; Martínez-Torres, D. Combination of molecular data support the existence of three main lineages in the phylogeny of aphids (Hemiptera: Aphididae) and the basal position of the subfamily Lachninae. Mol. Phylogenet. Evol. 2010, 55, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, S.; Jang, Y. Macroevolutionary patterns in the Aphidini aphids (Hemiptera: Aphididae): Diversification, host association, and biogeographic origins. PLoS ONE 2011, 6, e24749. [Google Scholar] [CrossRef] [PubMed]
- Foottit, R.G.; Maw, H.E.L.; Von Dohlen, C.D.; Hebert, P.D.N. Species identification of aphids (Insecta: Hemiptera: Aphididae) through DNA barcodes. Mol. Ecol. Res. 2008, 8, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Normark, B.B. Phylogeny and evolution of parthenogenetic weevils of the Aramigus tessellatus species complex (Coleoptera: Curculionidae: Naupactini): Evidence from mitochondrial DNA sequences. Evolution 1996, 50, 734–745. [Google Scholar] [CrossRef]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef]
- Simon, C.; Frati, F.; Beckenbach, A.T.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc.Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- NCBI Web Site. Available online: http://blast.ncbi.nlm.nih.gov/Blast (accessed on 8 October 2015).
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Mfold Web Server. Available online: http://www.bioinfo.rpi.edu/applications/mfold/ (accessed on 8 October 2015).
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, G. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Chen, J.; Jiang, L.-Y.; Qiao, G.-X. The Complete Mitochondrial Genome of Mindarus keteleerifoliae (Insecta: Hemiptera: Aphididae) and Comparison with Other Aphididae Insects. Int. J. Mol. Sci. 2015, 16, 30091-30102. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226219
Wang Y, Chen J, Jiang L-Y, Qiao G-X. The Complete Mitochondrial Genome of Mindarus keteleerifoliae (Insecta: Hemiptera: Aphididae) and Comparison with Other Aphididae Insects. International Journal of Molecular Sciences. 2015; 16(12):30091-30102. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226219
Chicago/Turabian StyleWang, Yuan, Jing Chen, Li-Yun Jiang, and Ge-Xia Qiao. 2015. "The Complete Mitochondrial Genome of Mindarus keteleerifoliae (Insecta: Hemiptera: Aphididae) and Comparison with Other Aphididae Insects" International Journal of Molecular Sciences 16, no. 12: 30091-30102. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226219