
An Overview of the Prediction of Protein DNA-Binding Sites

Abstract
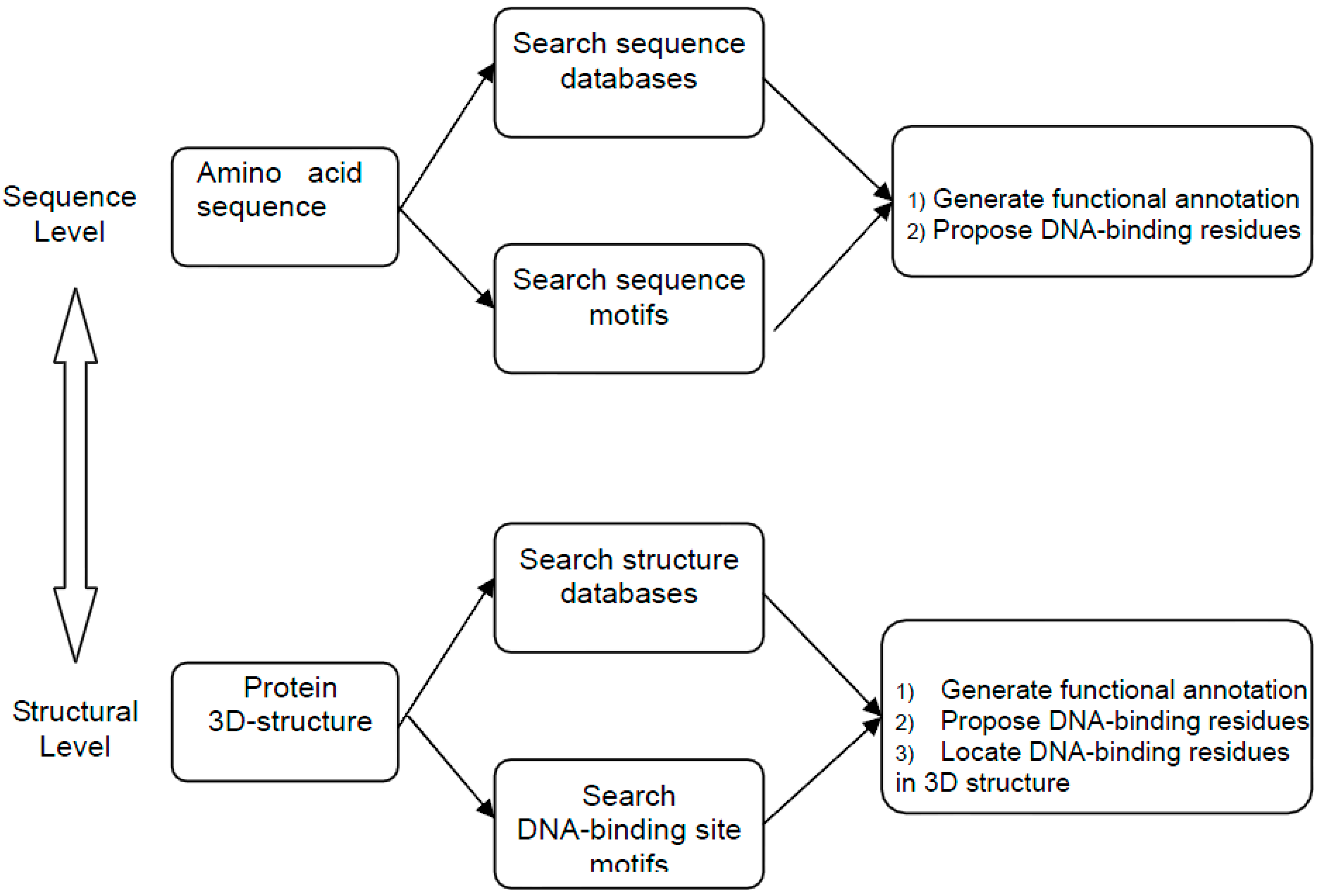
:1. Introduction

2. Method Development of DNA-Binding Site Prediction
2.1. Benchmark Data Set

{kind=link}
| ID | Ref. No. | Notes |
|---|---|---|
| DB179 | [33] | 179 DNA-binding proteins, almost entirely nonredundant at 40% sequence identity |
| NB3797 | [33] | 3797 nonbinding proteins, significant redundancy at 35% sequence identity level (only 3482 independent clusters) |
| PD138 | [27] | 138 DNA-binding proteins, almost entirely nonredundant at 35% sequence identity, divided into seven structural classes |
| DISIS | [3] | 78 DNA-binding proteins, close to nonredundant at 20% sequence identity |
| PDNA62 | [34] | 62 DNA-binding proteins, 78 chains, 57 nonredundant sequences at 30% identity. |
| NB110 | [34] | 110 nonbinding proteins, nonredundant at 30% sequence identity level, derived from the RS126 secondary structure data set by removing entries related to DNA |
| BIND54 | [35] | Reported as 54 binding proteins, actually 58 chains, nonredundant at 30% sequence identity, original list of proteins was reported in [1] |
| NB250 | [35] | 250 nonbinding proteins, mostly nonredundant at a 35% sequence identity |
| DBP374 | [18] | 374 DNA-binding proteins, significant redundancy at a 25% sequence identity level |
| TS75 | [18] | 75 DNA-binding proteins, designed to be independent from DBP374 and PDNA62 but has some redundant entries in both at a 35% sequence identity level |
| PDNA-316 | [29] | 316 target proteins used in metaDBSite Web server, at 30% sequence identity |
| DNABindR171 | [16] | 171 proteins with mutual sequence identity ≤30% and each protein has at least 40 amino acid residues. All the structures have resolution better than 3.0 Å and an R factor less than 0.3 |
2.2. Different Residue Properties Used in Developing Predictors
2.2.1. Sequence-Based Features
2.2.2. Structural-Based Features
2.2.3. Physical and Chemical Features
2.3. Prediction Methods
2.3.1. Prediction Based on Sequences
2.3.2. Prediction Based on Protein Structures
2.3.3. Homology Modeling and Threading
2.4. Prediction Algorithms
2.4.1. Prediction Algorithms Based on Individual Descriptors
2.4.2. Prediction Algorithms Based on Simple Statistical Methods
2.4.3. Prediction Algorithms Based on Machine Learning Methods
2.4.4. Hybrid Learning and Meta-Prediction Methods
2.5. Performance and Evaluation of Different Predictors
2.5.1. Performance Measures
| Parameter | Meaning | Expression |
|---|---|---|
| Accuracy (ACC) | Percentage of correct prediction | a |
| Sensitivity | Percentage of correctly predicted positive | |
| Specificity | Percentage of correctly predicted negative | |
| Strength | Mean value of the sum of sensitivity and specificity | |
| MCC | Matthews correlation coefficient | |
| Precision | Positive predictive rate | |
| F-measure | The harmonic mean of sensitivity and specificity | |
| AUC b | Probability that a classifier will rank a randomly chosen positive instance higher than a randomly chosen negative one |
2.5.2. Comparison of Different Prediction Methods
2.5.3. Selected Web Servers of DNA-Binding Site Predictors
| Author & Year | Data set (own/PDNA-316) | Performance | Alogrithm b | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | SEN | SPE | AUC | MCC | Strength | F-Measure | Precision | ||||
| Jones 2003 | own | 0.680 | 1 | [36] | |||||||
| Ahmad 2004 | own | 0.664 | 0.682 | 0.660 | 2 | [34] | |||||
| a Ahmad 2004 | PDNA-316 | 0.750 | 0.530 | 0.760 | 0.170 | 0.650 | 0.230 | 3 | [29] | ||
| Ferrer-Costa 2005 | own | 0.835 | 4 | [87] | |||||||
| Kuznetsov 2006 | own | 0.760 | 0.769 | 0.747 | 0.830 | 0.450 | 3 | [38] | |||
| Wang 2006 | own | 0.703 | 0.694 | 0.704 | 0.750 | 3 | [88] | ||||
| Wang 2006 | PDNA-316 | 0.780 | 0.540 | 0.800 | 0.210 | 0.670 | 0.260 | 3 | [29] | ||
| Yan 2006 | own | 0.710 | 0.530 | 0.350 | 5 | [16] | |||||
| Yan 2006 | PDNA-316 | 0.730 | 0.660 | 0.740 | 0.230 | 0.700 | 0.260 | 3 | [29] | ||
| Tjong 2007 | own | 0.680 | 2 | [61] | |||||||
| Ofran 2007 | own | 0.890 | 2/3 | [3] | |||||||
| Ofran 2007 | PDNA-316 | 0.920 | 0.190 | 0.980 | 0.250 | 0.590 | 0.270 | 3 | [29] | ||
| Hwang 2007 | own | 0.772 | 0.764 | 0.766 | 3 | [15] | |||||
| Hwang 2007 | PDNA-316 | 0.780 | 0.690 | 0.790 | 0.290 | 0.740 | 0.310 | 3 | [29] | ||
| Nimrod 2009 | own | 0.900 | 0.900 | 0.350 | 6 | [48,89] | |||||
| Wang 2009 | own | 0.800 | 0.731 | 0.806 | 0.850 | 6 | [80] | ||||
| Wang 2009 | PDNA-316 | 0.820 | 0.670 | 0.830 | 0.320 | 0.750 | 0.340 | 3 | [29] | ||
| Wu 2009 | own | 0.914 | 0.766 | 0.944 | 6 | [18] | |||||
| Carson 2010 | own | 0.785 | 0.797 | 0.772 | 0.860 | 0.570 | 7 | [19] | |||
| Ozbek 2010 | own | 0.960 | 0.360 | 0.990 | 8 | [84] | |||||
| Si 2011 | PDNA-316 | 0.770 | 0.770 | 0.770 | 0.320 | 0.770 | 0.330 | 3 | [29] | ||
| Methods | URLs | References | Publication Year |
|---|---|---|---|
| newDNA-Prot | http://sourceforge.net/projects/newdnaprot/ | [90] | 2014 |
| DNABind | http://mleg.cse.sc.edu/DNABind/ | [91] | 2013 |
| DNABR | http://www.cbi.seu.edu.cn/DNABR/ | [92] | 2012 |
| DR_bind | http://dnasite.limlab.ibms.sinica.edu.tw | [93] | 2012 |
| MetaDBSite | http://projects.biotec.tu-dresden.de/metadbsite/ http://sysbio.zju.edu.cn/metadbsite | [29] | 2011 |
| DNABINDPROT | http://www.prc.boun.edu.tr/appserv/prc/dnabindprot/ | [84] | 2010 |
| bindn-rf | http://bioinfo.ggc.org/bindn-rf/ | [80] | 2009 |
| DBindR | http://www.cbi.seu.edu.cn/DBindR/DBindR.htm | [18] | 2009 |
| DP-Bind | http://lcg.rit.albany.edu/dp-bind | [15,38] | 2007 |
| BindN | http://bioinfo.ggc.org/bindn/ | [88] | 2006 |
2.6. Status of the Prediction of Protein-Binding Sites in DNA Sequence
3. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Luscombe, N.M.; Austin, S.E.; Berman, H.M.; Thornton, J.M. An overview of the structures of protein–DNA complexes. Genome Biol. 2000, 1, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.C.; Rattei, T.; Arnold, R.; Guldener, U.; Munsterkotter, M.; Nenova, K.; Kastenmuller, G.; Tischler, P.; Wolling, A.; Volz, A.; et al. PEDANT covers all complete RefSeq genomes. Nucleic Acids Res. 2009, 37, D408–D411. [Google Scholar] [CrossRef]
- Ofran, Y.; Mysore, V.; Rost, B. Prediction of DNA-binding residues from sequence. Bioinformatics 2007, 23, i347–i353. [Google Scholar] [CrossRef] [PubMed]
- Ptashne, M. Regulation of transcription: From lambda to eukaryotes. Trends Biochem. Sci. 2005, 30, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; van Heyningen, P.; Berman, H.M.; Thornton, J.M. protein–DNA interactions: A structural analysis. J. Mol. Biol. 1999, 287, 877–896. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Barker, J.A.; Nobeli, I.; Thornton, J.M. Using structural motif templates to identify proteins with DNA binding function. Nucleic Acids Res. 2003, 31, 2811–2823. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Sarai, A. Structure-based prediction of DNA target sites by regulatory proteins. Proteins 1999, 35, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, N.M.; Laskowski, R.A.; Thornton, J.M. Amino acid-base interactions: A three-dimensional analysis of protein–DNA interactions at an atomic level. Nucleic Acids Res. 2001, 29, 2860–2874. [Google Scholar] [CrossRef] [PubMed]
- Mandel-Gutfreund, Y.; Margalit, H. Quantitative parameters for amino acid-base interaction: Implications for prediction of protein–DNA binding sites. Nucleic Acids Res. 1998, 26, 2306–2312. [Google Scholar] [CrossRef] [PubMed]
- Olson, W.K.; Gorin, A.A.; Lu, X.J.; Hock, L.M.; Zhurkin, V.B. DNA sequence-dependent deformability deduced from protein–DNA crystal complexes. Proc. Natl. Acad. Sci. USA 1998, 95, 11163–11168. [Google Scholar] [CrossRef] [PubMed]
- Orengo, C.A.; Michie, A.D.; Jones, S.; Jones, D.T.; Swindells, M.B.; Thornton, J.M. CATH—A hierarchic classification of protein domain structures. Structure 1997, 5, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Schultz, J.; Milpetz, F.; Bork, P. SMART: Identification and annotation of domains from signalling and extracellular protein sequences. Nucleic Acids Res. 1999, 27, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Stormo, G.D. DNA binding sites: Representation and discovery. Bioinformatics 2000, 16, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Gou, Z.; Kuznetsov, I.B. DP-Bind: A web server for sequence-based prediction of DNA-binding residues in DNA-binding proteins. Bioinformatics 2007, 23, 634–636. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Terribilini, M.; Wu, F.; Jernigan, R.L.; Dobbs, D.; Honavar, V. Predicting DNA-binding sites of proteins from amino acid sequence. BMC Bioinform. 2006, 7. [Google Scholar] [CrossRef]
- Ahmad, S.; Sarai, A. PSSM-based prediction of DNA binding sites in proteins. BMC Bioinform. 2005, 6. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Liu, H.; Duan, X.; Ding, Y.; Wu, H.; Bai, Y.; Sun, X. Prediction of DNA-binding residues in proteins from amino acid sequences using a random forest model with a hybrid feature. Bioinformatics 2009, 25, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.B.; Langlois, R.; Lu, H. NAPS: A residue-level nucleic acid-binding prediction server. Nucleic Acids Res. 2010, 38, W431–W435. [Google Scholar] [CrossRef] [PubMed]
- Alibes, A.; Serrano, L.; Nadra, A.D. Structure-based DNA-binding prediction and design. Methods Mol. Biol. 2010, 649, 77–88. [Google Scholar] [PubMed]
- Li, B.Q.; Feng, K.Y.; Ding, J.; Cai, Y.D. Predicting DNA-binding sites of proteins based on sequential and 3D structural information. Mol. Genet. Genomics 2014, 289, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, Q.Z.; Liu, S.; Fan, G.L.; Zuo, Y.C.; Peng, Y. PreDNA: Accurate prediction of DNA-binding sites in proteins by integrating sequence and geometric structure information. Bioinformatics 2013, 29, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Xia, J.; Zhang, W.; Liu, J. Exploiting a reduced set of weighted average features to improve prediction of DNA-binding residues from 3D structures. PLoS One 2011, 6, e28440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tang, Y.; Sheng, Z.; Zhao, D. An overview of the De Novo prediction of enzyme catalytic residues. Curr. Bioinform. 2009, 4, 197–206. [Google Scholar] [CrossRef]
- Nagano, N.; Orengo, C.A.; Thornton, J.M. One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 2002, 321, 741–765. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.V.; Havranek, J.J.; Baker, D.; Siggia, E.D. protein–DNA binding specificity predictions with structural models. Nucleic Acids Res. 2005, 33, 5781–5798. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, A.; Skolnick, J. Efficient prediction of nucleic acid binding function from low-resolution protein structures. J. Mol. Biol. 2006, 358, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Skolnick, J. A threading-based method for the prediction of DNA-binding proteins with application to the human genome. PLoS Comput. Biol. 2009, 5, e1000567. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Zhang, Z.; Lin, B.; Schroeder, M.; Huang, B. MetaDBSite: A meta approach to improve protein DNA-binding sites prediction. BMC Syst. Biol. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liu, J.S. Extracting sequence features to predict protein–DNA interactions: A comparative study. Nucleic Acids Res. 2008, 36, 4137–4148. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Moreira, B. 3D-footprint: A database for the structural analysis of protein–DNA complexes. Nucleic Acids Res. 2010, 38, D91–D97. [Google Scholar] [CrossRef] [PubMed]
- Norambuena, T.; Melo, F. The protein–DNA Interface database. BMC Bioinform. 2010, 11. [Google Scholar] [CrossRef]
- Gao, M.; Skolnick, J. DBD-Hunter: A knowledge-based method for the prediction of DNA–protein interactions. Nucleic Acids Res. 2008, 36, 3978–92. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Gromiha, M.M.; Sarai, A. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics 2004, 20, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Stawiski, E.W.; Gregoret, L.M.; Mandel-Gutfreund, Y. Annotating nucleic acid-binding function based on protein structure. J. Mol. Biol. 2003, 326, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Shanahan, H.P.; Berman, H.M.; Thornton, J.M. Using electrostatic potentials to predict DNA-binding sites on DNA-binding proteins. Nucleic Acids Res. 2003, 31, 7189–7198. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Kinoshita, K.; Nakamura, H. PreDs: A server for predicting dsDNA-binding site on protein molecular surfaces. Bioinformatics 2005, 21, 1721–1723. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, I.B.; Gou, Z.; Li, R.; Hwang, S. Using evolutionary and structural information to predict DNA-binding sites on DNA-binding proteins. Proteins 2006, 64, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Dunbrack, R.L., Jr. PISCES: Recent improvements to a PDB sequence culling server. Nucleic Acids Res. 2005, 33, W94–W98. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Linden, A. Measuring diagnostic and predictive accuracy in disease management: An introduction to receiver operating characteristic (ROC) analysis. J. Eval. Clin. Pract. 2006, 12, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, G.J.; Porter, C.T.; Borkakoti, N.; Thornton, J.M. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 2002, 324, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.V.; Wu, C.H. Prediction of catalytic residues using Support Vector Machine with selected protein sequence and structural properties. BMC Bioinform. 2006, 7. [Google Scholar] [CrossRef]
- Kauffman, C.; Karypis, G. An analysis of information content present in protein–DNA interactions. Pac. Symp. Biocomput. 2008, 13, 477–488. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Valdar, W.S. Scoring residue conservation. Proteins 2002, 48, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Keskin, O.; Sarai, A.; Nussinov, R. protein–DNA interactions: Structural, thermodynamic and clustering patterns of conserved residues in DNA-binding proteins. Nucleic Acids Res. 2008, 36, 5922–5932. [Google Scholar] [CrossRef] [PubMed]
- Nimrod, G.; Szilagyi, A.; Leslie, C.; Ben-Tal, N. Identification of DNA-binding proteins using structural, electrostatic and evolutionary features. J. Mol. Biol. 2009, 387, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Horst, J.A.; Cheng, G.; Nickle, D.C.; Samudrala, R. Protein meta-functional signatures from combining sequence, structure, evolution, and amino acid property information. PLoS Comput. Biol. 2008, 4, e1000181. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gromiha, M.M.; Raghava, G.P. Identification of DNA-binding proteins using support vector machines and evolutionary profiles. BMC Bioinform. 2007, 8. [Google Scholar] [CrossRef]
- Harrison, S.C. A structural taxonomy of DNA-binding domains. Nature 1991, 353, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, H.P.; Garcia, M.A.; Jones, S.; Thornton, J.M. Identifying DNA-binding proteins using structural motifs and the electrostatic potential. Nucleic Acids Res. 2004, 32, 4732–4741. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Zhao, J.; Wang, Z.X. Flexibility analysis of enzyme active sites by crystallographic temperature factors. Protein Eng. 2003, 16, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, A.; Bartlett, G.J.; Thornton, J.M. Using a neural network and spatial clustering to predict the location of active sites in enzymes. J. Mol. Biol. 2003, 330, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.R.; Sheng, Z.Y.; Chen, Y.Z.; Zhang, Z. An improved prediction of catalytic residues in enzyme structures. Protein Eng. Des. Sel. 2008, 21, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 2007, 23, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Karypis, G. YASSPP: Better kernels and coding schemes lead to improvements in protein secondary structure prediction. Proteins 2006, 64, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.; Andersen, C.A.; Rost, B. DSSPcont: Continuous secondary structure assignments for proteins. Nucleic Acids Res. 2003, 31, 3293–3295. [Google Scholar] [CrossRef] [PubMed]
- Tjong, H.; Zhou, H.X. DISPLAR: An accurate method for predicting DNA-binding sites on protein surfaces. Nucleic Acids Res. 2007, 35, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- SJ, H.; JM, T. NACCESS Computer program. In Department of Biochemistry and Molecular Biology; University College of London: London, UK, 1993. [Google Scholar]
- Faucher, J.; Pliska, V. Hydrophobic parameters pi of amino acid side chains from the partitioning of N-acetyl-amino-acid amides. Eur. J. Med. Chem. 1983, 18, 369–375. [Google Scholar]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Kinoshita, K.; Nakamura, H. Structure-based prediction of DNA-binding sites on proteins using the empirical preference of electrostatic potential and the shape of molecular surfaces. Proteins 2004, 55, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Shazman, S.; Celniker, G.; Haber, O.; Glaser, F.; Mandel-Gutfreund, Y. Patch Finder Plus (PFplus): A web server for extracting and displaying positive electrostatic patches on protein surfaces. Nucleic Acids Res. 2007, 35, W526–W530. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM—A program for macromolecular energy, minimization and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Ahmad, S.; Sarai, A. Moment-based prediction of DNA-binding proteins. J. Mol. Biol. 2004, 341, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.D.; Mayer, C.E.; Soding, J. Prediction of protein functional residues from sequence by probability density estimation. Bioinformatics 2008, 24, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.M.; Pan, X.Y.; Xu, C.; Shen, H.B. Computational prediction of DNA–protein interactions: A review. Curr. Comput. Aided Drug Des. 2010, 6, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, N.; Langlois, R.E.; Zhao, G.; Lu, H. Kernel-based machine learning protocol for predicting DNA-binding proteins. Nucleic Acids Res. 2005, 33, 6486–6493. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar]
- Chu, W.Y.; Huang, Y.F.; Huang, C.C.; Cheng, Y.S.; Huang, C.K.; Oyang, Y.J. ProteDNA: A sequence-based predictor of sequence-specific DNA-binding residues in transcription factors. Nucleic Acids Res. 2009, 37, W396–W401. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, N.; Langlois, R.; Zhao, G.; Lu, H. Structure based prediction of binding residues on DNA-binding proteins. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2005, 3, 2611–2614. [Google Scholar] [PubMed]
- Shao, X.; Tian, Y.; Wu, L.; Wang, Y.; Jing, L.; Deng, N. Predicting DNA- and RNA-binding proteins from sequences with kernel methods. J. Theor. Biol. 2009, 258, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.F.; Fan, X.D.; Li, Y.D. Identifying splicing sites in eukaryotic RNA: Support vector machine approach. Comput. Biol. Med. 2003, 33, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, X.; Chen, X.; Zhao, G. Computational methods for DNA-binding protein and binding residue prediction. Protein Pept. Lett. 2013, 20, 346–351. [Google Scholar] [PubMed]
- Lou, W.; Wang, X.; Chen, F.; Chen, Y.; Jiang, B.; Zhang, H. Sequence based prediction of DNA-binding proteins based on hybrid feature selection using random forest and Gaussian naive Bayes. PLoS One 2014, 9, e86703. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, M.Q.; Yang, J.Y. Prediction of DNA-binding residues from protein sequence information using random forests. BMC Genomics 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.K. A Data complexity analysis of comparative advantages of decision forest constructors. Pattern Anal. Appl. 2002, 5, 102–112. [Google Scholar] [CrossRef]
- Martin-Galiano, A.J.; Smialowski, P.; Frishman, D. Predicting experimental properties of integral membrane proteins by a naive Bayes approach. Proteins 2008, 70, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Tomlins, S.A.; Varambally, S.; Mahavisno, V.; Barrette, T.; Kalyana-Sundaram, S.; Ghosh, D.; Pandey, A.; Chinnaiyan, A.M. Probabilistic model of the human protein–protein interaction network. Nat. Biotechnol. 2005, 23, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Ozbek, P.; Soner, S.; Erman, B.; Haliloglu, T. DNABINDPROT: Fluctuation-based predictor of DNA-binding residues within a network of interacting residues. Nucleic Acids Res. 2010, 38, W417–W423. [Google Scholar] [CrossRef] [PubMed]
- Bujnicki, J.M.; Elofsson, A.; Fischer, D.; Rychlewski, L. LiveBench-1: Continuous benchmarking of protein structure prediction servers. Protein Sci. 2001, 10, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Schroeder, M. Using protein binding site prediction to improve protein docking. Gene 2008, 422, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Costa, C.; Shanahan, H.P.; Jones, S.; Thornton, J.M. HTHquery: A method for detecting DNA-binding proteins with a helix-turn-helix structural motif. Bioinformatics 2005, 21, 3679–3680. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brown, S.J. BindN: A web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 2006, 34, W243–W248. [Google Scholar] [CrossRef] [PubMed]
- Nimrod, G.; Schushan, M.; Szilagyi, A.; Leslie, C.; Ben-Tal, N. iDBPs: A web server for the identification of DNA binding proteins. Bioinformatics 2010, 26, 692–693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J.; Zheng, W.; Zhang, C.; Qiu, X.; Chen, K.; Ruan, J. newDNA-Prot: Prediction of DNA-binding proteins by employing support vector machine and a comprehensive sequence representation. Comput. Biol. Chem. 2014, 52, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Hu, J. DNABind: A hybrid algorithm for structure-based prediction of DNA-binding residues by combining machine learning- and template-based approaches. Proteins 2013, 81, 1885–1899. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Guo, J.; Liu, H.D.; Xie, J.M.; Sun, X. Sequence-based prediction of DNA-binding residues in proteins with conservation and correlation information. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Wright, J.D.; Lim, C. DR_bind: A web server for predicting DNA-binding residues from the protein structure based on electrostatics, evolution and geometry. Nucleic Acids Res. 2012, 40, W249–W256. [Google Scholar] [CrossRef] [PubMed]
- Matthew, S.; Tianyin, Z.; Lin, Y.; Ana, C.D.M.; Raluca, G.; Remo, R. Absence of a simple code: How transcription factors read the genome. Trends Biochem. Sci. 2014, 39, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, T.Y.; Iris, D.; Anthony, M.; Wyeth, W.W.; Raluca, G.; Remo, R. NAR breakthrough article: TFBSshape: A motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2014, 42, 148–155. [Google Scholar] [CrossRef]
- Ghersi, D.; Sanchez, R. Improving accuracy and efficiency of blind protein-ligand docking by focusing on predicted binding sites. Proteins 2009, 74, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, C.; Rangwala, H.; Karypis, G. Improving homology models for protein-ligand binding sites. Comput. Syst. Bioinform. Conf. 2008, 7, 211–222. [Google Scholar]
- Schroder, A.; Eichner, J.; Supper, J.; Eichner, J.; Wanke, D.; Henneges, C.; Zell, A. Predicting DNA-binding specificities of eukaryotic transcription factors. PLoS One 2010, 5, e13876. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; He, J.; Li, X.; Lu, L.; Yang, X.; Feng, K.; Lu, W.; Kong, X. A novel computational approach to predict transcription factor DNA binding preference. J. Proteome Res. 2009, 8, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Lu, L.; Liu, X.; Cai, Y.D.; Li, Y. An approach to predict transcription factor DNA binding site specificity based upon gene and transcription factor functional categorization. Bioinformatics 2007, 23, 2449–2454. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Cai, Y.D.; Li, Y. A novel computational method to predict transcription factor DNA binding preference. Biochem. Biophys. Res. Commun. 2006, 348, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yang, J.; Song, J.N.; Chou, K.C.; Shen, H.B. Improving the accuracy of predicting disulfide connectivity by feature selection. J. Comput. Chem. 2010, 31, 1478–1485. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Si, J.; Zhao, R.; Wu, R. An Overview of the Prediction of Protein DNA-Binding Sites. Int. J. Mol. Sci. 2015, 16, 5194-5215. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16035194
Si J, Zhao R, Wu R. An Overview of the Prediction of Protein DNA-Binding Sites. International Journal of Molecular Sciences. 2015; 16(3):5194-5215. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16035194
Chicago/Turabian StyleSi, Jingna, Rui Zhao, and Rongling Wu. 2015. "An Overview of the Prediction of Protein DNA-Binding Sites" International Journal of Molecular Sciences 16, no. 3: 5194-5215. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16035194