
Whole Exome Sequencing for a Patient with Rubinstein-Taybi Syndrome Reveals de Novo Variants besides an Overt CREBBP Mutation

Abstract
:1. Introduction
2. Results
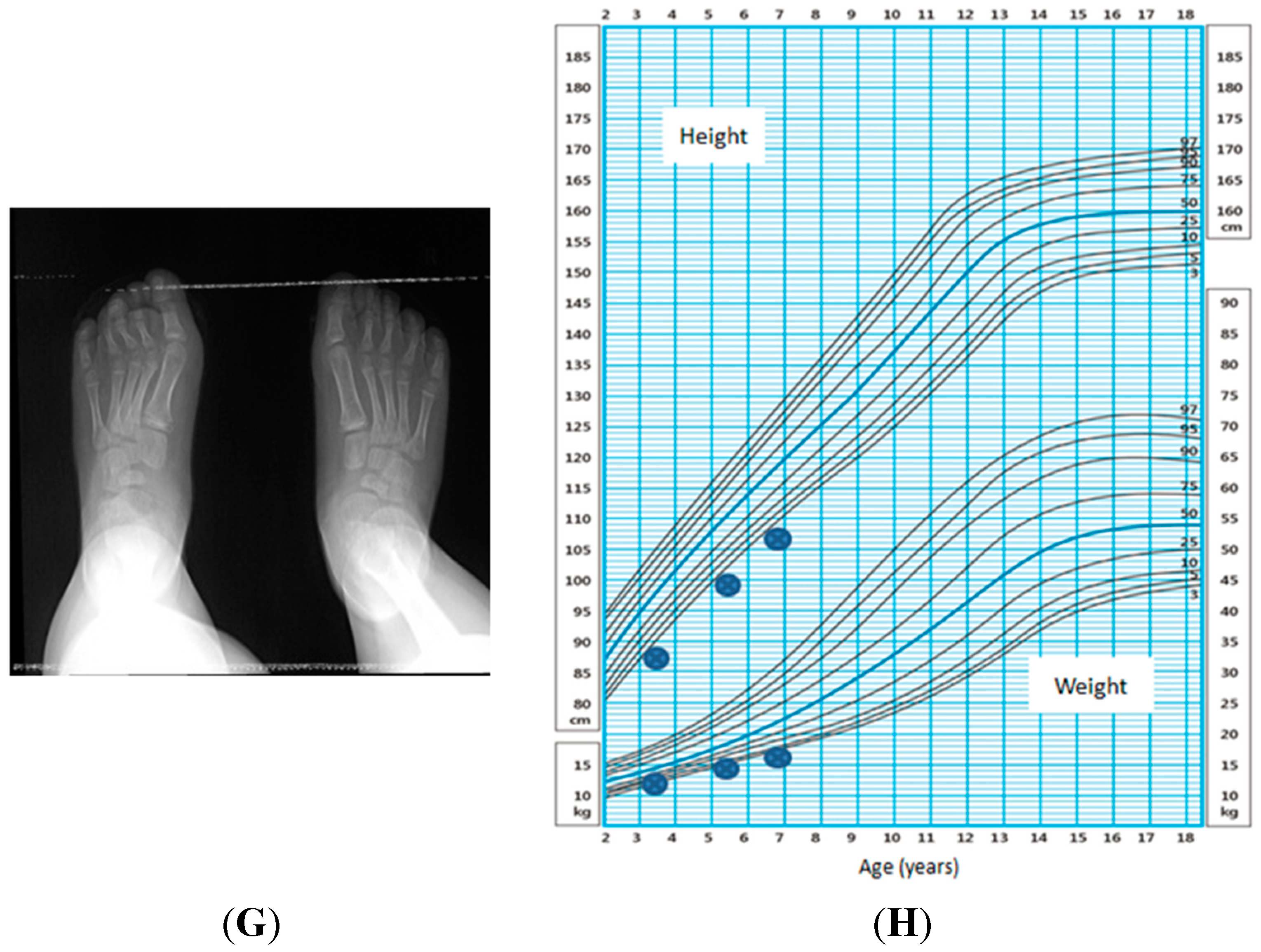
2.1. Physical Characteristics
2.2. Neuropsychiatric and Behavioral Features

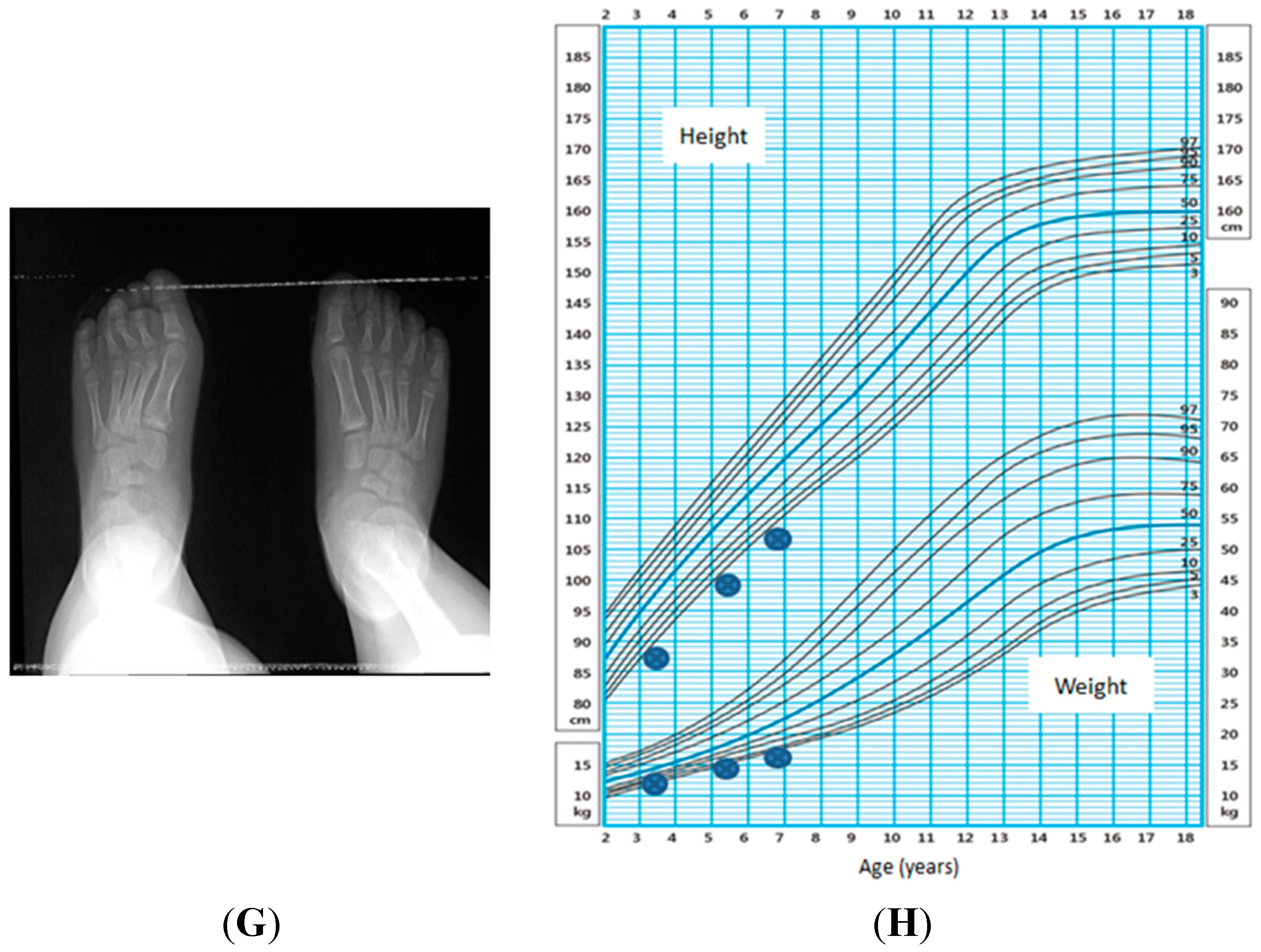
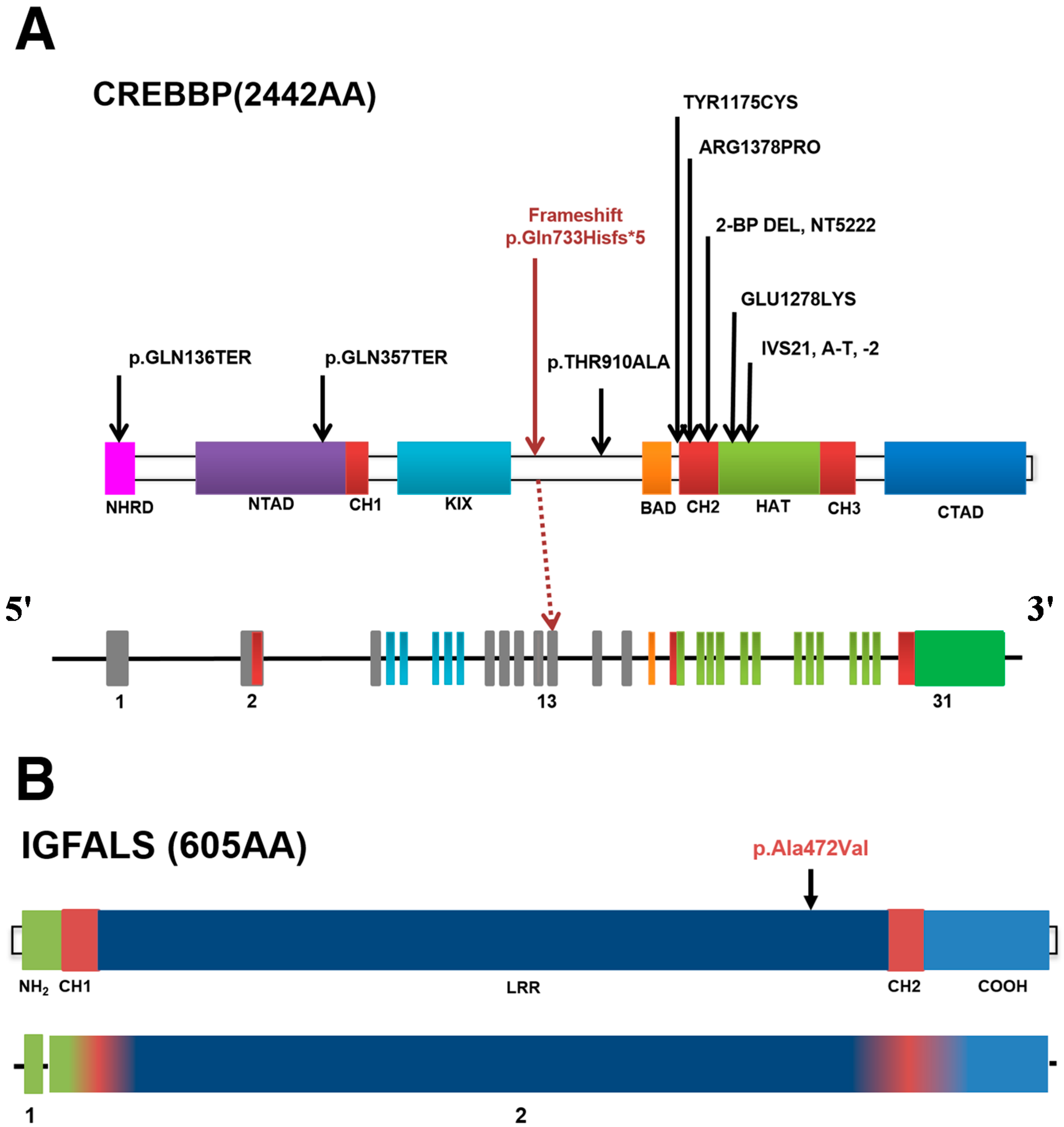
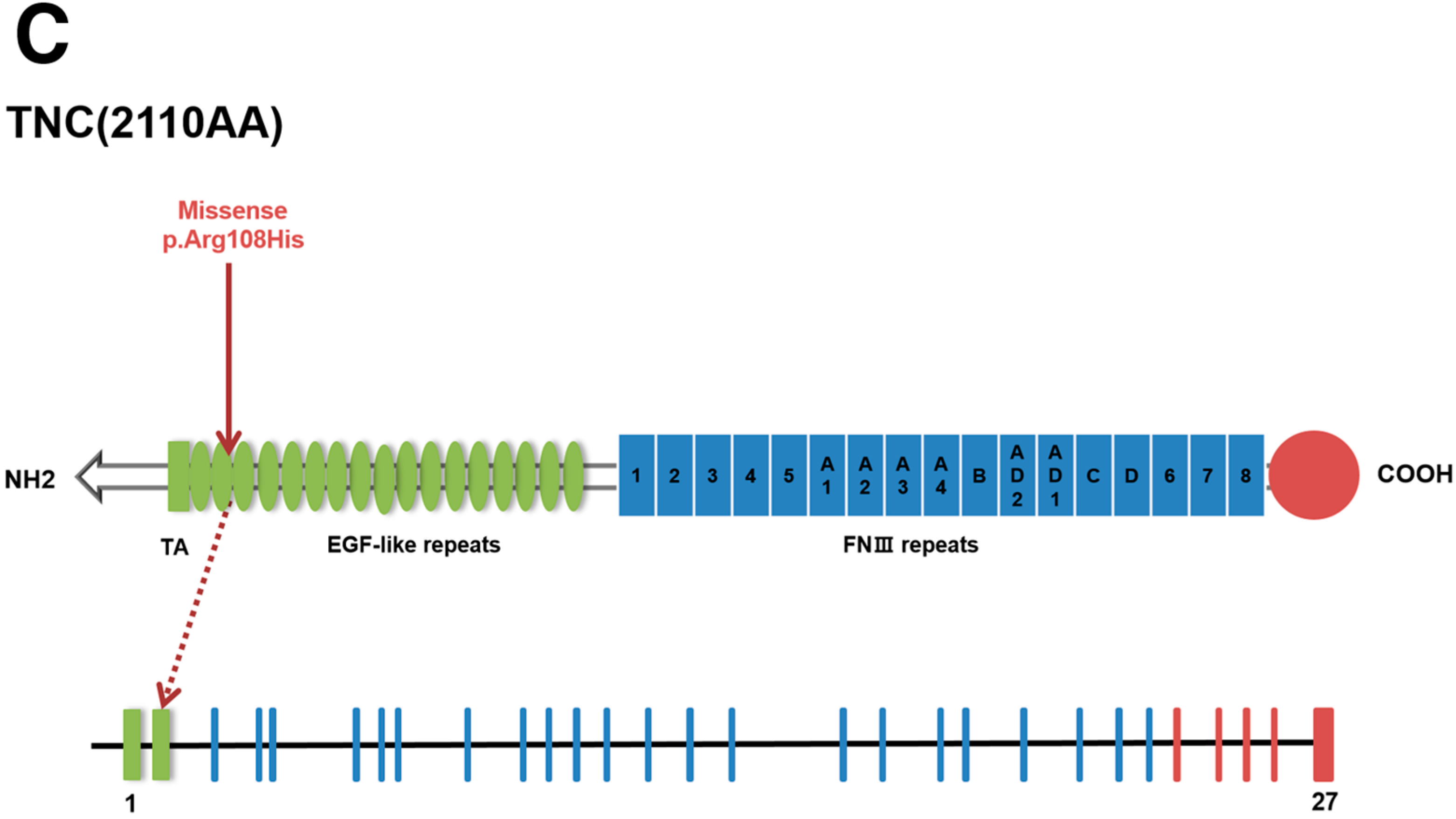
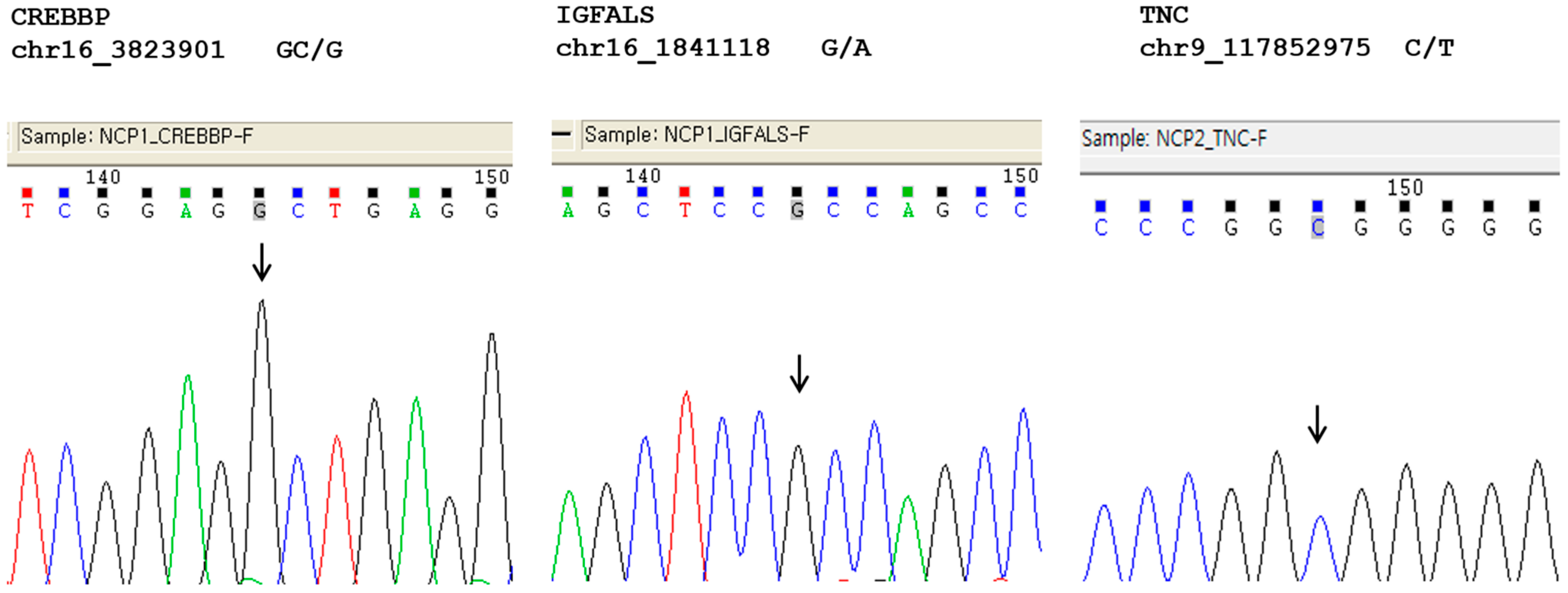
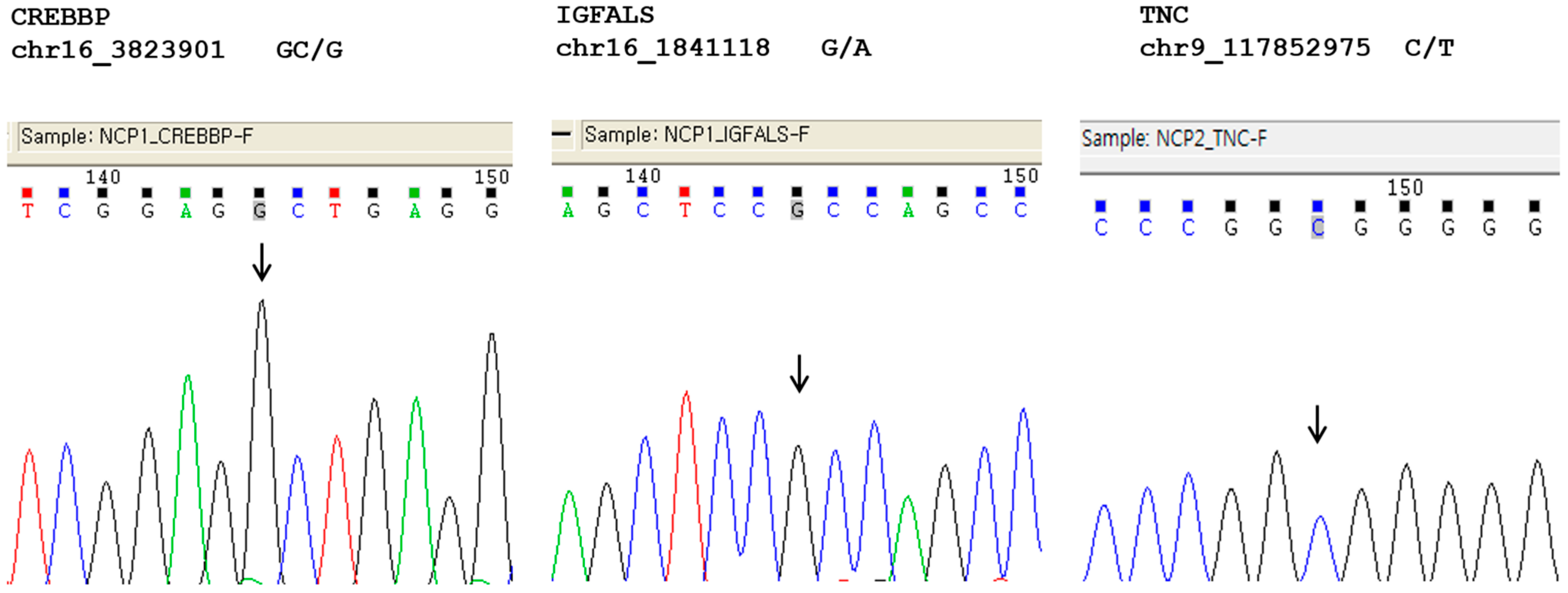
2.3. Genetic Variants
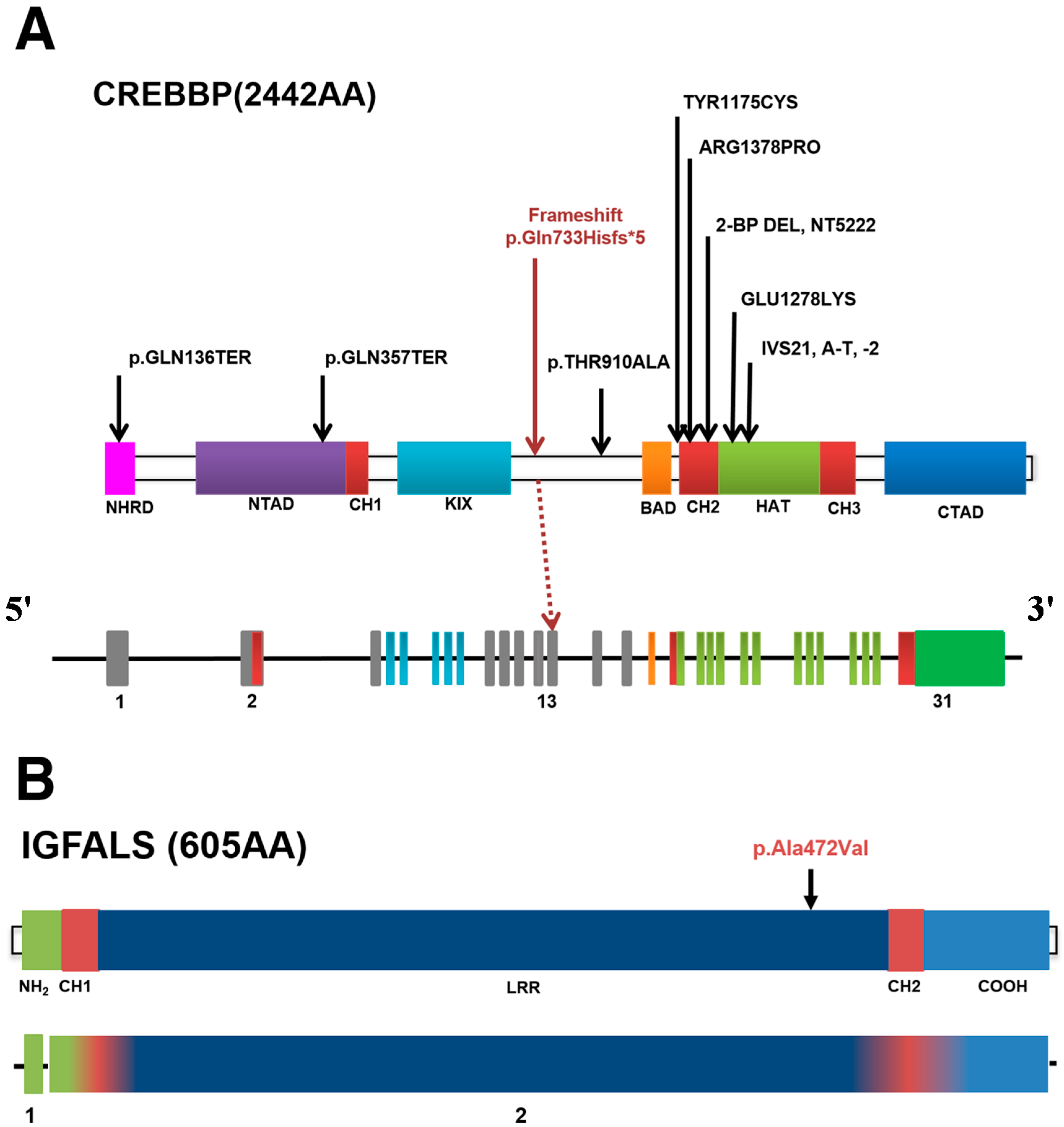
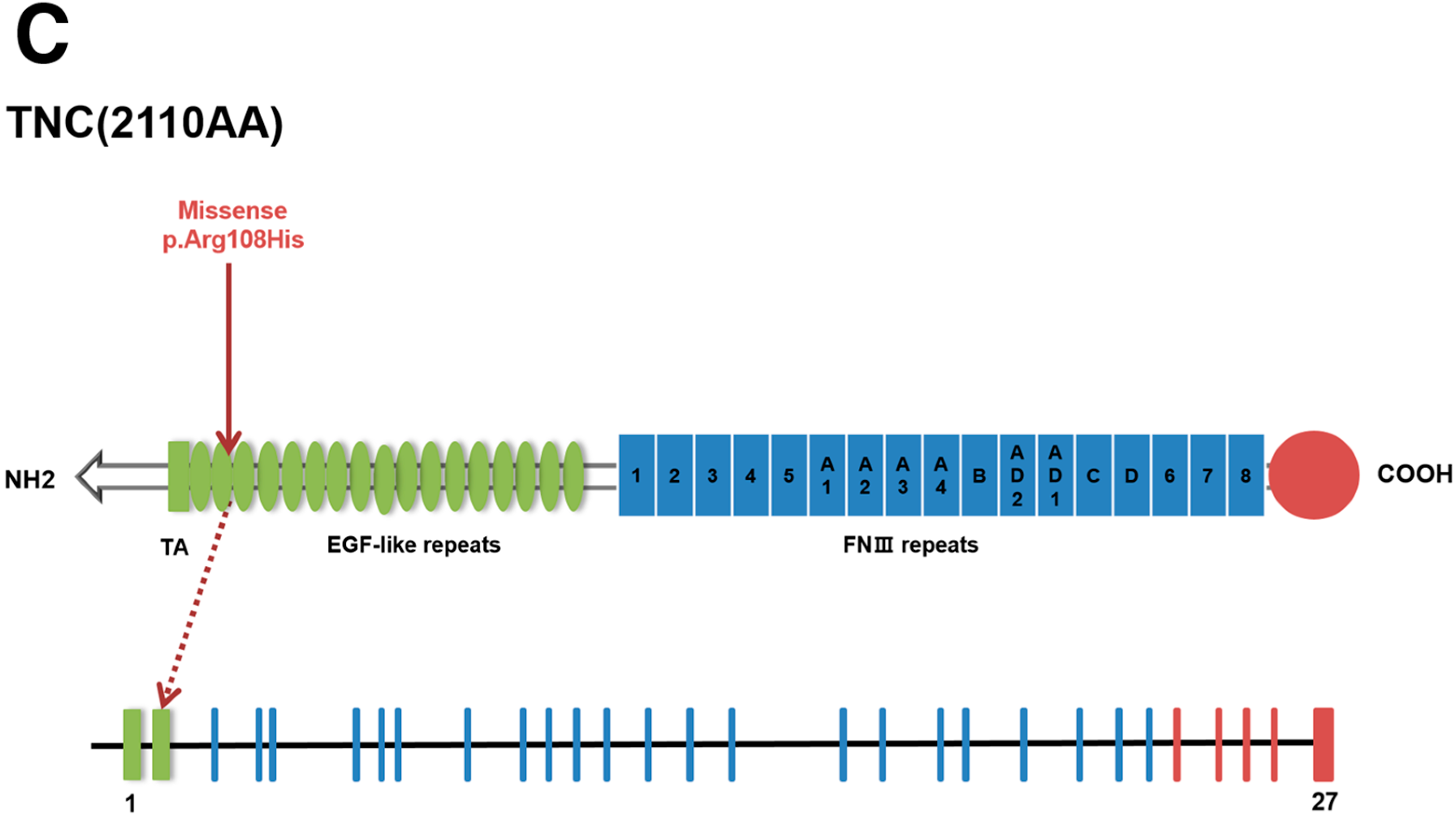

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | TNC | IGFALS | CREBBP |
|---|---|---|---|
| Chromosome | chr9 | chr16 | chr16 |
| Position | 117852975 | 1841118 | 3823901 |
| Reference Allele | C | G | C |
| Alternative Allele | T | A | – |
| Mutation Type | Missense | Missense | Frameshift |
| Sequence Variant | c.323G>A | c.1415C>T | c.2199delG |
| Protein Variant | p.Arg108His | p.Ala472Val | p.Gln733Hisfs*5 |
| phyloP Score | 2.369 | 0.598 | 1.492 |
| GERP Score | 1200.8 | 2536.4 | 476.2 |
| SIFT Score | 0 | 0.30 | – |
| Mutation Assessor | 1.87 | 0.995 | – |
| Classification 1 | Likely pathogenic 2 | Variants of unknown significance | Pathogenic |

3. Discussion
4. Methods
4.1. Ethics Statement
4.2. Subject
4.3. Clinical Evaluation
4.4. Genetic Analyses Using Bioinformatics Tools
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hennekam, R.C.; Stevens, C.A.; van de Kamp, J.J. Etiology and recurrence risk in Rubinstein-Taybi syndrome. Am. J. Med. Genet. Suppl. 1990, 6, 56–64. [Google Scholar] [PubMed]
- Hallam, T.M.; Bourtchouladze, R. Rubinstein-Taybi syndrome: Molecular findings and therapeutic approaches to improve cognitive dysfunction. Cell. Mol. Life Sci. 2006, 63, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, J.H.; Taybi, H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am. J. Dis. Child. 1963, 105, 588–608. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.; Swayne, S.; Rubinstein, J.H.; Lanphear, N.E.; Stevens, C.A. Rubinstein-Taybi syndrome medical guidelines. Am. J. Med. Genet. Part A 2003, 119A, 101–110. [Google Scholar] [CrossRef]
- Miller, R.W.; Rubinstein, J.H. Tumors in Rubinstein-Taybi syndrome. Am. J. Med. Genet. 1995, 56, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Hennekam, R.C.; Baselier, A.C.; Beyaert, E.; Bos, A.; Blok, J.B.; Jansma, H.B.; Thorbecke-Nilsen, V.V.; Veerman, H. Psychological and speech studies in Rubinstein-Taybi syndrome. Am. J. Mental Retard. 1992, 96, 645–660. [Google Scholar]
- Hennekam, R.C. Rubinstein-Taybi syndrome. Eur. J. Hum. Genet. 2006, 14, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Roelfsema, J.H.; White, S.J.; Ariyurek, Y.; Bartholdi, D.; Niedrist, D.; Papadia, F.; Bacino, C.A.; den Dunnen, J.T.; van Ommen, G.J.; Breuning, M.H.; et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: Mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 2005, 76, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, O.; Schmidt, S.; Richter, M.; Morlot, S.; Seemanova, E.; Wiebe, G.; Rasi, S. DNA sequencing of CREBBP demonstrates mutations in 56% of patients with Rubinstein-Taybi syndrome (RSTS) and in another patient with incomplete RSTS. Hum. Genet. 2005, 117, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Bentivegna, A.; Milani, D.; Gervasini, C.; Castronovo, P.; Mottadelli, F.; Manzini, S.; Colapietro, P.; Giordano, L.; Atzeri, F.; Divizia, M.T. Rubinstein-Taybi syndrome: Spectrum of CREBBP mutations in Italian patients. BMC Med. Genet. 2006, 7, 77. [Google Scholar] [CrossRef] [PubMed]
- Negri, G.; Milani, D.; Colapietro, P.; Forzano, F.; Monica, M.D.; Rusconi, D.; Consonni, L.; Caffi, L.G.; Finelli, P.; Scarano, G.; et al. Clinical and molecular characterization of Rubinstein-Taybi syndrome patients carrying distinct novel mutations of the EP300 gene. Clin. Genet. 2015, 87, 148–154. [Google Scholar] [CrossRef]
- Spena, S.; Milani, D.; Rusconi, D.; Negri, G.; Colapietro, P.; Elcioglu, N.; Bedeschi, F.; Pilotta, A.; Spaccini, L.; Ficcadenti, A. Insights into genotype–phenotype correlations from CREBBP point mutation screening in a cohort of 46 Rubinstein-Taybi syndrome patients. Clin. Genet. 2014. [Google Scholar] [CrossRef]
- Petrij, F.; Giles, R.H.; Dauwerse, H.G.; Saris, J.J.; Hennekam, R.C.; Masuno, M.; Tommerup, N.; van Ommen, G.J.; Goodman, R.H.; Peters, D.J.; et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 1995, 376, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.C.-H.; Dossett, C.J.; Walton, C.S.; Cramer, A.E.; Eng, P.A.; Nowakowska, B.A.; Pursley, A.N.; Stankiewicz, P.; Wiszniewska, J.; Cheung, S.W. Exon deletions of the EP300 and CREBBP genes in two children with Rubinstein-Taybi syndrome detected by aCGH. Eur. J. Hum. Genet. 2011, 19, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Kim, H.J.; Kim, Y.J.; Kwon, J.Y.; Kim, J.W.; Kim, S.H. Cryptic microdeletion of the CREBBP gene from t(1;16) (p36.2;p13.3) as a novel genetic defect causing Rubinstein-Taybi syndrome. Ann. Clin. Lab. Sci. 2013, 43, 450–456. [Google Scholar] [PubMed]
- Blough, R.I.; Petrij, F.; Dauwerse, J.G.; Milatovich-Cherry, A.; Weiss, L.; Saal, H.M.; Rubinstein, J.H. Variation in microdeletions of the cyclic AMP-responsive element-binding protein gene at chromosome band 16p13. 3 in the Rubinstein-Taybi syndrome. Am. J. Med. Genet. 2000, 90, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Gervasini, C.; Castronovo, P.; Bentivegna, A.; Mottadelli, F.; Faravelli, F.; Giovannucci-Uzielli, M.L.; Pessagno, A.; Lucci-Cordisco, E.; Pinto, A.M.; Salviati, L. High frequency of mosaic CREBBP deletions in Rubinstein–Taybi syndrome patients and mapping of somatic and germ-line breakpoints. Genomics 2007, 90, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.A.; Hennekam, R.C.; Blackburn, B.L. Growth in the Rubinstein-Taybi syndrome. Am. J. Med. Genet. Suppl. 1990, 6, 51–55. [Google Scholar] [PubMed]
- Galera, C.; Taupiac, E.; Fraisse, S.; Naudion, S.; Toussaint, E.; Rooryck-Thambo, C.; Delrue, M.A.; Arveiler, B.; Lacombe, D.; Bouvard, M.P. Socio-behavioral characteristics of children with Rubinstein-Taybi syndrome. J. Autism Dev. Disord. 2009, 39, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Calì, F.; Failla, P.; Chiavetta, V.; Ragalmuto, A.; Ruggeri, G.; Schinocca, P.; Schepis, C.; Romano, V.; Romano, C. Multiplex ligation-dependent probe amplification detection of an unknown large deletion of the CREB-binding protein gene in a patient with Rubinstein-Taybi syndrome. CEP 2013, 14025, 220. [Google Scholar]
- Hellings, J.A.; Hossain, S.; Martin, J.K.; Baratang, R.R. Psychopathology, GABA, and the Rubinstein-Taybi syndrome: A review and case study. Am. J. Med. Genet. 2002, 114, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Levitas, A.S.; Reid, C.S. Rubinstein-Taybi syndrome and psychiatric disorders. J. Intellect. Disabil. Res. 1998, 42, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Waite, J.; Moss, J.; Beck, S.R.; Richards, C.; Nelson, L.; Arron, K.; Burbidge, C.; Berg, K.; Oliver, C. Repetitive behavior in Rubinstein-Taybi syndrome: Parallels with Autism spectrum phenomenology. J. Autism Dev. Disord. 2014. [Google Scholar] [CrossRef]
- Lopez-Atalaya, J.P.; Valor, L.M.; Barco, A. Epigenetic factors in intellectual disability: The Rubinstein-Taybi syndrome as a paradigm of neurodevelopmental disorder with epigenetic origin. Prog. Mol. Biol. Transl. Sci. 2014, 128, 139. [Google Scholar] [PubMed]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar] [PubMed]
- Marzuillo, P.; Grandone, A.; Coppola, R.; Cozzolino, D.; Festa, A.; Messa, F.; Luongo, C.; del Giudice, E.M.; Perrone, L. Novel cAMP binding protein-BP (CREBBP) mutation in a girl with Rubinstein-Taybi syndrome, GH deficiency, Arnold Chiari malformation and pituitary hypoplasia. BMC Med. Genet. 2013, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Nies, D.E.; Hemesath, T.J.; Kim, J.-H.; Gulcher, J.R.; Stefansson, K. The complete cDNA sequence of human hexabrachion (Tenascin). A multidomain protein containing unique epidermal growth factor repeats. J. Biol. Chem. 1991, 266, 2818–2823. [Google Scholar] [PubMed]
- Wiese, S.; Karus, M.; Faissner, A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front. Pharmacol. 2012, 3, 120. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.C.; Pauls, D.L.; Lange, C.; Sasanfar, R.; Santangelo, S.L. Sex-specific association of a common variant of the XG gene with Autism spectrum disorders. Am. J. Med. Genet. Part B 2013, 162, 742–750. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, F.; Zong, L.; Zhang, P.; Guan, L.; Zhang, J.; Wang, D.; Wang, J.; Chai, W.; Lan, L.; et al. Exome sequencing and linkage analysis identified tenascin-C (TNC) as a novel causative gene in nonsyndromic hearing loss. PLoS One 2013, 8, e69549. [Google Scholar] [CrossRef] [PubMed]
- Rosenhall, U.; Nordin, V.; Sandstrom, M.; Ahlsen, G.; Gillberg, C. Autism and hearing loss. J. Autism Dev. Disord. 1999, 29, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Schorry, E.; Keddache, M.; Lanphear, N.; Rubinstein, J.; Srodulski, S.; Fletcher, D.; Blough-Pfau, R.; Grabowski, G. Genotype–phenotype correlations in Rubinstein-Taybi syndrome. Am. J. Med. Genet. Part A 2008, 146, 2512–2519. [Google Scholar] [CrossRef]
- Bourtchouladze, R.; Lidge, R.; Catapano, R.; Stanley, J.; Gossweiler, S.; Romashko, D.; Scott, R.; Tully, T. A mouse model of Rubinstein-Taybi syndrome: Defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc. Natl. Acad. Sci. USA 2003, 100, 10518–10522. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.R.; Baxter, R.C.; Camerato, T.; Dai, J.; Wood, W.I. Structure and functional expression of the acid-labile subunit of the insulin-like growth factor-binding protein complex. Mol. Endocrinol. 1992, 6, 870–876. [Google Scholar] [PubMed]
- Courtland, H.W.; DeMambro, V.; Maynard, J.; Sun, H.; Elis, S.; Rosen, C.; Yakar, S. Sex-specific regulation of body size and bone slenderness by the acid labile subunit. J. Bone Miner. Res. 2010, 25, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Domene, H.; Bengolea, S.; Jasper, H.; Boisclair, Y. Acid-labile subunit deficiency: Phenotypic similarities and differences between human and mouse. J. Endocrinol. Investig. 2004, 28, 43–46. [Google Scholar]
- Domené, H.M.; Scaglia, P.A.; Lteif, A.; Mahmud, F.H.; Kirmani, S.; Frystyk, J.; Bedecarrás, P.; Gutiérrez, M.; Jasper, H.G. Phenotypic effects of null and haploinsufficiency of acid-labile subunit in a family with two novel IGFALS gene mutations. J. Clin. Endocrinol. Metab. 2007, 92, 4444–4450. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kwak, K.J.; Park, G.B. Korean Wechsler Preschool and Primary Scale of Intelligence; Special Education Publishing: Seoul, Korean, 2002. [Google Scholar]
- Sparrow, S.; Balla, D.; Cichetti, D. Vineland Adaptive Behavior Scales; American Guidance Services: Circle Pines, MN, USA, 1984. [Google Scholar]
- Aylward, G. Practitioner’s Guide to Developmental and Psychological Testing; Springer: New York, NY, USA, 1994. [Google Scholar]
- Lord, C.; Rutter, M.; DiLavore, P.D.; Risi, S. Autism Diagnostic Observation Schedule; Western Psychological Services: Los Angeles, CA, USA, 2001. [Google Scholar]
- Lord, C.; Rutter, M.; Goode, S.; Heemsbergen, J.; Jordan, H.; Mawhood, L.; Schopler, E. Autism diagnostic observation schedule: A standardized observation of communicative and social behavior. J. Autism Dev. Disord. 1989, 19, 185–212. [Google Scholar] [CrossRef] [PubMed]
- Le Couteur, A.; Lord, C.; Rutter, M. The Autism Diagnostic Interview-Revised (ADI-R); Western Psychological Services: Los Angeles, CA, USA, 2003. [Google Scholar]
- Lord, C.; Rutter, M.; Le Couteur, A. Autism diagnostic interview-revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 1994, 24, 659–685. [Google Scholar] [CrossRef] [PubMed]
- Constantino, J.; Gruber, C.P. Social Responsiveness Scale (SRS); Western Psychological Services: Los Angeles, CA, USA, 2005. [Google Scholar]
- Rutter, M.; Bailey, A.; Lord, C. Social Communication Questionnaire (SCQ); Western Psychological Services: Los Angeles, CA, USA, 2003. [Google Scholar]
- Buysse, D.J.; Reynolds, C.F., III; Monk, T.H.; Berman, S.R.; Kupfer, D.J. The Pittsburgh sleep quality index: A new instrument for psychiatric practice and research. Psychiatry Res. 1989, 28, 193–213. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.; Yegneswaran, B.; Liao, P.; Chung, S.A.; Vairavanathan, S.; Islam, S.; Khajehdehi, A.; Shapiro, C.M. Stop questionnaire: A tool to screen patients for obstructive sleep apnea. Anesthesiology 2008, 108, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Kurokawa, R.; Krones, A.; Tatsumi, K.; Ishii, M.; Taki, T.; Masuno, M.; Ohashi, H.; Yanagisawa, M.; Rosenfeld, M,G.; et al. Defect of histone acetyltransferase activity of the nuclear transcriptional coactivator CBP in Rubinstein-Taybi syndrome. Hum. Mol. Genet. 2001, 10, 1071–1076. [Google Scholar] [CrossRef]
- Bartsch, O.; Locher, K.; Meinecke, P.; Kress, W.; Seemanova, E.; Wagner, A.; Ostermann, K.; Rodel, G. Molecular studies in 10 cases of Rubinstein-Taybi syndrome, including a mild variant showing a missense mutation in codon 1175 of CREBBP. J. Med. Genet. 2002, 39, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Kalkhoven, E.; Roelfsema, J.H.; Teunissen, H.; den Boer, A.; Ariyurek, Y.; Zantema, A.; Breuning, M.H.; Hennekam, R.C.; Peters, D.J. Loss of CBP acetyltransferase activity by PHD finger mutations in Rubinstein-Taybi syndrome. Hum. Mol. Genet. 2003, 12, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Coupry, I.; Roudaut, C.; Stef, M.; Delrue, M.A.; Marche, M.; Burgelin, I.; Taine, L.; Cruaud, C.; Lacombe, D.; Arveiler, B. Molecular analysis of the CBP gene in 60 patients with Rubinstein-Taybi syndrome. J. Med. Genet. 2002, 39, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Kalkhoven, E. CBP and p300: HATs for different occasions. Biochem. Pharmacol. 2004, 68, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Udaka, T.; Samejima, H.; Kosaki, R.; Kurosawa, K.; Okamoto, N.; Mizuno, S.; Makita, Y.; Numabe, H.; Toral, J.F.; Takahashi, T.; et al. Comprehensive screening of CREB-binding protein gene mutations among patients with Rubinstein-Taybi syndrome using denaturing high-performance liquid chromatography. Congenit. Anom. (Kyoto) 2005, 45, 125–131. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, H.J.; Kim, K.; Kim, I.H.; Rho, S.-H.; Park, J.-E.; Lee, K.Y.; Kim, S.A.; Choi, B.Y.; Kim, N. Whole Exome Sequencing for a Patient with Rubinstein-Taybi Syndrome Reveals de Novo Variants besides an Overt CREBBP Mutation. Int. J. Mol. Sci. 2015, 16, 5697-5713. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16035697
Yoo HJ, Kim K, Kim IH, Rho S-H, Park J-E, Lee KY, Kim SA, Choi BY, Kim N. Whole Exome Sequencing for a Patient with Rubinstein-Taybi Syndrome Reveals de Novo Variants besides an Overt CREBBP Mutation. International Journal of Molecular Sciences. 2015; 16(3):5697-5713. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16035697
Chicago/Turabian StyleYoo, Hee Jeong, Kyung Kim, In Hyang Kim, Seong-Hwan Rho, Jong-Eun Park, Ki Young Lee, Soon Ae Kim, Byung Yoon Choi, and Namshin Kim. 2015. "Whole Exome Sequencing for a Patient with Rubinstein-Taybi Syndrome Reveals de Novo Variants besides an Overt CREBBP Mutation" International Journal of Molecular Sciences 16, no. 3: 5697-5713. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16035697