
Role of Pancreatic Transcription Factors in Maintenance of Mature β-Cell Function

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
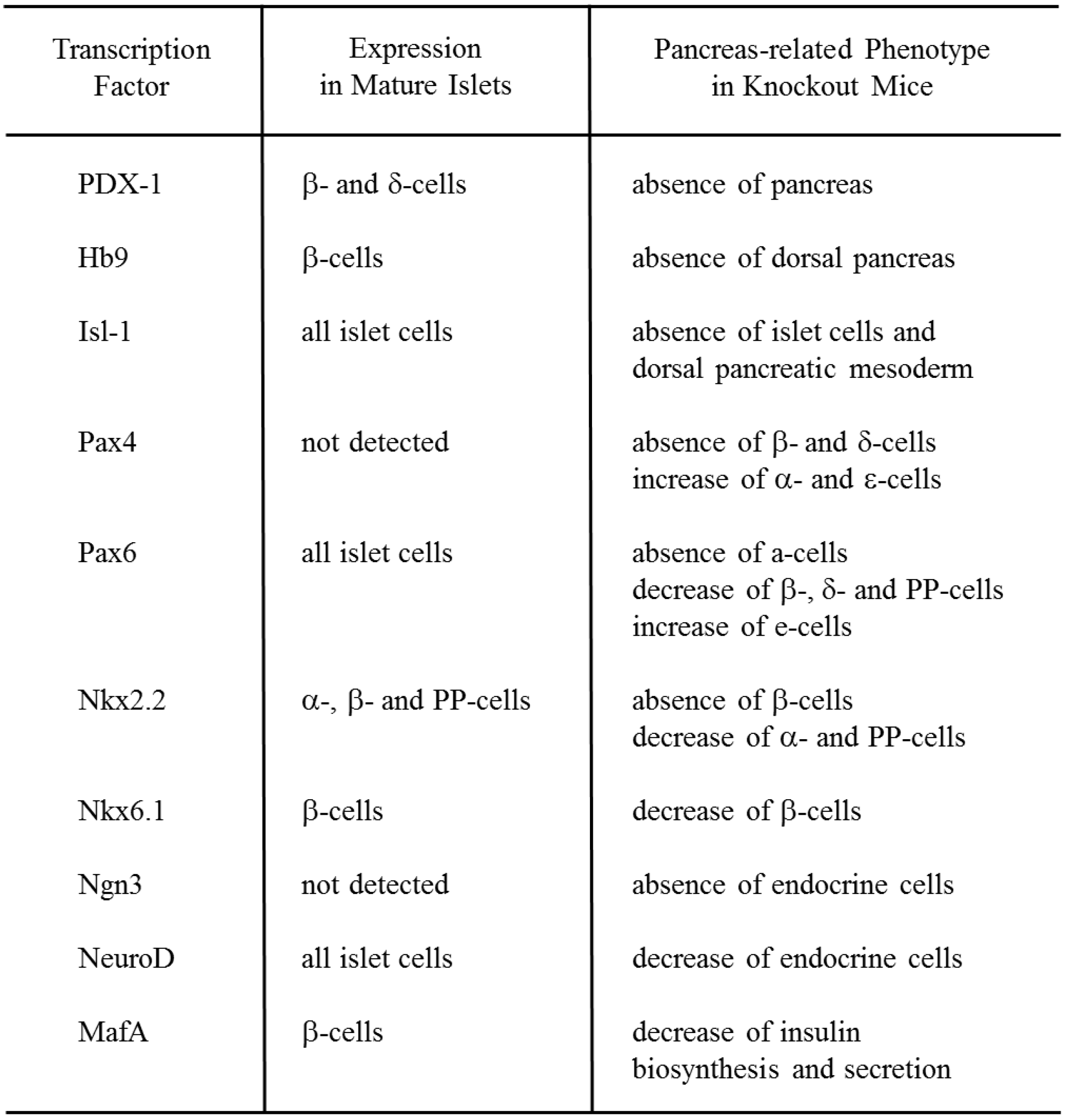
:1. Role of Pancreatic Transcription Factors in the Pancreas


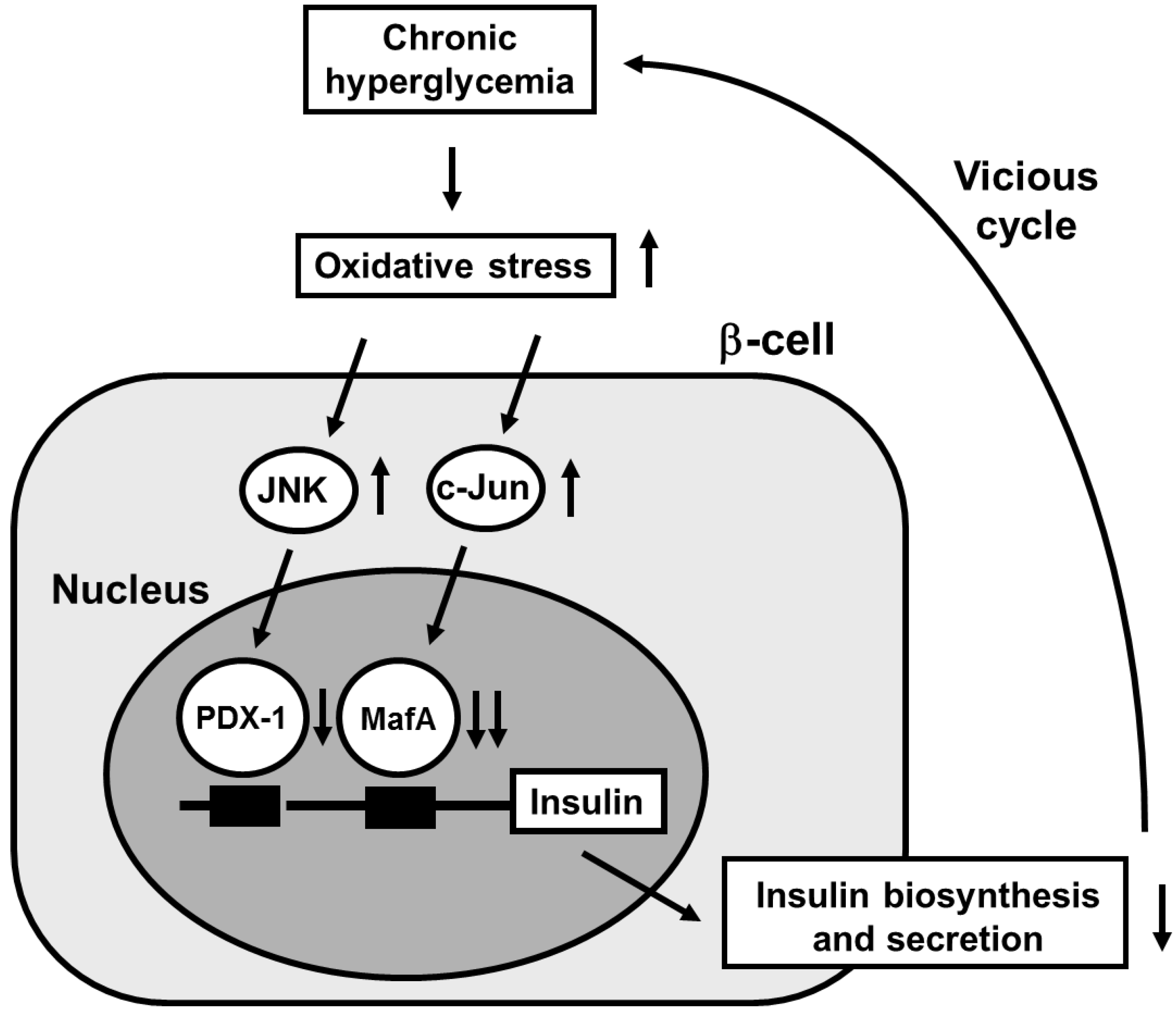
2. Involvement of Pancreatic Transcription Factors in β-Cell Glucose Toxicity

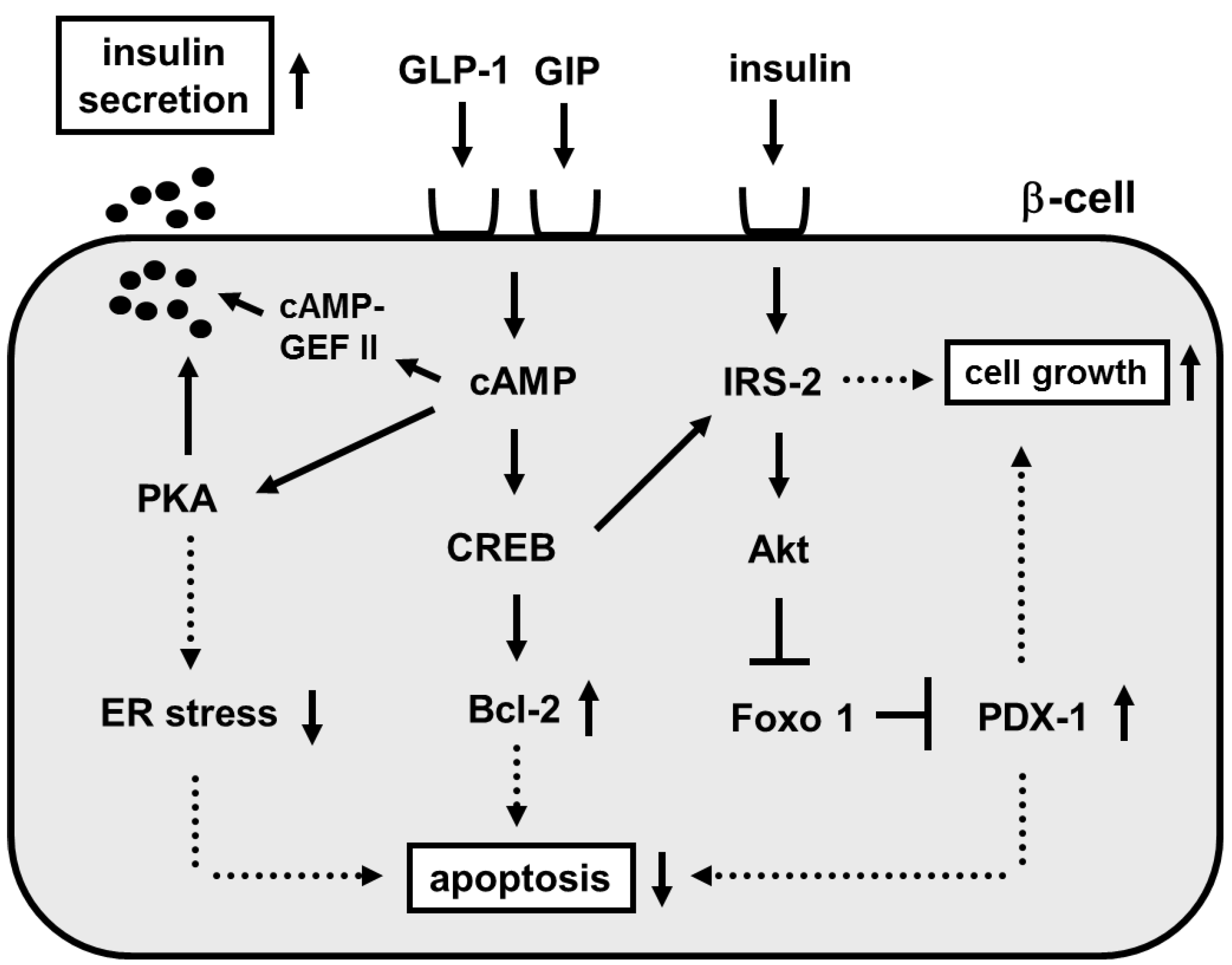
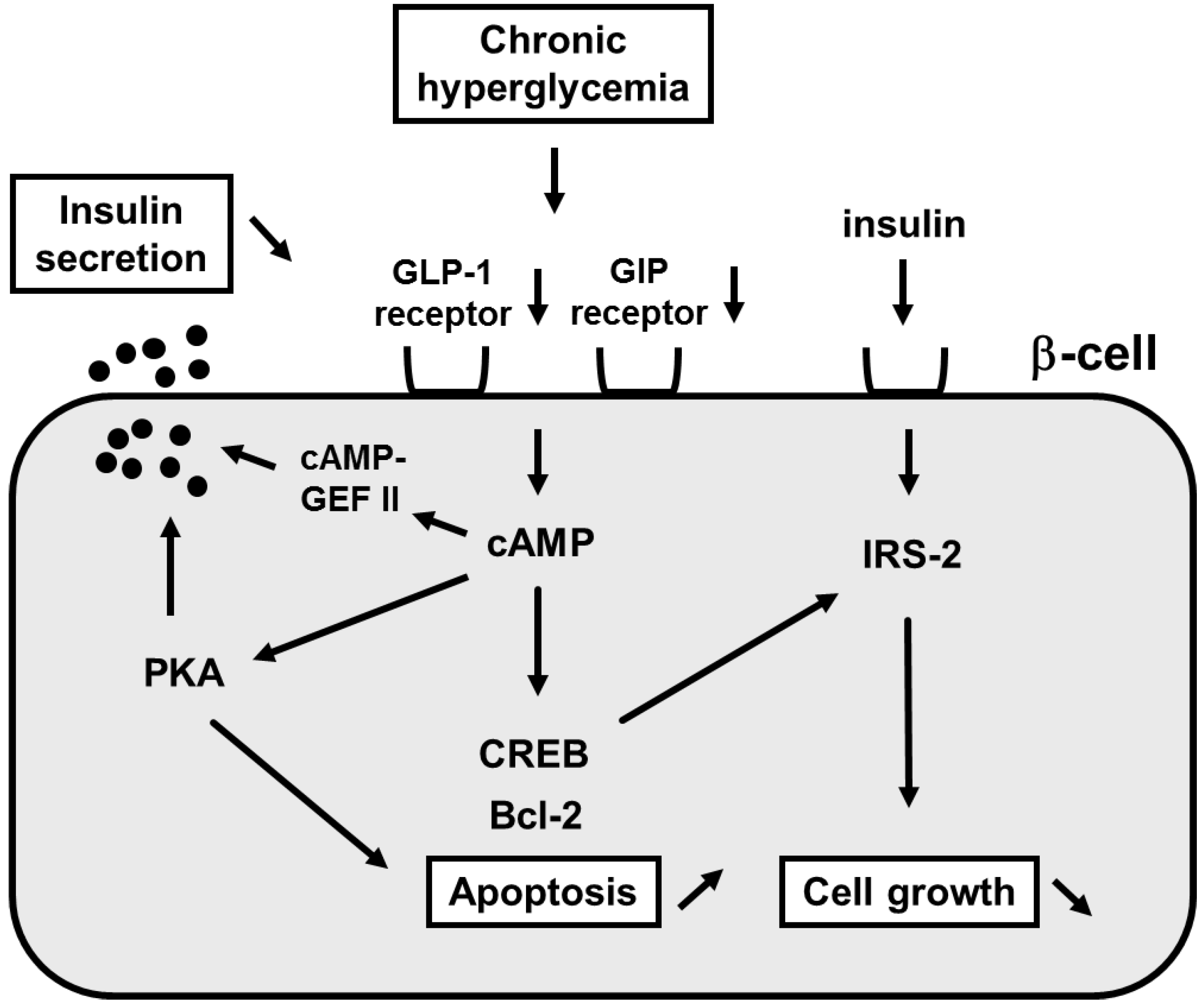
3. Involvement of Incretin Signaling in β-Cell Glucose Toxicity


Author Contributions
Conflicts of Interest
References
- Ohlsson, H.; Karlsson, K.; Edlund, T. IPF1, a homeodomain-containing-transactivator of the insulin gene. EMBO J. 1993, 2, 4251–4259. [Google Scholar]
- Miller, C.P.; McGehee, R.E.; Habener, J.F. IDX-1: A new homeodomain transcription factor expressed in rat pancreatic islets and duodenum that transactivates the somatostatin gene. EMBO J. 1994, 13, 1145–1156. [Google Scholar] [PubMed]
- Leonard, J.; Peers, B.; Johnson, T.; Ferreri, K.; Lee, S.; Montminy, M.R. Characterization of somatostatin transactivating factor-1, a novel homeobox factor that stimulates somatostatin expression in pancreatic islet cells. Mol. Endocrinol. 1993, 7, 1275–1283. [Google Scholar] [PubMed]
- Jonsson, J.; Carlsson, L.; Edlund, T.; Edlund, H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1993, 37, 606–609. [Google Scholar]
- Guz, Y.; Montminy, M.R.; Stein, R.; Leonard, J.; Gamer, L.W.; Wright, C.V.; Teitelmen, G. Expression of murine STF-1, a putative insulin gene transcription factor, in beta cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development 1995, 121, 11–18. [Google Scholar] [PubMed]
- Ahlgren, U.; Jonsson, J.; Edlund, H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development 1996, 122, 1409–1416. [Google Scholar] [PubMed]
- Offield, M.F.; Jetton, T.L.; Labosky, P.; Ray, M.; Stein, R.W.; Magnuson, M.A.; Hogan, B.L.; Wright, C.V. PDX-1 is required for pancreas outgrowth and differentiation of the rostral duodenum. Development 1996, 122, 983–985. [Google Scholar]
- Kaneto, H.; Miyagawa, J.; Kajimoto, Y.; Yamamoto, K.; Watada, H.; Umayahara, Y.; Hanafusa, Y.; Matsuzawa, Y.; Yamasaki, Y.; Higashiyama, S.; et al. Expression of heparin-binding epidermal growth factor-like growth factor during pancreas development: A potential role of PDX-1 in the transcriptional activation. J. Biol. Chem. 1997, 272, 29137–29143. [Google Scholar] [CrossRef] [PubMed]
- Stoffers, D.A.; Zinkin, N.T.; Stanojevic, V.; Clarke, W.L.; Habener, J.F. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet. 1997, 15, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Bonner-Weir, S.; Montminy, M.; Wright, C. Regulatory factor linked to late-onset diabetes? Nature 1998, 392, 560. [Google Scholar] [CrossRef] [PubMed]
- Stoffers, D.A.; Heller, R.S.; Miller, C.P.; Habener, J.F. Developmental expression of the homeodomain protein IDX-1 mice transgenic for an IDX-1 promoter/LacZ transcriptional reporter. Endocrinology 1999, 140, 5374–5381. [Google Scholar] [PubMed]
- Holland, A.M.; Hale, M.A.; Kagami, H.; Hammer, R.E.; MacDonald, R.J. Experimental control of pancreatic development and maintenance. Proc. Natl. Acad. Sci. USA 2002, 99, 12236–12241. [Google Scholar] [CrossRef] [PubMed]
- Melloul, D. Transcription factors in islet development and physiology: Role of PDX-1 in beta-cell function. Ann. N. Y. Acad. Sci. 2004, 1014, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.A.; Thaler, J.; Pfaff, S.L.; Gu, H.; Kehrl, J.H. Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9-deficient mice. Nat. Genet. 1999, 23, 71–75. [Google Scholar] [PubMed]
- Li, H.; Arber, S.; Jessell, T.M.; Edlund, H. Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat. Genet. 1999, 23, 67–70. [Google Scholar] [PubMed]
- Ahlgren, U.; Pfaff, S.L.; Jessell, T.M.; Edlund, T.; Edlund, H. Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature 1997, 385, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Pineda, B.; Chowdhury, K.; Torres, M.; Oliver, G.; Gruss, P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature 1997, 386, 399–402. [Google Scholar] [CrossRef] [PubMed]
- St-Onge, L.; Sosa-Pineda, B.; Chowdhury, K.; Mansouri, A.; Gruss, P. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature 1997, 387, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Sander, M.; Neubuser, A.; Kalamaras, J.; Ee, H.C.; Martin, G.R.; German, M.S. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev. 1997, 11, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Sander, M.; Sussel, L.; Conners, J.; Scheel, D.; Kalamaras, J.; Dela Cruz, F.; Schwitzgebel, V.; Hayes-Jordan, A.; German, M. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 2000, 127, 5533–5540. [Google Scholar] [PubMed]
- Sussel, L.; Kalamaras, J.; Hartigan-O’Connor, D.J.; Meneses, J.J.; Pedersen, R.A.; Rubenstein, J.L.; German, M.S. Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development 1998, 125, 2213–2221. [Google Scholar] [PubMed]
- Fujitani, Y.; Kajimoto, Y.; Yasuda, T.; Matsuoka, T.; Kaneto, H.; Umayahara, Y.; Fujita, N.; Watada, H.; Miyazaki, J.; Yamasaki, Y.; et al. Identification of a portable repression domain and an E1A-responsive activation domain in Pax 4: A possible role of Pax 4 as a transcriptional repressor in the pancreas. Mol. Cell. Biol. 1999, 19, 8281–8291. [Google Scholar] [PubMed]
- Smith, S.B.; Ee, H.C.; Conners, J.R.; German, M.S. Paired-homeodomain transcription factor PAX4 acts as a transcriptional repressor in early pancreatic development. Mol. Cell. Biol. 1999, 19, 8272–8280. [Google Scholar] [PubMed]
- Prado, C.L.; Pugh-Bernard, A.E.; Elghazi, L.; Sosa-Pineda, B.; Sussel, L. Ghrelin cells replace insulin-producing β cells in two mouse model of pancreas development. Proc. Natl. Acad. Sci. USA 2004, 101, 2924–2929. [Google Scholar] [CrossRef] [PubMed]
- Heller, R.S.; Jenny, M.; Collombat, P.; Mansouri, A.; Tomasetto, C.; Madsen, O.D.; Mellitzer, G.; Gradwohl, G.; Serup, P. Genetic determinants of pancreatic β-cell development. Dev. Biol. 2005, 286, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.K.; Nelson, S.B.; Jorgensen, M.C.; Henseleit, K.D.; Fujitani, Y.; Wright, C.V.; Sander, M.; Serup, P. Endodermal expression of Nkx6 genes depends differentially on Pdx1. Dev. Biol. 2005, 288, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Collombat, P.; Mansouri, A.; Hecksher-Sorensen, J.; Serup, P.; Krull, J.; Gradwohl, G.; Gruss, P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003, 17, 2591–2603. [Google Scholar] [CrossRef] [PubMed]
- Ferber, S.; Halkin, A.; Cohen, H.; Ber, I.; Einav, Y.; Goldberg, I.; Barshack, I.; Seijffers, R.; Kopolovic, J.; Kaiser, N.; et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat. Med. 2000, 6, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Heller, R.S.; Stoffers, D.A.; Bock, T.; Svenstrup, K.; Jensen, J.; Horn, T.; Miller, C.P.; Habener, J.F.; Madsen, O.D.; Serup, P. Improved glucose tolerance and acinar dysmorphogenesis by targeted expression of transcription factor PDX-1 to the exocrine pancreas. Diabetes 2001, 50, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Nakamura, T.; Fujita, Y.; Kishi, A.; Fujimiya, M.; Yamada, S.; Kudo, M.; Nishio, Y.; Maegawa, H.; Haneda, M.; et al. Combined expression of pancreatic duodenal homeobox 1 and islet factor 1 induces immature enterocytes to produce insulin. Diabetes 2002, 51, 1398–1408. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Kajimoto, Y.; Yasuda, T.; Watada, H.; Fujitani, Y.; Kosaka, H.; Gotow, T.; Miyatsuka, T.; Umayahara, Y.; Yamasaki, Y.; et al. PDX-1 induces differentiation of intestinal epithelioid IEC-6 into insulin-producing cells. Diabetes 2002, 51, 2505–2513. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Kaneto, H.; Weir, G.C.; Bonner-Weir, S. PDX-1 protein containing its own Antennapedia-like protein transduction domain can transduce pancreatic duct and islet cells. Diabetes 2003, 52, 1732–1737. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Yamato, E.; Tashiro, F.; Ikegami, H.; Ogihara, T.; Miyazaki, J. β-Cell neogenesis induced by adenovirus-mediated gene delivery of transcription factor pdx-1 into mouse pancreas. Gene Ther. 2003, 10, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.-Q.; Cao, L.-Z.; Burkhardt, B.R.; Xia, C.-Q.; Litherland, S.A.; Atkinson, M.A.; Yang, L.-J. In vivo and in vitro characterization of insulin-producing cells obtained from murine bone marrow. Diabetes 2004, 53, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Miyatsuka, T.; Kaneto, H.; Kajimoto, Y.; Hirota, S.; Arakawa, Y.; Fujitani, Y.; Umayahara, Y.; Watada, H.; Yamasaki, Y.; Magnuson, M.A.; et al. Ectopically expressed PDX-1 in liver initiates endocrine and exocrine pancreas differentiation but causes dysmorphogenesis. Biochem. Biophys. Res. Commun. 2003, 310, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Nakatani, Y.; Miyatsuka, T.; Matsuoka, T.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. PDX-1/VP16 fusion protein, together with NeuroD or Ngn3, markedly induces insulin gene transcription and ameliorates glucose tolerance. Diabetes 2005, 54, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.-Z.; Tang, D.-Q.; Horb, M.E.; Li, S.-W.; Yang, L.-J. High glucose is necessary for complete maturation of Pdx 1-VP16-expressing hepatic cells into functional insulin-producing cells. Diabetes 2004, 53, 3168–3178. [Google Scholar] [CrossRef] [PubMed]
- Imai, J.; Katagiri, H.; Yamada, T.; Ishigaki, Y.; Ogihara, T.; Uno, K.; Hasegawa, Y.; Gao, J.; Ishihara, H.; Sasano, H.; et al. Constitutively active PDX1 induced efficient insulin production in adult murine liver. Biochem. Biophys. Res. Commun. 2005, 326, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Petersen, H.V.; Serup, P.; Leonard, J.; Michelsen, B.K.; Madsen, O.D. Transcriptional regulation of the human insulin gene is dependent on the homeodomain protein STF1/IPF1 acting through the CT boxes. Proc. Natl. Acad. Sci. USA 1994, 91, 10465–10469. [Google Scholar] [CrossRef] [PubMed]
- Peers, B.; Leonard, J.; Sharma, S.; Teitelman, G.; Montminy, M.R. Insulin expression in pancreatic islet cells relies on cooperative interactions between the helix loop helix factor E47 and the homeobox factor STF-1. Mol. Endocrinol. 1994, 8, 1798–1806. [Google Scholar] [PubMed]
- Waeber, G.; Thompson, N.; Nicod, P.; Bonny, C. Transcriptional activation of the GLUT2 gene by the IPF-1/STF-1/IDX-1 homeobox factor. Mol. Endocrinol. 1996, 10, 1327–1334. [Google Scholar] [PubMed]
- Watada, H.; Kajimoto, Y.; Umayahara, Y.; Matsuoka, T.; Kaneto, H.; Fujitani, Y.; Kamada, T.; Kawamori, R.; Yamasaki, Y. The human glucokinase gene β-cell-type promoter: An essential role of insulin promoter factor 1 (IPF1)/PDX-1 in its activation in HIT-T15 cells. Diabetes 1996, 45, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, U.; Jonsson, J.; Jonsson, L.; Simu, K.; Edlund, H. β-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the β-cell phenotype and maturity onset diabetes. Genes Dev. 1998, 12, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Maechler, P.; Ritz-Laser, B.; Hagenfeldt, K.A.; Ishihara, H.; Philippe, J.; Wollheim, C.B. Pdx1 level defines pancreatic gene expression pattern and cell lineage differentiation. J. Biol. Chem. 2001, 276, 25279–25286. [Google Scholar] [CrossRef] [PubMed]
- Brissova, M.; Shiota, M.; Nicholson, W.E.; Gannon, M.; Knobel, S.M.; Piston, D.W.; Wright, C.V.; Powers, A.C. Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J. Biol. Chem. 2002, 277, 1125–1132. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Jhala, U.S.; Winnay, J.N.; Krajewski, S.; Montminy, M.; Kahn, C.R. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J. Clin. Investig. 2004, 114, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.M.; Gonez, L.J.; Naselli, G.; MacDonald, R.J.; Harrison, L.C. Conditional expression demonstrates the role of the homeodomain transcription factor Pdx1 in maintenance and regeneration of beta-cells in the adult pancreas. Diabetes 2005, 54, 2586–2595. [Google Scholar] [CrossRef] [PubMed]
- Stoffers, D.A.; Ferrer, J.; Clarke, W.L.; Habener, J.F. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat. Genet. 1997, 17, 138–139. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, W.M.; Frayling, T.M.; Ellard, S.; Evans, J.C.; Allen, L.I.S.; Bulman, M.P.; Ayres, S.; Shepherd, M.; Clark, P.; Millward, A.; et al. Missense mutations in the insulin promoter factor-1 gene predispose to type 2 diabetes. J. Clin. Investig. 1999, 104, R33–R39. [Google Scholar] [CrossRef] [PubMed]
- Hani, E.H.; Stoffers, D.A.; Chevre, J.-C.; Durand, E.; Stanojevic, V.; Dina, C.; Haebener, J.F.; Froguel, P. Defective mutations in the insulin promoter factor-1 (IPF-1) gene in late-onset type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, R41–R48. [Google Scholar] [CrossRef] [PubMed]
- Clocquet, A.R.; Egan, J.M.; Stoffers, D.A.; Muller, D.C.; Wideman, L.; Chin, G.A.; Clarke, W.L.; Hanks, J.B.; Habener, J.F.; Elahi, D. Impaired insulin secretion and increased insulin sensitivity in familial maturity-onset diabetes of the young 4 (insulin promoter factor 1 gene). Diabetes 2000, 49, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Macfarlane, W.M.; Lehto, M.; Gu, H.F.; Shepherd, L.M.; Ivarsson, S.A.; Wibell, L.; Smith, T.; Groop, L.C. Functional consequences of mutations in the MODY4 gene (IPF1) and coexistence with MODY3 mutations. Diabetologia 2001, 44, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Naya, F.J.; Stellrecht, C.M.M.; Tsai, M.-J. Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev. 1995, 9, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Naya, F.J.; Huang, H.; Qiu, Y.; Mutoh, H.; DeMayo, F.; Leiter, A.B.; Tsai, M.-J. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997, 11, 323–334. [Google Scholar] [CrossRef]
- Kojima, H.; Fujimiya, M.; Matsumura, K.; Younan, P.; Imaeda, H.; Maeda, M.; Chan, L. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat. Med. 2003, 9, 595–603. [Google Scholar] [CrossRef]
- Noguchi, H.; Bonner-Weir, S.; Wei, F.-Y.; Matsushita, M.; Matsumoto, S. BETA2/NeuroD protein can be transduced into cells due to an arginine- and lysine-rich sequence. Diabetes 2005, 54, 2859–2866. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Stein, R. Glucose-induced transcription of the insulin gene is mediated by factors required for β-cell-type-specific expression. Mol. Cell. Biol. 1994, 14, 871–879. [Google Scholar] [PubMed]
- German, M.S.; Wang, J. The insulin gene contains multiple transcriptional elements that respond to glucose. Mol. Cell. Biol. 1994, 14, 4067–4075. [Google Scholar] [PubMed]
- Grapin-Botton, A.; Majithiam, A.R.; Melton, D.A. Key events of pancreas formation are triggered in gut endoderm by ectopic expression of pancreatic regulatory genes. Genes Dev. 2001, 15, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Apelqvist, A.; Li, H.; Sommer, L.; Beatus, P.; Anderson, D.J.; Honjo, T.; de Angelis, M.H.; Lendahl, U.; Edlund, H. Notch signaling controls pancreatic cell differentiation. Nature 1999, 400, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Schwitzgebel, V.M.; Scheel, D.W.; Conners, J.R.; Kalamaras, J.; Lee, J.E.; Anderson, D.J.; Sussel, L.; Johnson, J.D.; German, M.S. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 2000, 127, 3533–3542. [Google Scholar] [PubMed]
- Gradwohl, G.; Dierich, A.; LeMeur, M.; Guillemot, F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci. USA 2000, 97, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar] [PubMed]
- Watada, H. Neurogenin 3 is a key transcription factor for differentiation of the endocrine pancreas. Endocr. J. 2004, 51, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bendala, J.; Klein, D.; Ribeiro, M.; Ricordi, C.; Inverardi, L.; Pastori, R.; Edlund, H. TAT-mediated neurogenin 3 protein transduction stimulates pancreatic endocrine differentiation in vitro. Diabetes 2005, 54, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Fusco-DeMane, D.; Henderson, E.; Efrat, S.; Stein, R. The role of the insulin control element and RIPE3b1 activators in glucose-stimulated transcription of the insulin gene. Mol. Endocrinol. 1995, 9, 1468–1488. [Google Scholar] [PubMed]
- Moran, A.; Zhang, H.-J.; Olson, L.K.; Harmon, J.S.; Poitout, V.; Robertson, R.P. Differentiation of glucose toxicity from beta cell exhaustion during the evolution of defective insulin gene expression in the pancreatic islet cell line, HIT-T15. J. Clin. Investig. 1997, 99, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Zhao, L.; Artner, I.; Jarrett, H.W.; Friedman, D.; Means, A.; Stein, R. Members of the large Maf transcription family regulate insulin gene transcription in islet beta cells. Mol. Cell. Biol. 2003, 23, 6049–6062. [Google Scholar] [CrossRef] [PubMed]
- Olbrot, M.; Rud, J.; Moss, L.G.; Sharma, A. Identification of β-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc. Natl. Acad. Sci. USA 2002, 99, 6737–6742. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Han, S.I.; Shioda, S.; Hirai, M.; Nishizawa, M.; Handa, H. MafA is a glucose-regulated and pancreatic β-cell-specific transcriptional activator for the insulin gene. J. Biol. Chem. 2002, 277, 49903–49910. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Artner, I.; Henderson, E.; Means, A.; Sander, M.; Stein, R. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc. Natl. Acad. Sci. USA 2004, 101, 2930–2933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Moriguchi, T.; Kajihara, M.; Esaki, R.; Harada, A.; Shimohata, H.; Oishi, H.; Hamada, M.; Morito, N.; Hasegawa, K.; et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol. 2005, 25, 4969–4976. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Matsuoka, T.; Nakatani, Y.; Miyatsuka, T.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. A crucial role of MafA as a novel therapeutic target for diabetes. J. Biol. Chem. 2005, 280, 15047–15052. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Kaneto, H.; Stein, R.; Miyatsuka, T.; Kawamori, D.; Henderson, E.; Kojima, I.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. MafA regulates expression of genes important to islet β cell function. Mol. Endocrinol. 2007, 21, 2764–2774. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Brun, T.; Kataoka, K.; Sharma, A.J.; Wollheim, C.B. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 2007, 50, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Laybutt, D.R.; Kaneto, H.; Bonner-Weir, S.; Sharma, A. β-Cell adaptation and decompensation during the progression of diabetes. Diabetes 2001, 50, S154–S159. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Nolan, C.J. Islet β cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Olson, L.K.; Robertson, R.P. Chronic exposure of βTC-6 cells to supraphysiologic concentrations of glucose decreases binding of the RIPE3b1 insulin gene transcription activator. J. Clin. Investig. 1996, 97, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Robertson, R.P. Minireview: Secondary beta-cell failure in type 2 diabetes—A convergence of glucotoxicity and lipotoxicity. Endocrinology 2002, 143, 339–342. [Google Scholar] [PubMed]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and β-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gorogawa, S.; Kajimoto, Y.; Umayahara, U.; Kaneto, H.; Watada, H.; Kuroda, A.; Kawamori, D.; Yasuda, T.; Matsuhisa, M.; Yamasaki, Y.; et al. Probucol preserves pancreatic β-cell function through reduction of oxidative stress in type 2 diabetes. Diabetes Res. Clin. Pract. 2002, 57, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Harmon, J.S.; Stein, R.; Robertson, R.P. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J. Biol. Chem. 2005, 280, 11107–11113. [Google Scholar] [CrossRef] [PubMed]
- Ihara, Y.; Toyokuni, S.; Uchida, K.; Odaka, H.; Tanaka, T.; Ikeda, H.; Hiai, H.; Seino, Y.; Yamada, Y. Hyperglycemia causes oxidative stress in pancreatic β-cells of GK rats, a model of type 2 diabetes. Diabetes 1999, 48, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Kajimoto, Y.; Matsuoka, T.; Kaneto, H.; Watada, H.; Fujitani, Y.; Kishimoto, M.; Sakamoto, K.; Matsuhisa, M.; Kawamori, R.; Yamasaki, Y.; et al. Induction of glycation suppresses glucokinase gene expression in HIT-T15 cells. Diabetologia 1999, 42, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Maechler, P.; Jornot, L.; Wollheim, C.B. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J. Biol. Chem. 1999, 274, 27905–27913. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Kajimoto, Y.; Miyagawa, J.; Matsuoka, T.; Fujitani, Y.; Umayahara, Y.; Hanafusa, T.; Matsuzawa, Y.; Yamasaki, Y.; Hori, M. Beneficial effects of antioxidants for diabetes: Possible protection of pancreatic β-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Xu, G.; Song, K.H.; Suzuma, K.; Bonner-Weir, S.; Sharma, A.; Weir, G.C. Activation of the hexosamine pathway leads to deterioration of pancreatic β-cell function by provoking oxidative stress. J. Biol. Chem. 2001, 276, 31099–31104. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Matsuoka, T.; Nakatani, Y.; Kawamori, D.; Miyatsuka, T.; Matsuhisa, M.; Yamasaki, Y. Oxidative stress, ER stress, and the JNK pathway in type 2 diabetes. J. Mol. Med. 2005, 83, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Kajimoto, Y.; Watada, H.; Kaneto, H.; Kishimoto, M.; Umayahara, Y.; Fujitani, Y.; Kamada, T.; Kawamori, R.; Yamasaki, Y. Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J. Clin. Investig. 1997, 99, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Tanaka, Y.; Takahashi, H. Glucose toxicity in β-cells: Type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003, 52, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J. Biol. Chem. 2004, 271, 42351–42354. [Google Scholar] [CrossRef]
- Tanaka, Y.; Gleason, C.E.; Tran, P.O.T.; Harmon, J.S.; Robertson, R.P. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc. Natl. Acad. Sci. USA 1999, 96, 10857–10862. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Tran, P.O.T.; Harmon, J.; Robertson, R.P. A role of glutathione peroxidase in protecting pancreatic β cells against oxidative stress in a model of glucose toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 12363–12368. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Matsumoto, K.; Nishikawa, T.; Suefuji, M.; Nakamaru, K.; Hirashima, Y.; Kawashima, J.; Shirotani, T.; Ichinose, K.; Brownlee, M.; et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic β-cells. Biochem. Biophys. Res. Commun. 2003, 300, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Fujii, J.; Myint, T.; Islam, K.N.; Miyazawa, N.; Suzuki, K.; Kawasaki, Y.; Nakamura, M.; Tatsumi, H.; Yamasaki, Y.; et al. Reducing sugar triggers oxidative modification and apoptosis in pancreatic β-cells by provoking oxidative stress through the glycation reaction. Biochem. J. 1996, 320, 855–863. [Google Scholar] [PubMed]
- Kaneto, H.; Xu, G.; Fujii, N.; Kim, S.; Bonner-Weir, S.; Weir, G.C. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J. Biol. Chem. 2002, 277, 30010–30018. [Google Scholar] [CrossRef] [PubMed]
- Kawamori, D.; Kajimoto, Y.; Kaneto, H.; Umayahara, Y.; Fujitani, Y.; Miyatsuka, T.; Watada, H.; Leibiger, I.B.; Yamasaki, Y.; Hori, M. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun N-terminal kinase. Diabetes 2003, 52, 2896–2904. [Google Scholar] [CrossRef] [PubMed]
- Devary, Y.; Gottlieb, R.A.; Lau, L.F.; Karin, M. Rapid and preferential activation of the c-jun gene during the mammalian UV response. Mol. Cell. Biol. 1991, 11, 2804–2811. [Google Scholar] [PubMed]
- Nose, K.; Shibanuma, M.; Kikuchi, K.; Kageyama, H.; Sakiyama, S.; Kuroki, T. Transcriptional activation of early-response genes by hydrogen peroxide in a mouse osteoblastic cell line. Eur. J. Biochem. 1991, 201, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Kaneto, H.; Miyatsuka, T.; Yamamoto, T.; Yamamoto, K.; Kato, K.; Shimomura, I.; Stein, R.; Matsuhisa, M. Regulation of MafA expression in pancreatic β-cells in db/db mice with diabetes. Diabetes 2010, 59, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Maekawa, T.; Sudo, T.; Ishii, S.; Seino, Y.; Imura, H. c-Jun represses the human insulin promoter activity that depends on multiple cAMP response elements. Proc. Natl. Acad. Sci. USA 1992, 89, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Henderson, E.; Stein, R. c-jun inhibits transcriptional activation by the insulin enhancer, and the insulin control element is the target of control. Mol. Cell. Biol. 1994, 14, 655–662. [Google Scholar] [PubMed]
- Nauck, M.A.; Heimesaat, M.M.; Orskov, C.; Holst, J.J.; Ebert, R.; Creutzfeldt, W. Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J. Clin. Investig. 1993, 91, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Kaneto, H.; Laybutt, D.R.; Duvivier-Kali, V.; Trivedi, N.; Suzuma, K.; King, G.L.; Weir, G.C.; Bonner-Weir, S. Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: Possible contribution to the impaired incretin effects in diabetes. Diabetes 2007, 56, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Matsuoka, T.; Kaneto, H.; Tochino, Y.; Kato, K.; Yamamoto, K.; Yamamoto, T.; Matsuhisa, M.; Shimomura, I. Effect of alogliptin, pioglitazone and glargine on pancreatic β-cells in diabetic db/db mice. Biochem. Biophys. Res. Commun. 2011, 404, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Matveyenko, A.V.; Kerr-Conte, J.; Cho, J.H.; McIntosh, C.H.; Maedler, K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum. Mol. Genet. 2009, 18, 2388–2399. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.F.; Deng, Y.; Zhou, Y.; Fan, R.R.; Chan, J.C.; Laybutt, D.R.; Luzuriaga, J.; Xu, G. Pharmacological reduction of NEFA restores the efficacy of incretin-based therapies through GLP-1 receptor signalling in the beta cell in mouse models of diabetes. Diabetologia 2013, 56, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Florez, J.C.; Jablonski, K.A.; Bayley, N.; Pollin, T.I.; de Bakker, P.I.; Shuldiner, A.R.; Knowler, W.C.; Nathan, D.M.; Altshuler, D. TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N. Engl. J. Med. 2006, 355, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Gianniny, L.; Burtt, N.P.; Lyssenko, V.; Giuducci, C.; Sjögren, M.; Florez, J.C.; Almgren, P.; Isomaa, B.; Orho-Melander, M.; et al. Common single nucleotide polymorphisms in TCF7L2 are reproducibly associated with type 2 diabetes and reduce the insulin response to glucose in nondiabetic individuals. Diabetes 2006, 55, 2890–2895. [Google Scholar] [CrossRef]
- Horikoshi, M.; Hara, K.; Ito, C.; Nagai, R.; Froguel, P.; Kadowaki, T. A genetic variation of the transcription factor 7-like 2 gene is associated with risk of type 2 diabetes in the Japanese population. Diabetologia 2007, 50, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Lyssenko, V.; Lupi, R.; Marchetti, P.; Lyssenko, V.; Lupi, R.; Marchetti, P.; del Guerra, S.; Orho-Melander, M.; Almgren, P.; Sjögren, M.; et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J. Clin. Investig. 2007, 117, 2155–2163. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; van Es, J.H.; Huch, M.; Li, V.S.; José, A.; Hatzis, P.; Mokry, M.; Haegebarth, A.; van den Born, M.; Chambon, P.; et al. Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell 2012, 151, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Villareal, D.T.; Robertson, H.; Bell, G.I.; Patterson, B.W.; Tran, H.; Wice, B.; Polonsky, K.S. TCF7L2 variant rs7903146 affects the risk of type 2 diabetes by modulating incretin action. Diabetes 2010, 59, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Habener, J.F. Glucagon-like peptide-1 activation of TCF7L2-dependent Wnt signaling enhances pancreatic beta cell proliferation. J. Biol. Chem. 2008, 283, 8723–8735. [Google Scholar] [CrossRef] [PubMed]
- Takamoto, I.; Kubota, N.; Nakaya, K.; Kumagai, K.; Hashimoto, S.; Kubota, T.; Inoue, M.; Kajiwara, E.; Katsuyama, H.; Obata, A.; et al. TCF7L2 in mouse pancreatic beta cells plays a crucial role in glucose homeostasis by regulating beta cell mass. Diabetologia 2014, 57, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.K.; Mondragon, A.; Chen, L.; McGinty, J.A.; French, P.M.; Ferrer, J.; Thorens, B.; Hodson, D.J.; Rutter, G.A.; Xavier, G.D. Selective disruption of Tcf7l2 in the pancreatic β cell impairs secretory function and lowers β cell mass. Hum. Mol. Genet. 2015, 24, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaneto, H.; Matsuoka, T.-a. Role of Pancreatic Transcription Factors in Maintenance of Mature β-Cell Function. Int. J. Mol. Sci. 2015, 16, 6281-6297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16036281
Kaneto H, Matsuoka T-a. Role of Pancreatic Transcription Factors in Maintenance of Mature β-Cell Function. International Journal of Molecular Sciences. 2015; 16(3):6281-6297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16036281
Chicago/Turabian StyleKaneto, Hideaki, and Taka-aki Matsuoka. 2015. "Role of Pancreatic Transcription Factors in Maintenance of Mature β-Cell Function" International Journal of Molecular Sciences 16, no. 3: 6281-6297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16036281