
Xeroderma Pigmentosum: Low Prevalence of Germline XPA Mutations in a Brazilian XP Population

Abstract
:1. Introduction
2. Results and Discussion
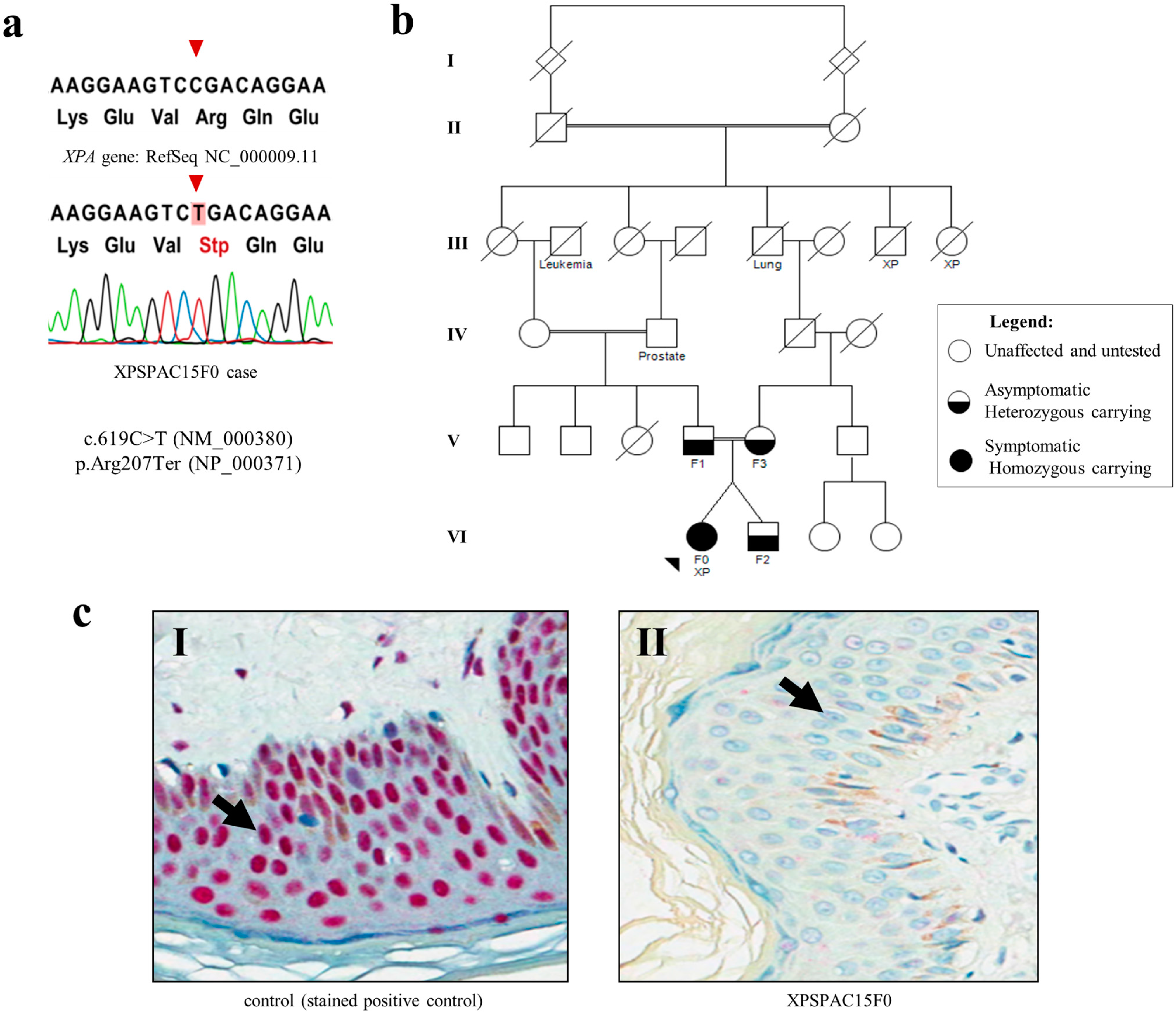
2.1. Germline Mutation Spectrum of XP Syndrome and Clinical Profile

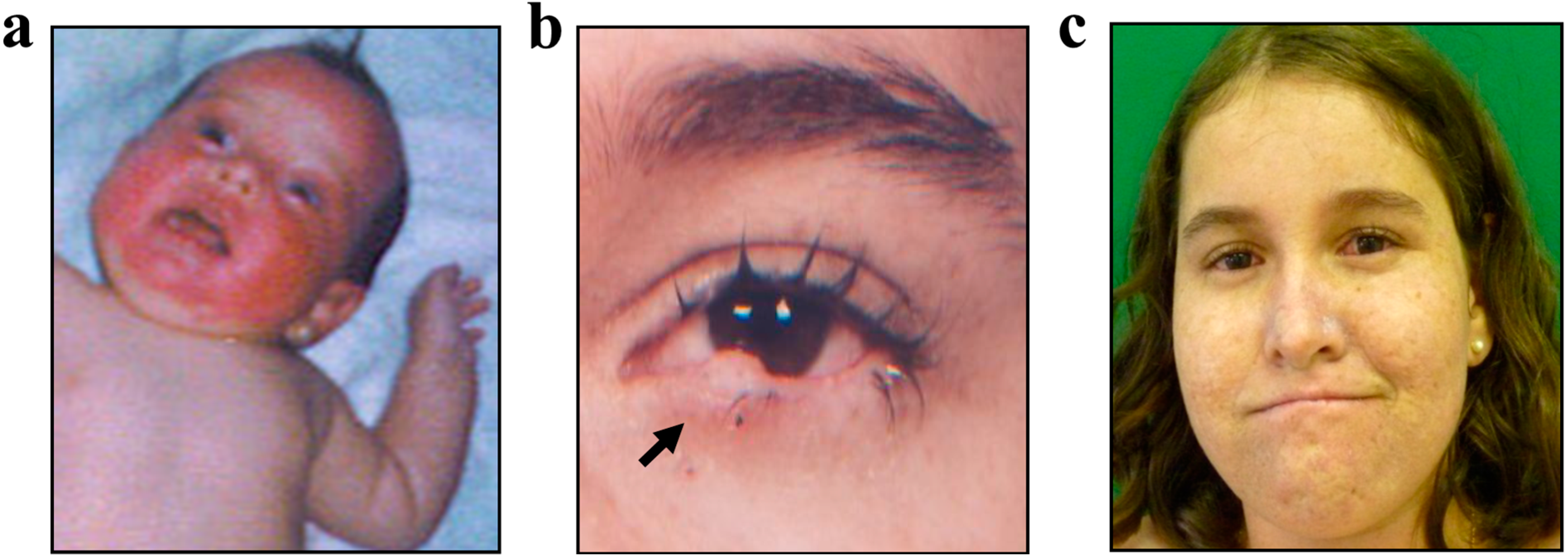
2.2. Case Report

3. Experimental Section
3.1. Enrollment of Patients and Ethics Statement
3.2. Genomic DNA Extraction
3.3. XPA Direct Sequencing

{kind=link}
{kind=link}
| Exon | Size (bp) | Forward Primer | Reverse Primer |
|---|---|---|---|
| 1 | 483 | AGGCGCTCTCACTCAGAAAG | GTGGACAGGACGCTTTGAC |
| 2 | 523 | AGACTAGCTGGGACCTTCAGT | AACAACAGAGAGCAGCAACC |
| 3 | 419 | GGCATTGCATACATGCTG | ACCATCGGCATCCTTCCTAT |
| 4 | 645 | CCTAGAGCCTTTTCCCTTGC | CCAGCCTGAGTGACAGAGTG |
| 4 | 337 | GCTGTGTGTGCCCCTAAGTTGC | AGCAAAAGCCAAACCAATTATGAC |
| 5 | 611 | GTGAGCCCACCACAGTTGAT | GGTTTGAGCTTAGTGCCTTG |
| 7 | 574 | CTCTTGTTTCACACTGCTCCAG | CCAGGTGACCTTCACTGAAAC |
| 7 | 594 | GTGAGGTAAGAAAGTAAGTTTGCCAAG | TCTAGCACTCAGCTCCCATCTCTG |
3.4. XPA Protein Expression by Immunohistochemistry
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fassihi, H. Spotlight on “xeroderma pigmentosum”. Photochem. Photobiol. Sci. 2013, 12, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E.; Lam, E.T.; Revet, I. Disorders of nucleotide excision repair: The genetic and molecular basis of heterogeneity. Nat. Rev. Genet. 2009, 10, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. The discovery that xeroderma pigmentosum (XP) results from defective nucleotide excision repair. DNA Repair (Amst.) 2004, 3. [Google Scholar] [CrossRef]
- Menck, C.F.; Munford, V. DNA repair diseases: What do they tell us about cancer and aging? Genet. Mol. Biol. 2014, 37, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, K.H.; Lee, M.M.; Scotto, J. Xeroderma pigmentosum: Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch. Dermatol. 1987, 123, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Anttinen, A.; Koulu, L.; Nikoskelainen, E.; Portin, R.; Kurki, T.; Erkinjuntti, M.; Jaspers, N.G.; Raams, A.; Green, M.H.; Lehmann, A.R.; et al. Neurological symptoms and natural course of xeroderma pigmentosum. Brain 2008, 131, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.; Lehmann, J.; Kalfon, L.; Slor, H.; Falik-Zaccai, T.C.; Emmert, S. Clinical utility gene card for: Xeroderma pigmentosum. Eur. J. Hum. Genet. 2013, 22. [Google Scholar] [CrossRef]
- Moriwaki, S.; Kraemer, K.H. Xeroderma pigmentosum—Bridging a gap between clinic and laboratory. Photodermatol. Photoimmunol. Photomed. 2001, 17, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Bradford, P.T.; Goldstein, A.M.; Tamura, D.; Khan, S.G.; Ueda, T.; Boyle, J.; Oh, K.S.; Imoto, K.; Inui, H.; Moriwaki, S.; et al. Cancer and neurologic degeneration in xeroderma pigmentosum: Long term follow-up characterises the role of DNA repair. J. Med. Genet. 2011, 48, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Cardoso, C.; Paes da Silva Ramos Fernandes, L.M.; Ferreira-Rocha, J.; Teixeira-Soares, C.; Antônio-Barreto, J.; Humberto-Damante, J. Xeroderma pigmentosum—A case report with oral implications. J. Clin. Exp. Dent. 2012, 4, e248–e251. [Google Scholar] [CrossRef] [PubMed]
- Leite, R.A.; Marchetto, M.C.; Muotri, A.R.; Vasconcelos, D.E.M.; de Oliveira, Z.N.; Machado, M.C.; Menck, C.F. Identification of XP complementation groups by recombinant adenovirus carrying DNA repair genes. J. Investig. Dermatol. 2009, 129, 502–506. [Google Scholar] [CrossRef] [PubMed]
- De Sá, B.C.; Rezze, G.G.; Scramim, A.P.; Landman, G.; Neves, R.I. Cutaneous melanoma in childhood and adolescence: Retrospective study of 32 patients. Melanoma Res. 2004, 14, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Procianoy, F.; Cruz, A.A.; Baccega, A.; Ferraz, V.; Chahud, F. Aggravation of eyelid and conjunctival malignancies following photodynamic therapy in desanctis-cacchione syndrome. Ophthal. Plast. Reconstr. Surg. 2006, 22, 498–499. [Google Scholar] [CrossRef] [PubMed]
- Mareddy, S.; Reddy, J.; Babu, S.; Balan, P. Xeroderma pigmentosum: Man deprived of his right to light. Sci. World J. 2013, 2013. [Google Scholar] [CrossRef]
- Kraemer, K.H.; Tamura, D.; Khan, S.G.; Digiovanna, J.J. Burning issues in the diagnosis of xeroderma pigmentosum. Br. J. Dermatol. 2013, 169. [Google Scholar] [CrossRef]
- Hananian, J.; Cleaver, J.E. Xeroderma pigmentosum exhibiting neurological disorders and systemic lupus erythematosus. Clin. Genet. 1980, 17, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Li, R.; Zhang, Y.; Zhong, R.; Pei, J.; Xiong, L.; Zhang, X.; Han, B. XPA gene rs1800975 single nucleotide polymorphism and lung cancer risk: A meta-analysis. Tumour Biol. 2014, 35, 6607–6617. [Google Scholar] [CrossRef] [PubMed]
- Bartels, C.L.; Lambert, M.W. Domains in the XPA protein important in its role as a processivity factor. Biochem. Biophys. Res. Commun. 2007, 356, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, K.M.; Christensen, J.; Li, L.; Finch, R.A.; Glazer, P.M. Human XPA and RPA DNA repair proteins participate in specific recognition of triplex-induced helical distortions. Proc. Natl Acad. Sci. USA 2002, 99, 5848–5853. [Google Scholar] [CrossRef] [PubMed]
- Satokata, I.; Tanaka, K.; Miura, N.; Narita, M.; Mimaki, T.; Satoh, Y.; Kondo, S.; Okada, Y. Three nonsense mutations responsible for group a xeroderma pigmentosum. Mutat. Res. 1992, 273, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Sidwell, R.U.; Sandison, A.; Wing, J.; Fawcett, H.D.; Seet, J.E.; Fisher, C.; Nardo, T.; Stefanini, M.; Lehmann, A.R.; Cream, J.J. A novel mutation in the XPA gene associated with unusually mild clinical features in a patient who developed a spindle cell melanoma. Br. J. Dermatol. 2006, 155, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Laposa, R.R.; Cleaver, J.E. DNA repair on the brain. Proc. Natl Acad. Sci. USA 2001, 98, 12860–12862. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Kanda, F.; Aoyama, N.; Fujii, M.; Nishigori, C.; Toda, T. Neuroimaging features of xeroderma pigmentosum group A. Brain Behav. 2012, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.P.; Liu, Y.C.; Alimchandani, M.; Liu, Q.; Aung, P.P.; Matsuda, K.; Lee, C.C.; Tsokos, M.; Hewitt, S.; Rushing, E.J.; et al. The influence of DNA repair on neurological degeneration, cachexia, skin cancer and internal neoplasms: Autopsy report of four xeroderma pigmentosum patients (XP-A, XP-C and XP-D). Acta. Neuropathol. Commun. 2013, 1. [Google Scholar] [CrossRef]
- Kuraoka, I.; Bender, C.; Romieu, A.; Cadet, J.; Wood, R.D.; Lindahl, T. Removal of oxygen free-radical-induced 5',8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl. Acad. Sci. USA 2000, 97, 3832–3837. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J. The case for 8,5'-cyclopurine-2'-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience 2007, 145, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.H.; Silva, A.A.; Mota, L.D.; Olivieri, E.R.; Prescinoti, V.C.; Patrão, D.; Camargo, L.P.; Brentani, H.; Carraro, D.M.; Brentani, R.R.; et al. The value of a tumor bank in the development of cancer research in Brazil: 13 Years of experience at the A.C. Camargo Hospital. Biopreserv. Biobank. 2012, 10, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santiago, K.M.; França de Nóbrega, A.; Rocha, R.M.; Rogatto, S.R.; Achatz, M.I. Xeroderma Pigmentosum: Low Prevalence of Germline XPA Mutations in a Brazilian XP Population. Int. J. Mol. Sci. 2015, 16, 8988-8996. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16048988
Santiago KM, França de Nóbrega A, Rocha RM, Rogatto SR, Achatz MI. Xeroderma Pigmentosum: Low Prevalence of Germline XPA Mutations in a Brazilian XP Population. International Journal of Molecular Sciences. 2015; 16(4):8988-8996. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16048988
Chicago/Turabian StyleSantiago, Karina Miranda, Amanda França de Nóbrega, Rafael Malagoli Rocha, Silvia Regina Rogatto, and Maria Isabel Achatz. 2015. "Xeroderma Pigmentosum: Low Prevalence of Germline XPA Mutations in a Brazilian XP Population" International Journal of Molecular Sciences 16, no. 4: 8988-8996. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16048988