
Anti-NR2A/B Antibodies and Other Major Molecular Mechanisms in the Pathogenesis of Cognitive Dysfunction in Systemic Lupus Erythematosus



Abstract
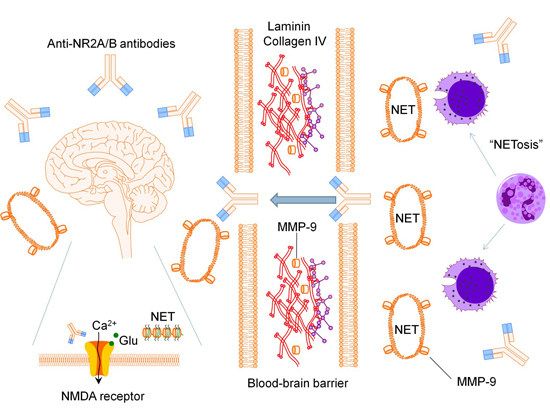
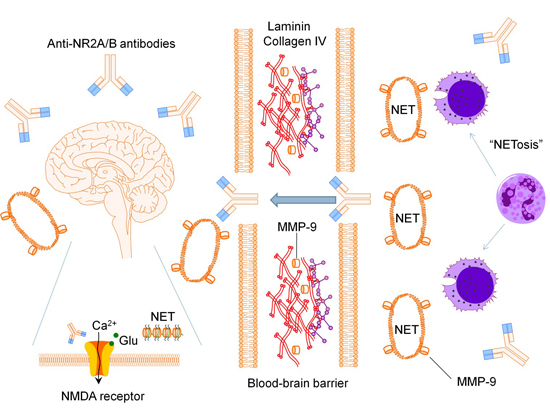
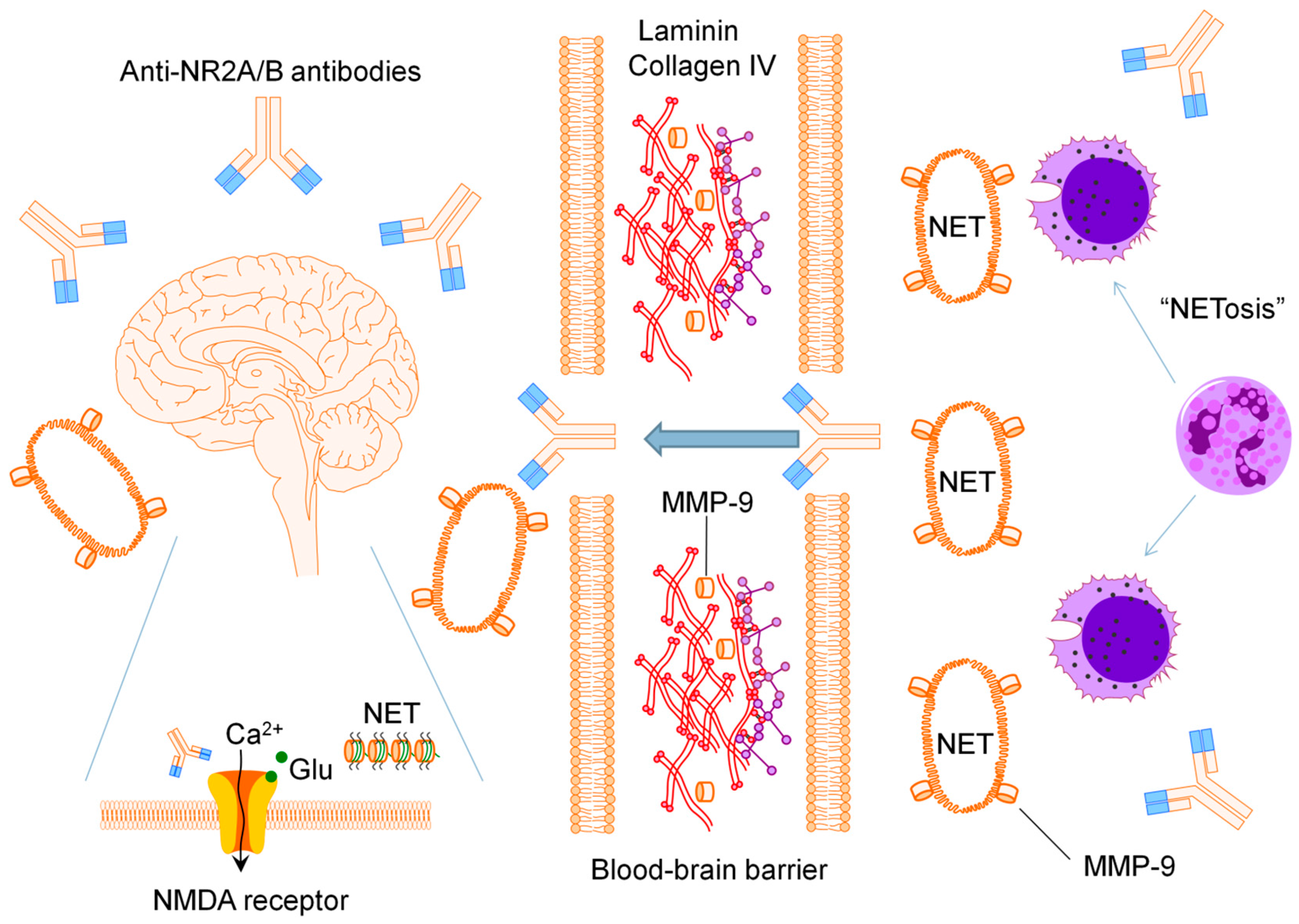
:
1. Introduction
2. Mechanisms of Cognitive Dysfunction
2.1. Anti-N-methyl-d-aspartate Receptor Subunit NR2A/B Antibodies

{kind=link}
{kind=link}
| References | Study Type | Number of Subjects | Amino Acid Sequence Used for Immunoassays | Reported Anti-NR2A/B Values | Essential Findings |
|---|---|---|---|---|---|
| [9] | Cross-sectional | 412 SLE patients | DWEYSVWLSN | Serum optical densities; 19% of SLE patients were anti-NR2/B antibodies positive | No association between anti-NR2A/B antibody status and cognitive impairment |
| [19] | Cross-sectional | 60 SLE patients | DWEYS | Serum optical densities; 33.3% of SLE patients were anti-NR2/B antibodies positive | No association between anti-NR2A/B antibody status and cognitive impairment |
| [31] | Longitudinal (18 months) | 40 pediatric SLE patients | DWEYSVWLSN | Serum concentration (U/mL) | Association between decline in working memory and an increase in anti-NR2A/B antibodies from baseline |
| [43] | Cross-sectional | 57 SLE patients | DWEYSVWLSN | Serum optical densities; 19% of SLE patients were anti-NR2/B antibodies positive | 7 of the 31 neuropsychological tests associated with positive anti-NR2A/B antibodies |
| [44] | Longitudinal (5 years) | 65 women with SLE | DWEYS | Serum optical densities; 35% of SLE patients were anti-NR2/B antibodies positive | No association between anti-NR2A/B antibody status and cognitive impairment; No association between rise in or persistently elevated anti-NR2A/B antibodies and cognitive function over 5 years |
| [45] | Cross-sectional | 93 SLE patients | DWEYSVWLSN | Serum optical densities; 25.8% of SLE patients were anti-NR2/B antibodies positive | No association between anti-NR2A/B antibody status and cognitive impairment |
| [46] | Cross-sectional | 43 SLE patients and 27 healthy controls | DWEYSVWLSN | Serum optical densities; 14% of SLE patients and 7.4% of healthy controls were anti-NR2/B antibodies positive | No association between anti-NR2A/B antibody status and cognitive impairment |
| [47] | Cross-sectional | 133 women with SLE | DWEYSVWLSN | Not available | Association with impaired performance in attention and executive function with positive anti-NR2A/B antibodies |
2.2. Matrix Metalloproteinase-9
2.3. Neutrophil Extracellular Traps
2.4. Pro-Inflammatory Mediators
3. Conclusions

Author Contributions
Conflicts of Interest
References
- Fragoso-Loyo, H.; Richaud-Patin, Y.; Orozco-Narvaez, A.; Davila-Maldonado, L.; Atisha-Fregoso, Y.; Llorente, L.; Sanchez-Guerrero, J. Interleukin-6 and chemokines in the neuropsychiatric manifestations of systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 1242–1450. [Google Scholar] [CrossRef] [PubMed]
- Faust, T.W.; Chang, E.H.; Kowal, C.; Berlin, R.; Gazaryan, I.G.; Bertini, E.; Zhang, J.; Sanchez-Guerrero, J.; Fragoso-Loyo, H.E.; Volpe, B.T.; et al. Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proc. Natl. Acad. Sci. USA 2010, 107, 18569–18574. [Google Scholar] [CrossRef] [PubMed]
- Borchers, A.T.; Naguwa, S.M.; Shoenfeld, Y.; Gershwin, M.E. The geoepidemiology of systemic lupus erythematosus. Autoimmun. Rev. 2010, 9, A277–A287. [Google Scholar] [CrossRef] [PubMed]
- Jakes, R.W.; Bae, S.C.; Louthrenoo, W.; Mok, C.C.; Navarra, S.V.; Kwon, N. Systematic review of the epidemiology of systemic lupus erythematosus in the Asia-Pacific region: Prevalence, incidence, clinical features, and mortality. Arthritis Care Res. 2012, 64, 159–168. [Google Scholar] [CrossRef]
- Hanly, J.G.; Urowitz, M.B.; Sanchez-Guerrero, J.; Bae, S.C.; Gordon, C.; Wallace, D.J.; Isenberg, D.; Alarcon, G.S.; Clarke, A.; Bernatsky, S.; et al. Neuropsychiatric events at the time of diagnosis of systemic lupus erythematosus: An international inception cohort study. Arthritis Rheum. 2007, 56, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Aranow, C.; Diamond, B.; Mackay, M. Glutamate receptor biology and its clinical significance in neuropsychiatric systemic lupus erythematosus. Rheum. Dis. Clin. N. Am. 2010, 36, 187–201. [Google Scholar] [CrossRef]
- Mak, A.; Cheung, M.W.; Chiew, H.J.; Liu, Y.; Ho, R.C. Global trend of survival and damage of systemic lupus erythematosus: Meta-analysis and meta-regression of observational studies from the 1950s to 2000s. Semin. Arthritis Rheum. 2012, 41, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Karassa, F.B.; Afeltra, A.; Ambrozic, A.; Chang, D.M.; de Keyser, F.; Doria, A.; Galeazzi, M.; Hirohata, S.; Hoffman, I.E.; Inanc, M.; et al. Accuracy of anti-ribosomal P protein antibody testing for the diagnosis of neuropsychiatric systemic lupus erythematosus: An international meta-analysis. Arthritis Rheum. 2006, 54, 312–324. [Google Scholar] [CrossRef]
- Hanly, J.G.; Urowitz, M.B.; Siannis, F.; Farewell, V.; Gordon, C.; Bae, S.C.; Isenberg, D.; Dooley, M.A.; Clarke, A.; Bernatsky, S.; et al. Autoantibodies and neuropsychiatric events at the time of systemic lupus erythematosus diagnosis: Results from an international inception cohort study. Arthritis Rheum. 2008, 58, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, T.; Piatti, A.; Luggen, M. Cognitive dysfunction in SLE: Development of a screening tool. Lupus 2011, 20, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Sanna, G.; Bertolaccini, M.L.; Khamashta, M.A. Neuropsychiatric involvement in systemic lupus erythematosus: Current therapeutic approach. Curr. Pharm. Des. 2008, 14, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Bertsias, G.K.; Ioannidis, J.P.; Aringer, M.; Bollen, E.; Bombardieri, S.; Bruce, I.N.; Cervera, R.; Dalakas, M.; Doria, A.; Hanly, J.G.; et al. EULAR recommendations for the management of systemic lupus erythematosus with neuropsychiatric manifestations: Report of a task force of the EULAR standing committee for clinical affairs. Ann. Rheum. Dis. 2010, 69, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Hanly, J.G.; Omisade, A.; Su, L.; Farewell, V.; Fisk, J.D. Assessment of cognitive function in systemic lupus erythematosus, rheumatoid arthritis, and multiple sclerosis by computerized neuropsychological tests. Arthritis Rheum. 2010, 62, 1478–1486. [Google Scholar] [CrossRef] [PubMed]
- Hanly, J.G. Diagnosis and management of neuropsychiatric SLE. Nat. Rev. Rheumatol. 2014, 10, 338–347. [Google Scholar] [CrossRef] [PubMed]
- The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999, 42, 599–608.
- Jeltsch-David, H.; Muller, S. Neuropsychiatric systemic lupus erythematosus: Pathogenesis and biomarkers. Nat. Rev. Neurol. 2014, 10, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Carbotte, R.M.; Denburg, S.D.; Denburg, J.A. Prevalence of cognitive impairment in systemic lupus erythematosus. J. Nerv. Ment. Dis. 1986, 174, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Denburg, S.D.; Carbotte, R.M.; Denburg, J.A. Cognitive impairment in systemic lupus erythematosus: A neuropsychological study of individual and group deficits. J. Clin. Exp. Neuropsychol. 1987, 9, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Lapteva, L.; Nowak, M.; Yarboro, C.H.; Takada, K.; Roebuck-Spencer, T.; Weickert, T.; Bleiberg, J.; Rosenstein, D.; Pao, M.; Patronas, N.; et al. Anti-N-methyl-d-aspartate receptor antibodies, cognitive dysfunction, and depression in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2505–2514. [Google Scholar] [CrossRef] [PubMed]
- Katz, P.; Julian, L.; Tonner, M.C.; Yazdany, J.; Trupin, L.; Yelin, E.; Criswell, L.A. Physical activity, obesity, and cognitive impairment among women with systemic lupus erythematosus. Arthritis Care Res. 2012, 64, 502–510. [Google Scholar] [CrossRef]
- Holliday, S.L.; Navarrete, M.G.; Hermosillo-Romo, D.; Valdez, C.R.; Saklad, A.R.; Escalante, A.; Brey, R.L. Validating a computerized neuropsychological test battery for mixed ethnic lupus patients. Lupus 2003, 12, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Roebuck-Spencer, T.M.; Yarboro, C.; Nowak, M.; Takada, K.; Jacobs, G.; Lapteva, L.; Weickert, T.; Volpe, B.; Diamond, B.; Illei, G.; et al. Use of computerized assessment to predict neuropsychological functioning and emotional distress in patients with systemic lupus erythematosus. Arthritis Rheum. 2006, 55, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Naqibuddin, M.; Carson, K.A.; Sampedro, M.; Wallace, D.J.; Weisman, M.H.; Holliday, S.L.; Padilla, P.A.; Brey, R.L. Cognitive function in a systemic lupus erythematosus inception cohort. J. Rheumatol. 2008, 35, 1776–1781. [Google Scholar] [CrossRef] [PubMed]
- Kozora, E.; Hanly, J.G.; Lapteva, L.; Filley, C.M. Cognitive dysfunction in systemic lupus erythematosus: Past, present, and future. Arthritis Rheum. 2008, 58, 3286–3298. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.I.; Ruth, N.M.; German, A.; Nelson, S.; Passo, M.H.; Roebuck-Spencer, T.; Ying, J.; Ris, D. Initial validation of the Pediatric Automated Neuropsychological Assessment Metrics for childhood-onset systemic lupus erythematosus. Arthritis Rheum. 2007, 57, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Guillen-Del Castillo, A.; Alonso, J.; Martinez-Valle, F.; Alonso-Vila, S.; Garrido-Castro, A.C.; Vilardell-Tarres, M.; Rovira, A.; Ordi-Ros, J. Increased myo-inositol in parietal white and gray matter as a biomarker of poor prognosis in neuropsychiatric lupus: A case report. Lupus 2014, 23, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Ainiala, H.; Dastidar, P.; Loukkola, J.; Lehtimaki, T.; Korpela, M.; Peltola, J.; Hietaharju, A. Cerebral MRI abnormalities and their association with neuropsychiatric manifestations in SLE: A population-based study. Scand. J. Rheumatol. 2005, 34, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller, S.; Bonilha, L.; Rio, P.A.; Min Li, L.; Costallat, L.T.; Cendes, F. Longitudinal analysis of gray and white matter loss in patients with systemic lupus erythematosus. Neuroimage 2007, 34, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Sarbu, N.; Alobeidi, F.; Toledano, P.; Espinosa, G.; Giles, I.; Rahman, A.; Yousry, T.; Capurro, S.; Jager, R.; Cervera, R.; et al. Brain abnormalities in newly diagnosed neuropsychiatric lupus: Systematic MRI approach and correlation with clinical and laboratory data in a large multicenter cohort. Autoimmun. Rev. 2015, 14, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Ramage, A.E.; Fox, P.T.; Brey, R.L.; Narayana, S.; Cykowski, M.D.; Naqibuddin, M.; Sampedro, M.; Holliday, S.L.; Franklin, C.; Wallace, D.J.; et al. Neuroimaging evidence of white matter inflammation in newly diagnosed systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3048–3057. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.I.; Klein-Gitelman, M.S.; Zelko, F.; Beebe, D.W.; Foell, D.; Lee, J.; Zaal, A.; Jones, J.; Roebuck-Spencer, T.; Ying, J. Blood-based candidate biomarkers of the presence of neuropsychiatric systemic lupus erythematosus in children. Lupus Sci. Med. 2014, 1. [Google Scholar] [CrossRef]
- Gono, T.; Takarada, T.; Fukumori, R.; Kawaguchi, Y.; Kaneko, H.; Hanaoka, M.; Katsumata, Y.; Yoneda, Y.; Yamanaka, H. NR2-reactive antibody decreases cell viability through augmentation of Ca2+ influx in systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3952–3959. [Google Scholar] [CrossRef] [PubMed]
- Senecal, J.L.; Raymond, Y. The pathogenesis of neuropsychiatric manifestations in systemic lupus erythematosus: A disease in search of autoantibodies, or autoantibodies in search of a disease? J. Rheumatol. 2004, 31, 2093–2098. [Google Scholar] [PubMed]
- Gaynor, B.; Putterman, C.; Valadon, P.; Spatz, L.; Scharff, M.D.; Diamond, B. Peptide inhibition of glomerular deposition of an anti-DNA antibody. Proc. Natl. Acad. Sci. USA 1997, 94, 1955–1960. [Google Scholar] [CrossRef] [PubMed]
- Diamond, B.; Bloom, O.; Al Abed, Y.; Kowal, C.; Huerta, P.T.; Volpe, B.T. Moving towards a cure: Blocking pathogenic antibodies in systemic lupus erythematosus. J. Intern. Med. 2011, 269, 36–44. [Google Scholar] [CrossRef] [PubMed]
- DeGiorgio, L.A.; Konstantinov, K.N.; Lee, S.C.; Hardin, J.A.; Volpe, B.T.; Diamond, B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat. Med. 2001, 7, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Diamond, B.; Volpe, B.T. A model for lupus brain disease. Immunol. Rev. 2012, 248, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Kowal, C.; Degiorgio, L.A.; Lee, J.Y.; Edgar, M.A.; Huerta, P.T.; Volpe, B.T.; Diamond, B. Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc. Natl. Acad. Sci. USA 2006, 103, 19854–19859. [Google Scholar] [CrossRef] [PubMed]
- Gono, T.; Kawaguchi, Y.; Kaneko, H.; Nishimura, K.; Hanaoka, M.; Kataoka, S.; Okamoto, Y.; Katsumata, Y.; Yamanaka, H. Anti-NR2A antibody as a predictor for neuropsychiatric systemic lupus erythematosus. Rheumatology (Oxford, England) 2011, 50, 1578–1585. [Google Scholar] [CrossRef]
- Yoshio, T.; Okamoto, H.; Hirohata, S.; Minota, S. IgG anti-NR2 glutamate receptor autoantibodies from patients with systemic lupus erythematosus activate endothelial cells. Arthritis Rheum. 2013, 65, 457–463. [Google Scholar] [CrossRef]
- Sharma, A.; Isenberg, D.; Diamond, B. Studies of human polyclonal and monoclonal antibodies binding to lupus autoantigens and cross-reactive antigens. Rheumatology (Oxford, England) 2003, 42, 453–463. [Google Scholar] [CrossRef]
- Neuhaus, W.; Freidl, M.; Szkokan, P.; Berger, M.; Wirth, M.; Winkler, J.; Gabor, F.; Pifl, C.; Noe, C.R. Effects of NMDA receptor modulators on a blood-brain barrier in vitro model. Brain Res. 2011, 1394, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Omdal, R.; Brokstad, K.; Waterloo, K.; Koldingsnes, W.; Jonsson, R.; Mellgren, S.I. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur. J. Neurol.: Off. J. Eur. Fed. Neurol. Soc. 2005, 12, 392–398. [Google Scholar]
- Hanly, J.G.; Robichaud, J.; Fisk, J.D. Anti-NR2 glutamate receptor antibodies and cognitive function in systemic lupus erythematosus. J. Rheumatol. 2006, 33, 1553–1558. [Google Scholar] [PubMed]
- Harrison, M.J.; Ravdin, L.D.; Lockshin, M.D. Relationship between serum NR2a antibodies and cognitive dysfunction in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2515–2522. [Google Scholar] [CrossRef] [PubMed]
- Kozora, E.; West, S.G.; Maier, S.F.; Filley, C.M.; Arciniegas, D.B.; Brown, M.; Miller, D.; Grimm, A.; Zhang, L. Antibodies against N-methyl-d-aspartate receptors in patients with systemic lupus erythematosus without major neuropsychiatric syndromes. J. Neurol. Sci. 2010, 295, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Massardo, L.; Bravo-Zehnder, M.; Calderon, J.; Flores, P.; Padilla, O.; Aguirre, J.M.; Scoriels, L.; Gonzalez, A. Anti-N-methyl-d-aspartate receptor and anti-ribosomal-P autoantibodies contribute to cognitive dysfunction in systemic lupus erythematosus. Lupus 2015, 24, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Naqibuddin, M.; Carson, K.A.; Wallace, D.J.; Weisman, M.H.; Holliday, S.L.; Sampedro, M.; Padilla, P.A.; Brey, R.L. Depression and cognitive impairment in newly diagnosed systemic lupus erythematosus. J. Rheumatol. 2010, 37, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Yoshio, T.; Onda, K.; Nara, H.; Minota, S. Association of IgG anti-NR2 glutamate receptor antibodies in cerebrospinal fluid with neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Fragoso-Loyo, H.; Cabiedes, J.; Orozco-Narvaez, A.; Davila-Maldonado, L.; Atisha-Fregoso, Y.; Diamond, B.; Llorente, L.; Sanchez-Guerrero, J. Serum and cerebrospinal fluid autoantibodies in patients with neuropsychiatric lupus erythematosus. Implications for diagnosis and pathogenesis. PLoS ONE 2008, 3, e3347. [Google Scholar] [CrossRef] [PubMed]
- Arinuma, Y.; Yanagida, T.; Hirohata, S. Association of cerebrospinal fluid anti-NR2 glutamate receptor antibodies with diffuse neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2008, 58, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Hirohata, S.; Arinuma, Y.; Yanagida, T.; Yoshio, T. Blood-brain barrier damages and intrathecal synthesis of anti-N-methyl-d-aspartate receptor NR2 antibodies in diffuse psychiatric/neuropsychological syndromes in systemic lupus erythematosus. Arthritis Res. Ther. 2014, 16. [Google Scholar] [CrossRef]
- Steup-Beekman, G.; Steens, S.; van Buchem, M.; Huizinga, T. Anti-NMDA receptor autoantibodies in patients with systemic lupus erythematosus and their first-degree relatives. Lupus 2007, 16, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Dambinova, S.A.; Khounteev, G.A.; Izykenova, G.A.; Zavolokov, I.G.; Ilyukhina, A.Y.; Skoromets, A.A. Blood test detecting autoantibodies to N-methyl-d-aspartate neuroreceptors for evaluation of patients with transient ischemic attack and stroke. Clin. Chem. 2003, 49, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Weissman, J.D.; Khunteev, G.A.; Heath, R.; Dambinova, S.A. NR2 antibodies: Risk assessment of transient ischemic attack (TIA)/stroke in patients with history of isolated and multiple cerebrovascular events. J. Neurol. Sci. 2011, 300, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; Yang, Y.; Rosenberg, G.A. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 2009, 158, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, T.; Nakajima, H.; Doi, Y.; Sugino, M.; Kimura, F.; Hanafusa, T.; Takahashi, T. Increased serum matrix metalloproteinase-9 in neuromyelitis optica: Implication of disruption of blood-brain barrier. J. Neuroimmunol. 2011, 236, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Yushchenko, M.; Weber, F.; Mader, M.; Scholl, U.; Maliszewska, M.; Tumani, H.; Felgenhauer, K.; Beuche, W. Matrix metalloproteinase-9 (MMP-9) in human cerebrospinal fluid (CSF): Elevated levels are primarily related to CSF cell count. J. Neuroimmunol. 2000, 110, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Moxon-Emre, I.; Schlichter, L.C. Neutrophil depletion reduces blood-brain barrier breakdown, axon injury, and inflammation after intracerebral hemorrhage. J. Neuropathol. Exp. Neurol. 2011, 70, 218–235. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.L.; Latour, L.L.; Lee, K.Y.; Schaewe, T.J.; Luby, M.; Chang, G.S.; El-Zammar, Z.; Alam, S.; Hallenbeck, J.M.; Kidwell, C.S.; et al. Blood-brain barrier disruption in humans is independently associated with increased matrix metalloproteinase-9. Stroke J. Cereb. Circ. 2010, 41, e123–e128. [Google Scholar] [CrossRef]
- Rosell, A.; Cuadrado, E.; Ortega-Aznar, A.; Hernandez-Guillamon, M.; Lo, E.H.; Montaner, J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke J. Cereb. Circ. 2008, 39, 1121–1126. [Google Scholar] [CrossRef]
- Robak, E.; Wierzbowska, A.; Chmiela, M.; Kulczycka, L.; Sysa-Jedrejowska, A.; Robak, T. Circulating total and active metalloproteinase-9 and tissue inhibitor of metalloproteinases-1 in patients with systemic lupus erythomatosus. Mediat. Inflam. 2006, 2006, 17898. [Google Scholar] [CrossRef]
- Ainiala, H.; Hietaharju, A.; Dastidar, P.; Loukkola, J.; Lehtimaki, T.; Peltola, J.; Korpela, M.; Heinonen, T.; Nikkari, S.T. Increased serum matrix metalloproteinase 9 levels in systemic lupus erythematosus patients with neuropsychiatric manifestations and brain magnetic resonance imaging abnormalities. Arthritis Rheum. 2004, 50, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Matache, C.; Stefanescu, M.; Dragomir, C.; Tanaseanu, S.; Onu, A.; Ofiteru, A.; Szegli, G. Matrix metalloproteinase-9 and its natural inhibitor TIMP-1 expressed or secreted by peripheral blood mononuclear cells from patients with systemic lupus erythematosus. J. Autoimmun. 2003, 20, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Lesiak, A.; Narbutt, J.; Sysa-Jedrzejowska, A.; Lukamowicz, J.; McCauliffe, D.P.; Wozniacka, A. Effect of chloroquine phosphate treatment on serum MMP-9 and TIMP-1 levels in patients with systemic lupus erythematosus. Lupus 2010, 19, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Bahrehmand, F.; Vaisi-Raygani, A.; Kiani, A.; Rahimi, Z.; Tavilani, H.; Ardalan, M.; Vaisi-Raygani, H.; Shakiba, E.; Pourmotabbed, T. Matrix metalloproteinase 9 polymorphisms and systemic lupus erythematosus: Correlation with systemic inflammatory markers and oxidative stress. Lupus 2014. [Google Scholar] [CrossRef]
- Faber-Elmann, A.; Sthoeger, Z.; Tcherniack, A.; Dayan, M.; Mozes, E. Activity of matrix metalloproteinase-9 is elevated in sera of patients with systemic lupus erythematosus. Clin. Exp. Immunol. 2002, 127, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Trysberg, E.; Blennow, K.; Zachrisson, O.; Tarkowski, A. Intrathecal levels of matrix metalloproteinases in systemic lupus erythematosus with central nervous system engagement. Arthritis Res. Ther. 2004, 6, R551–R556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrera, C.; Fadeel, B. Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, B.E.; Grinstein, S. Unconventional roles of the NADPH oxidase: Signaling, ion homeostasis, and cell death. Sci. STKE 2007, 2007, pe11. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Goosmann, C.; Kuhn, L.I.; Zychlinsky, A. Automatic quantification of in vitro NET formation. Front. Immunol. 2012, 3, 413. [Google Scholar] [PubMed]
- Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps: Is immunity the second function of chromatin? J. Cell Biol. 2012, 198, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Ruhnau, J.; Schulze, K.; Gaida, B.; Langner, S.; Kessler, C.; Broker, B.; Dressel, A.; Vogelgesang, A. Stroke alters respiratory burst in neutrophils and monocytes. Stroke J. Cereb. Circ. 2014, 45, 794–800. [Google Scholar] [CrossRef]
- Allen, C.; Thornton, P.; Denes, A.; McColl, B.W.; Pierozynski, A.; Monestier, M.; Pinteaux, E.; Rothwell, N.J.; Allan, S.M. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J. Immunol. 2012, 189, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Kaplan, M.J. Neutrophils in the pathogenesis and manifestations of SLE. Nat. Rev. Rheumatol. 2011, 7, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Kaplan, M.J. Lupus neutrophils: “NET” gain in understanding lupus pathogenesis. Curr. Opin. Rheumatol. 2012, 24, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rivera, C.; Kaplan, M.J. Low-density granulocytes: A distinct class of neutrophils in systemic autoimmunity. Semin. Immunopathol. 2013, 35, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Hasler, P.; Holzgreve, W.; Gebhardt, S.; Hahn, S. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum. Immunol. 2005, 66, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Onuora, S. Connective tissue diseases: A NET of peril for endothelial cells in SLE? Nat. Rev. Rheumatol. 2014, 10. [Google Scholar] [CrossRef]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tyden, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Zychlinsky, A. Beneficial suicide: Why neutrophils die to make NETs. Nat. Rev. Microbiol. 2007, 5, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2014. [Google Scholar] [CrossRef]
- James, W.G.; Bullard, D.C.; Hickey, M.J. Critical role of the α 4 integrin/VCAM-1 pathway in cerebral leukocyte trafficking in lupus-prone MRL/fas(lpr) mice. J. Immunol. 2003, 170, 520–527. [Google Scholar] [CrossRef] [PubMed]
- James, W.G.; Hutchinson, P.; Bullard, D.C.; Hickey, M.J. Cerebral leucocyte infiltration in lupus-prone MRL/MpJ-fas lpr mice—Roles of intercellular adhesion molecule-1 and P-selectin. Clin. Exp. Immunol. 2006, 144, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Lapter, S.; Marom, A.; Meshorer, A.; Elmann, A.; Sharabi, A.; Vadai, E.; Neufeld, A.; Sztainberg, Y.; Gil, S.; Getselter, D.; et al. Amelioration of brain pathology and behavioral dysfunction in mice with lupus following treatment with a tolerogenic peptide. Arthritis Rheum. 2009, 60, 3744–3754. [Google Scholar] [CrossRef] [PubMed]
- Hopia, L.; Thangarajh, M.; Khademi, M.; Laveskog, A.; Wallstrom, E.; Svenungsson, E.; Andersson, M. Cerebrospinal fluid levels of a proliferation-inducing ligand (APRIL) are increased in patients with neuropsychiatric systemic lupus erythematosus. Scand. J. Rheumatol. 2011, 40, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch-David, H.; Muller, S. Neuropsychiatric systemic lupus erythematosus and cognitive dysfunction: The MRL-lpr mouse strain as a model. Autoimmun. Rev. 2014, 13, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Vo, A.; Volpe, B.T.; Tang, C.C.; Schiffer, W.K.; Kowal, C.; Huerta, P.T.; Ulug, A.M.; Dewey, S.L.; Eidelberg, D.; Diamond, B. Regional brain metabolism in a murine systemic lupus erythematosus model. J. Cereb. Blood Flow Metab. 2014, 34, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Arinuma, Y.; Kikuchi, H.; Wada, T.; Nagai, T.; Tanaka, S.; Oba, H.; Hirohata, S. Brain MRI in patients with diffuse psychiatric/neuropsychological syndromes in systemic lupus erythematosus. Lupus Sci. Med. 2014, 1, e000050. [Google Scholar] [CrossRef] [PubMed]
- Postal, M.; Pelicari, K.O.; Sinicato, N.A.; Marini, R.; Costallat, L.T.; Appenzeller, S. Th1/Th2 cytokine profile in childhood-onset systemic lupus erythematosus. Cytokine 2013, 61, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.; Tang, C.S.; Ho, R.C. Serum tumour necrosis factor-alpha is associated with poor health-related quality of life and depressive symptoms in patients with systemic lupus erythematosus. Lupus 2013, 22, 254–261. [Google Scholar] [CrossRef]
- Pisetsky, D.S. Fulfilling Kochʼs postulates of autoimmunity: Anti-NR2 antibodies in mice and men. Arthritis Rheum. 2006, 54, 2349–2352. [Google Scholar] [CrossRef] [PubMed]
- Kowal, C.; DeGiorgio, L.A.; Nakaoka, T.; Hetherington, H.; Huerta, P.T.; Diamond, B.; Volpe, B.T. Cognition and immunity; antibody impairs memory. Immunity 2004, 21, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Huerta, P.T.; Kowal, C.; DeGiorgio, L.A.; Volpe, B.T.; Diamond, B. Immunity and behavior: Antibodies alter emotion. Proc. Natl. Acad. Sci. USA 2006, 103, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.X.; Sanders, E.; Tieng, A.T.; Putterman, C. Sex and autoantibody titers determine the development of neuropsychiatric manifestations in lupus-prone mice. J. Neuroimmunol. 2010, 229, 112–122. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tay, S.H.; Mak, A. Anti-NR2A/B Antibodies and Other Major Molecular Mechanisms in the Pathogenesis of Cognitive Dysfunction in Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2015, 16, 10281-10300. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160510281
Tay SH, Mak A. Anti-NR2A/B Antibodies and Other Major Molecular Mechanisms in the Pathogenesis of Cognitive Dysfunction in Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2015; 16(5):10281-10300. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160510281
Chicago/Turabian StyleTay, Sen Hee, and Anselm Mak. 2015. "Anti-NR2A/B Antibodies and Other Major Molecular Mechanisms in the Pathogenesis of Cognitive Dysfunction in Systemic Lupus Erythematosus" International Journal of Molecular Sciences 16, no. 5: 10281-10300. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160510281