
A Comprehensive Analysis of Codon Usage Patterns in Blunt Snout Bream (Megalobrama amblycephala) Based on RNA-Seq Data


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
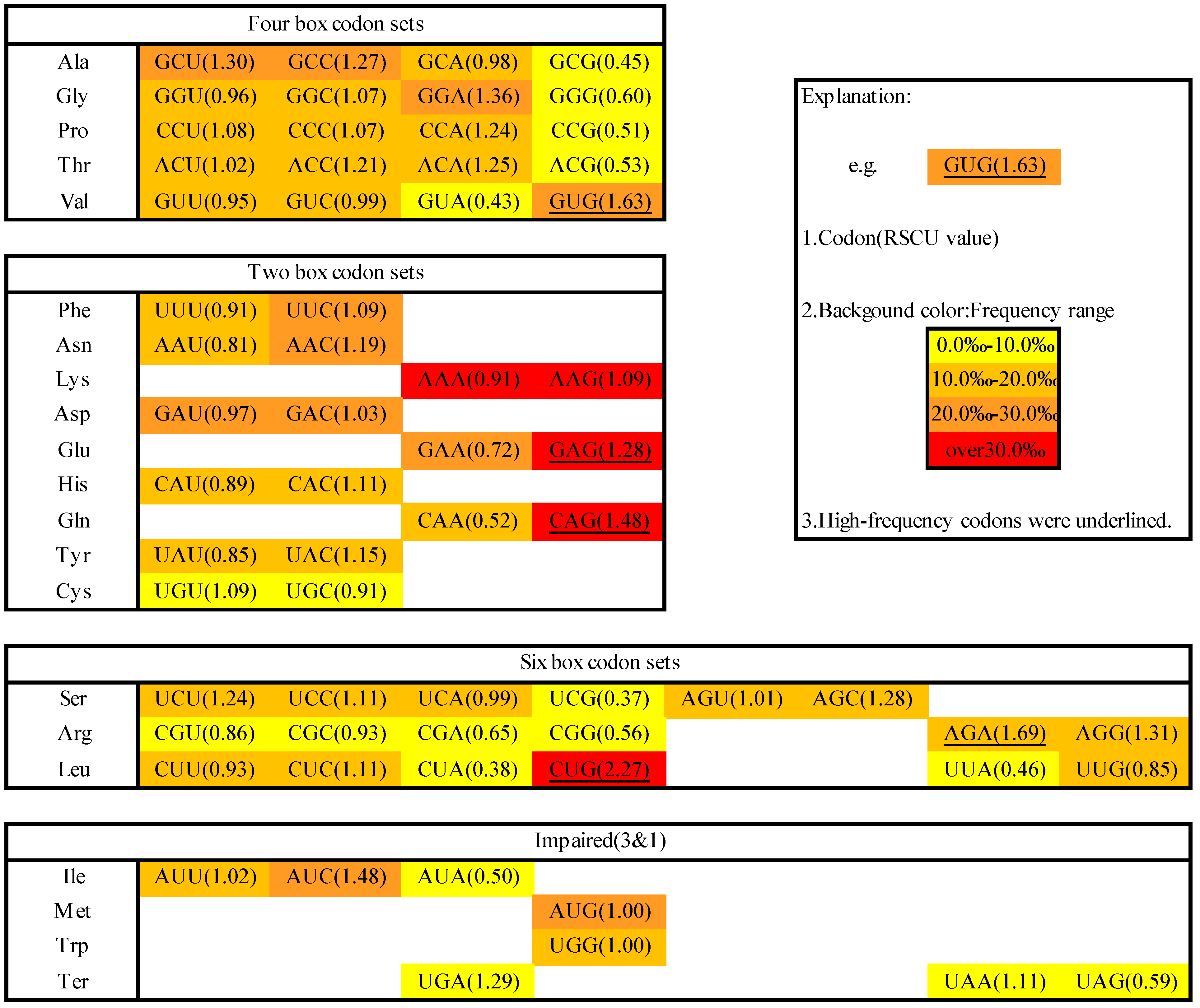
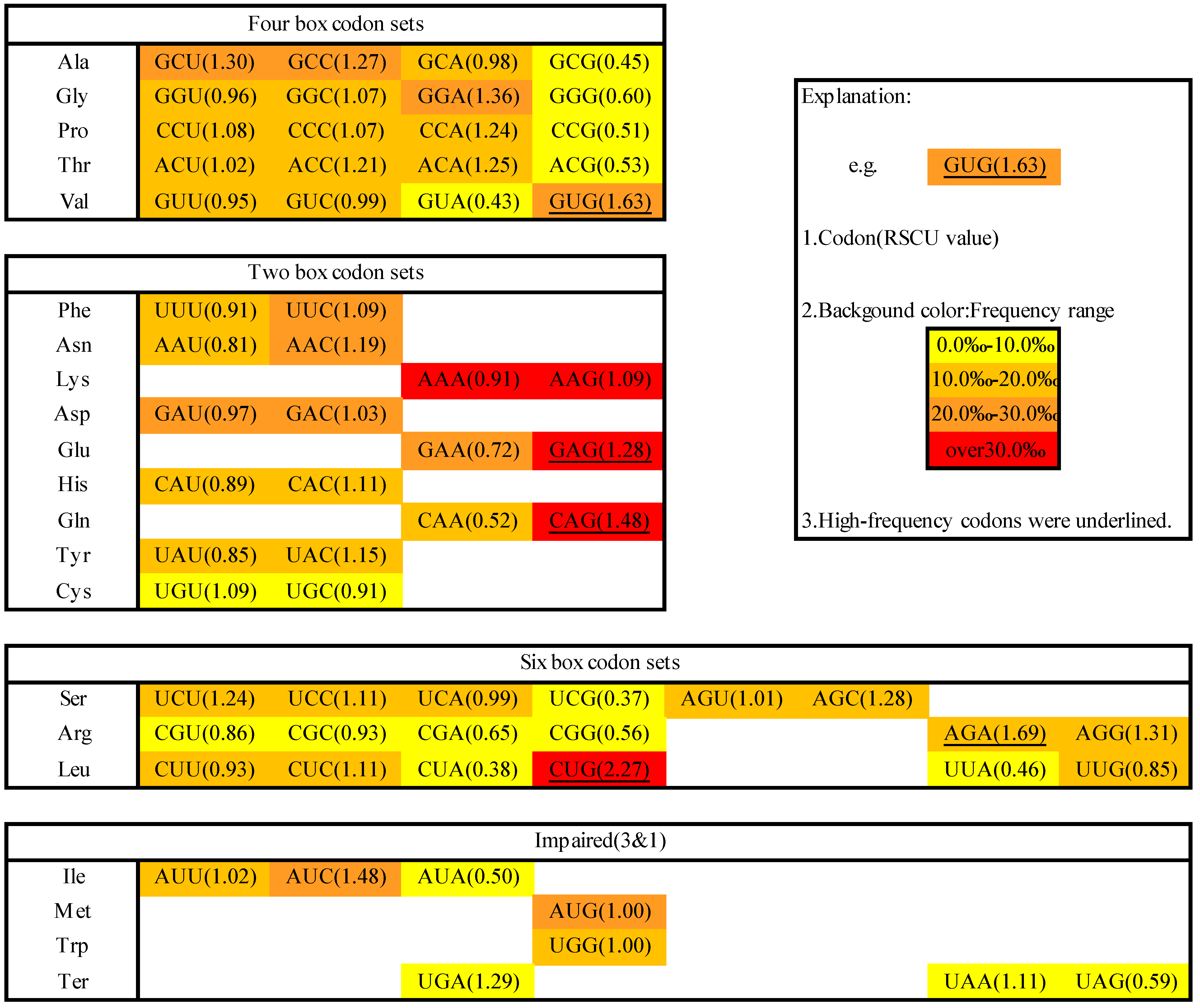
2.1. Codon Usage in M. amblycephala
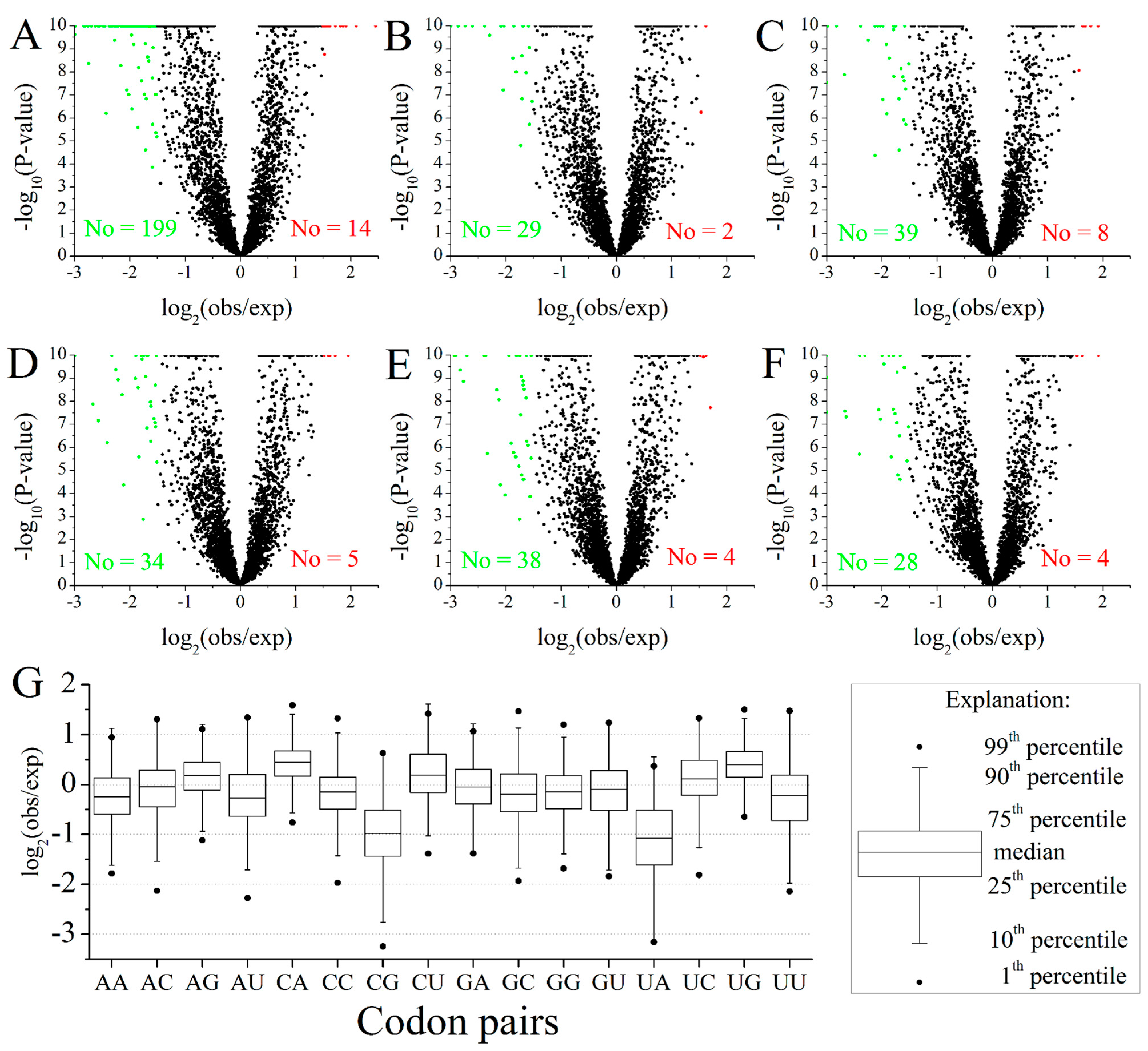
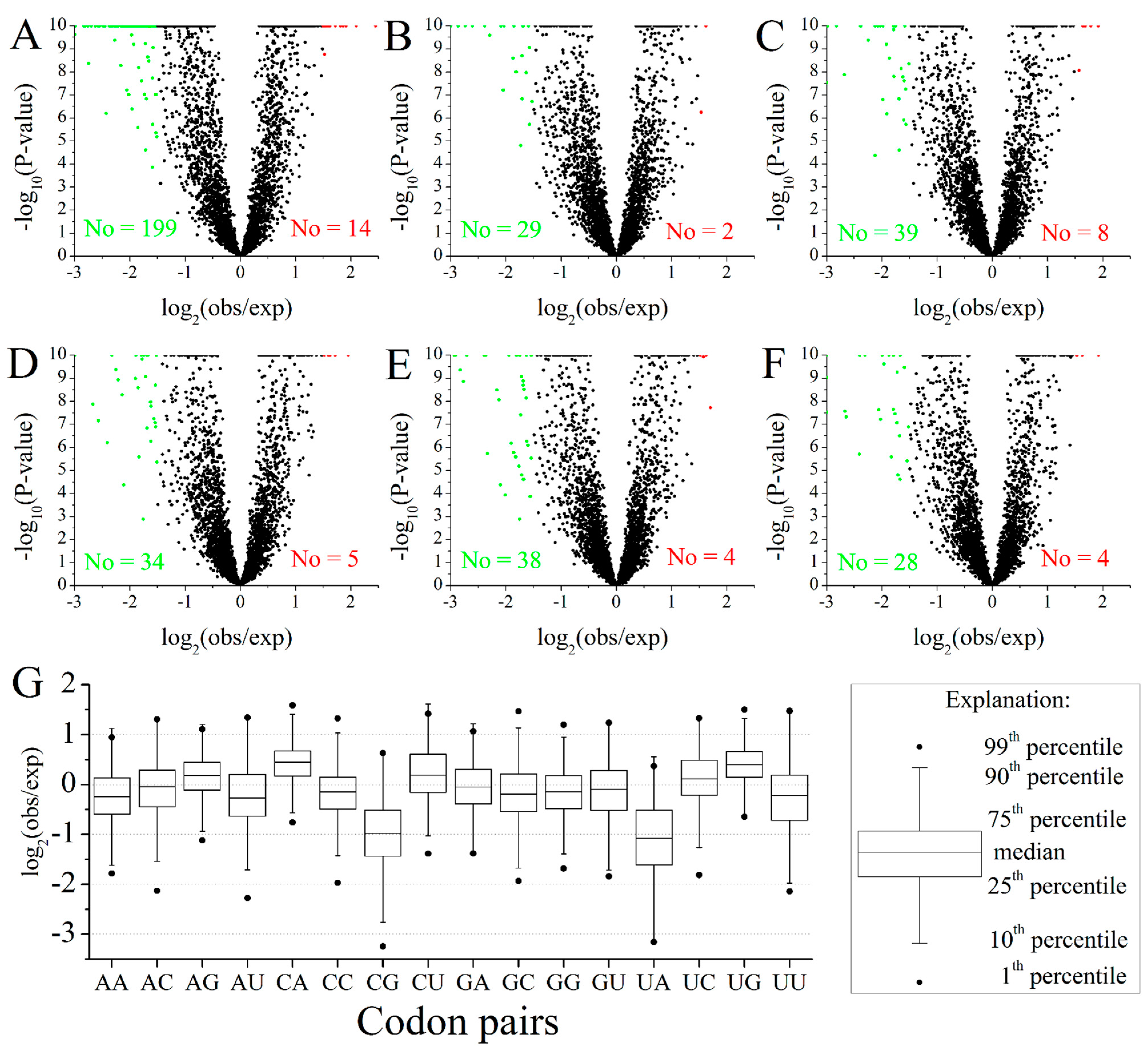
2.2. Codon Pairs in M. amblycephala
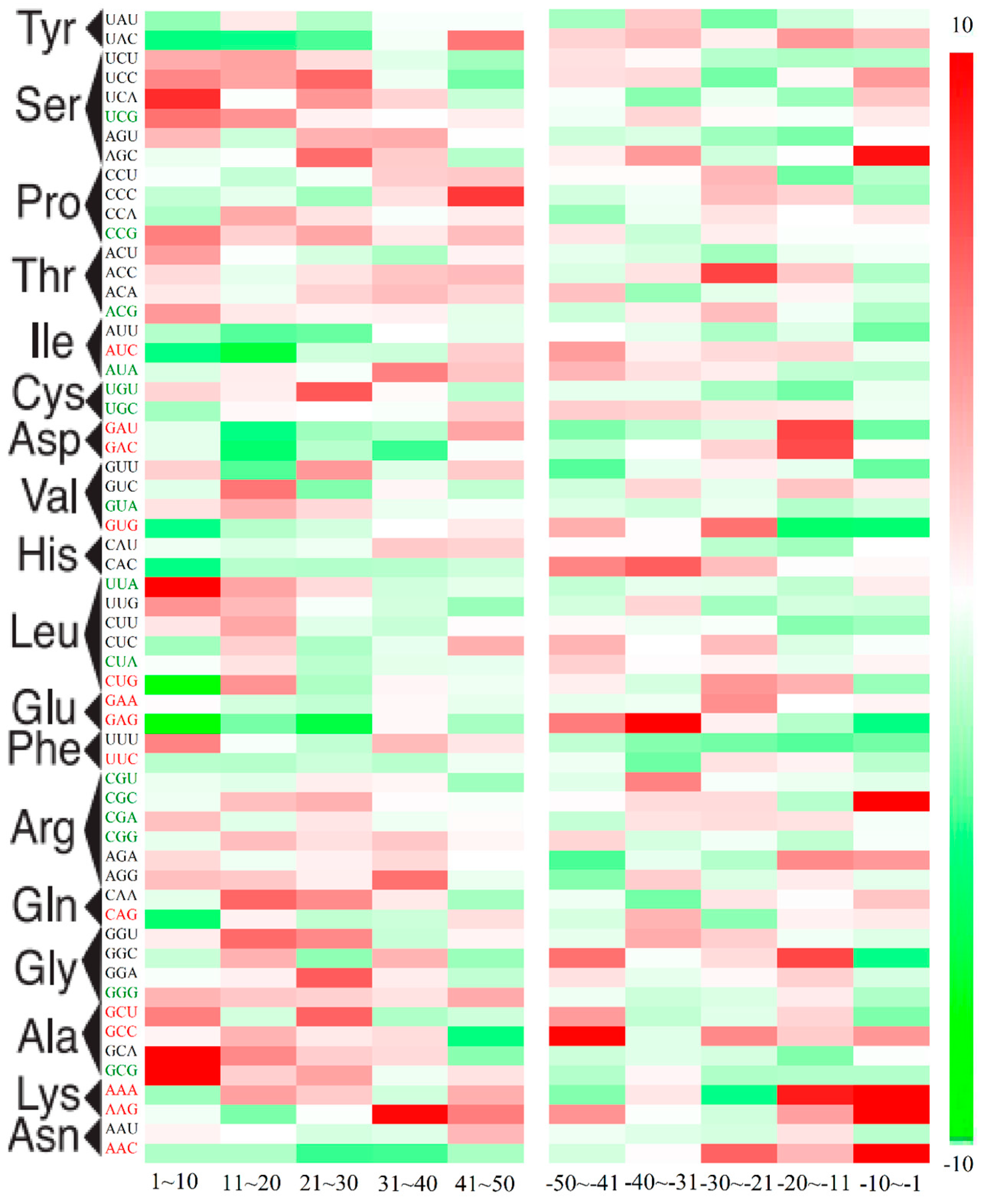
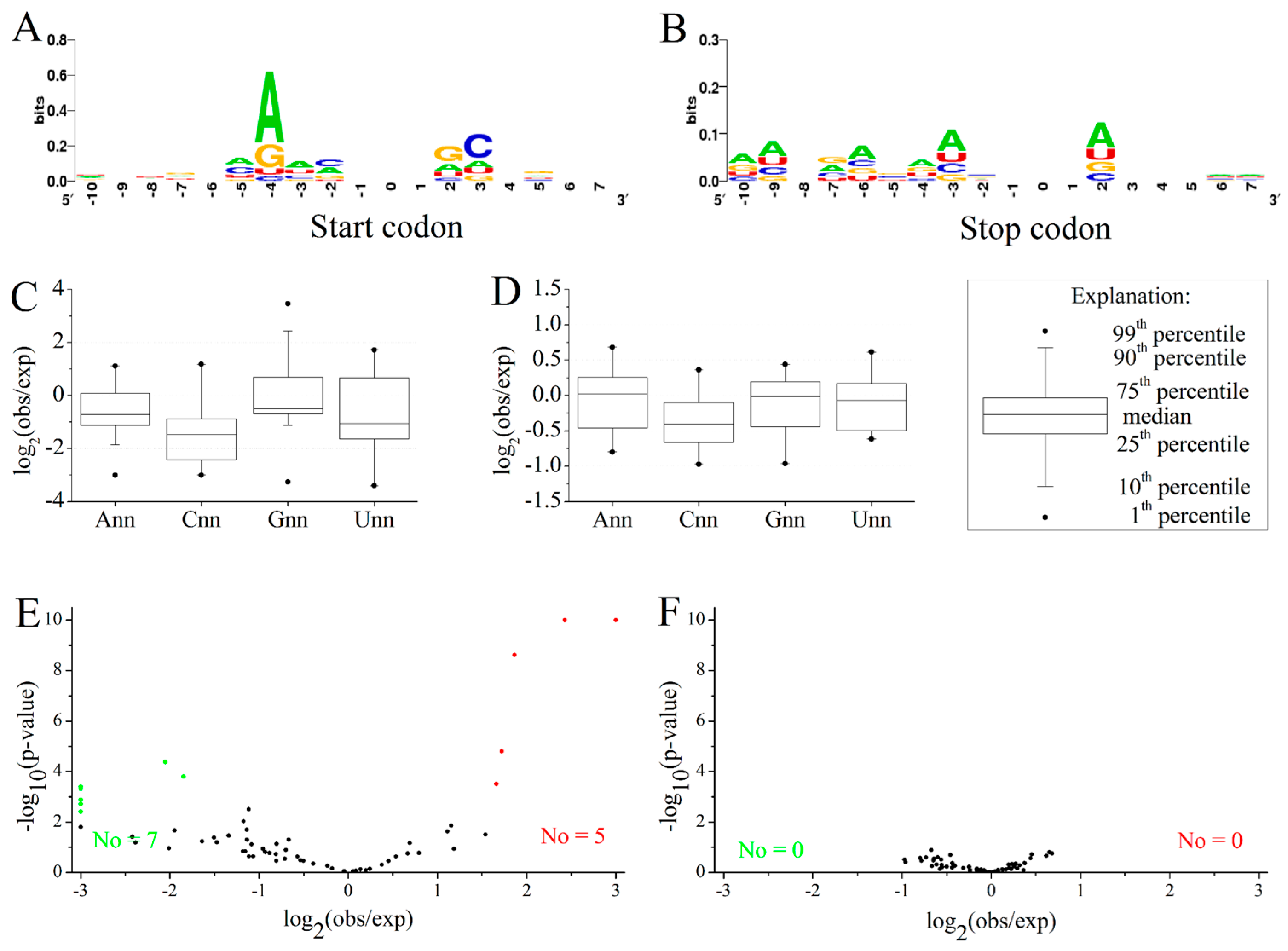
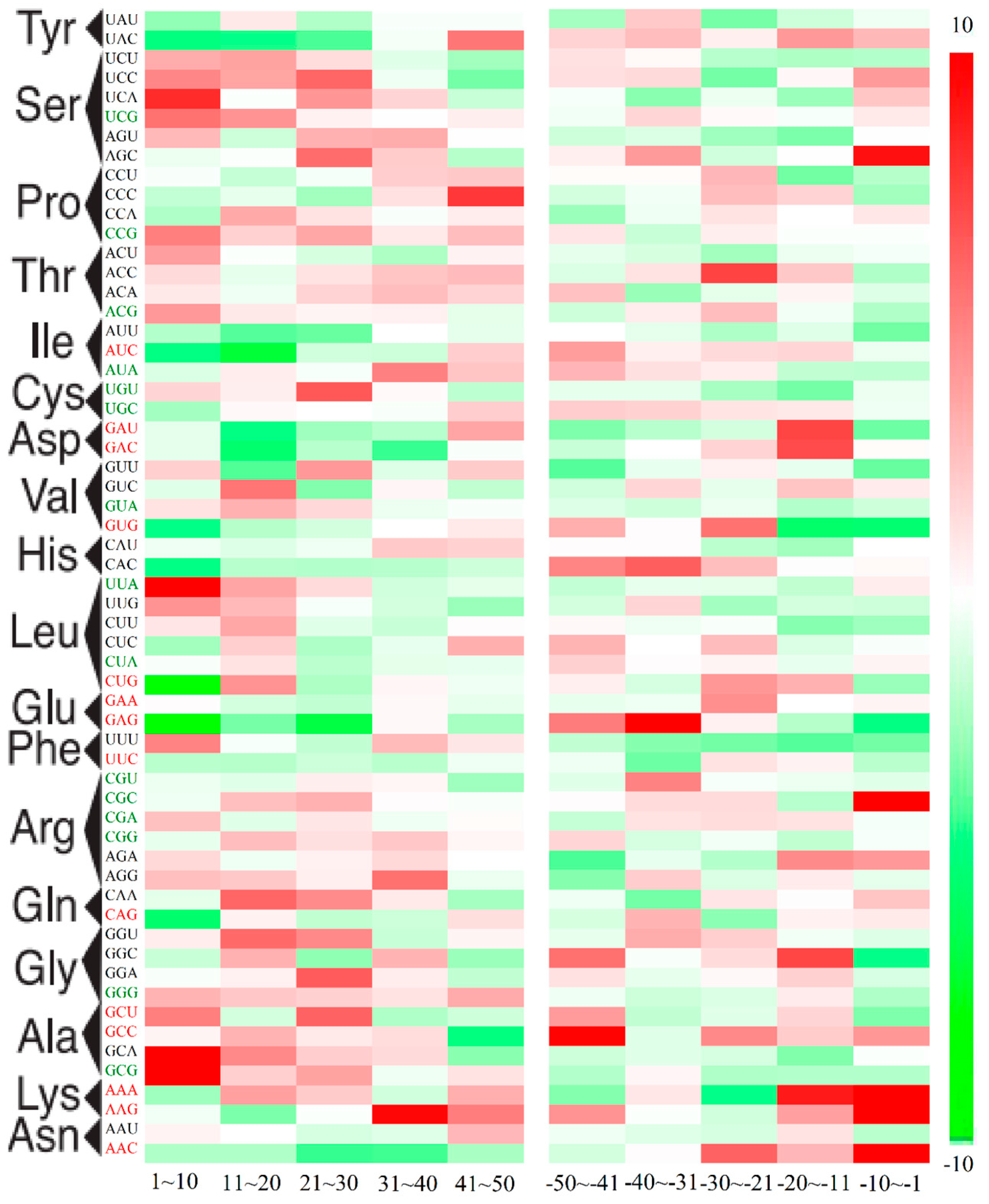
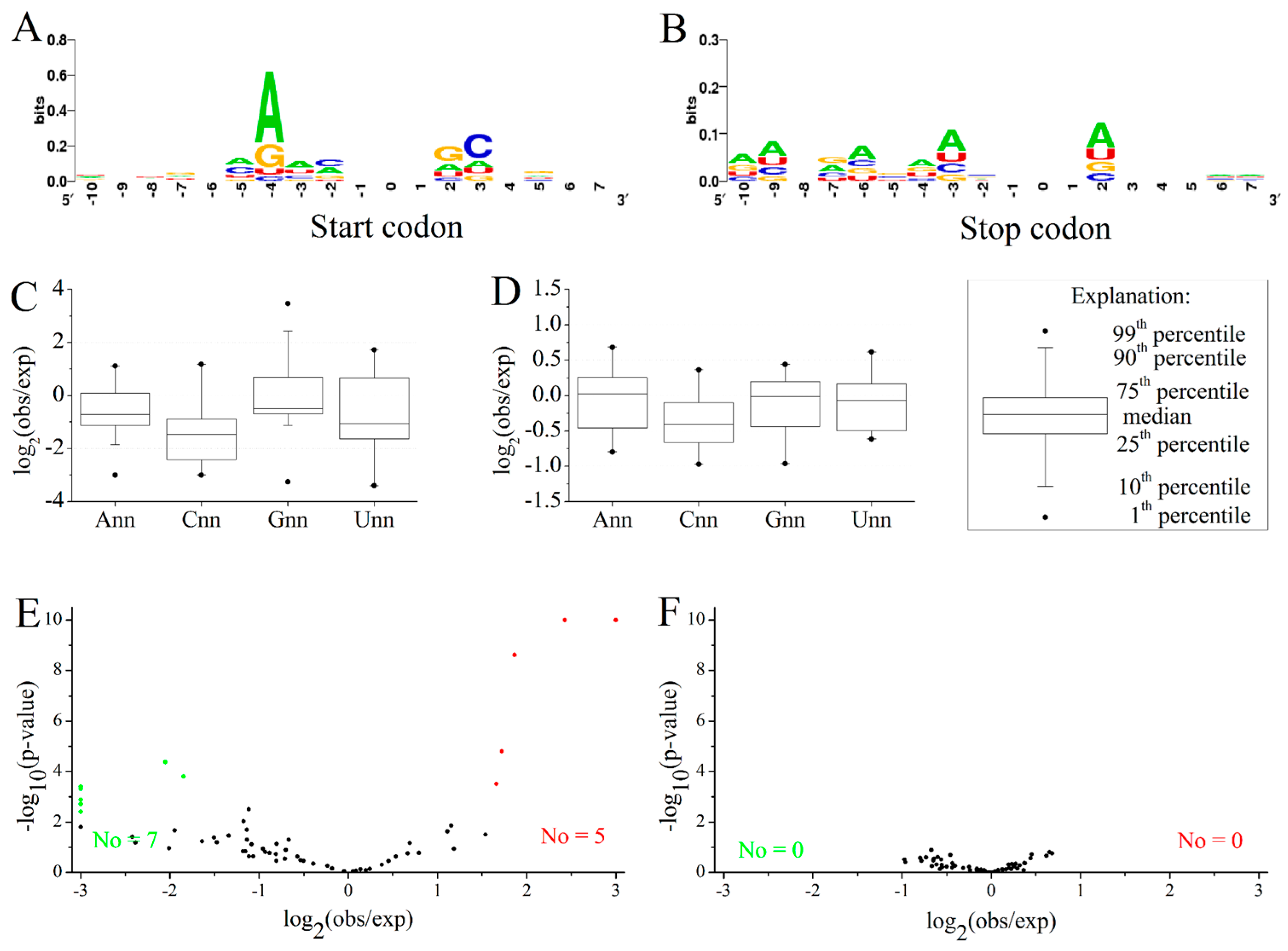
2.3. Position-Dependent Codon Usage Bias in M. amblycephala
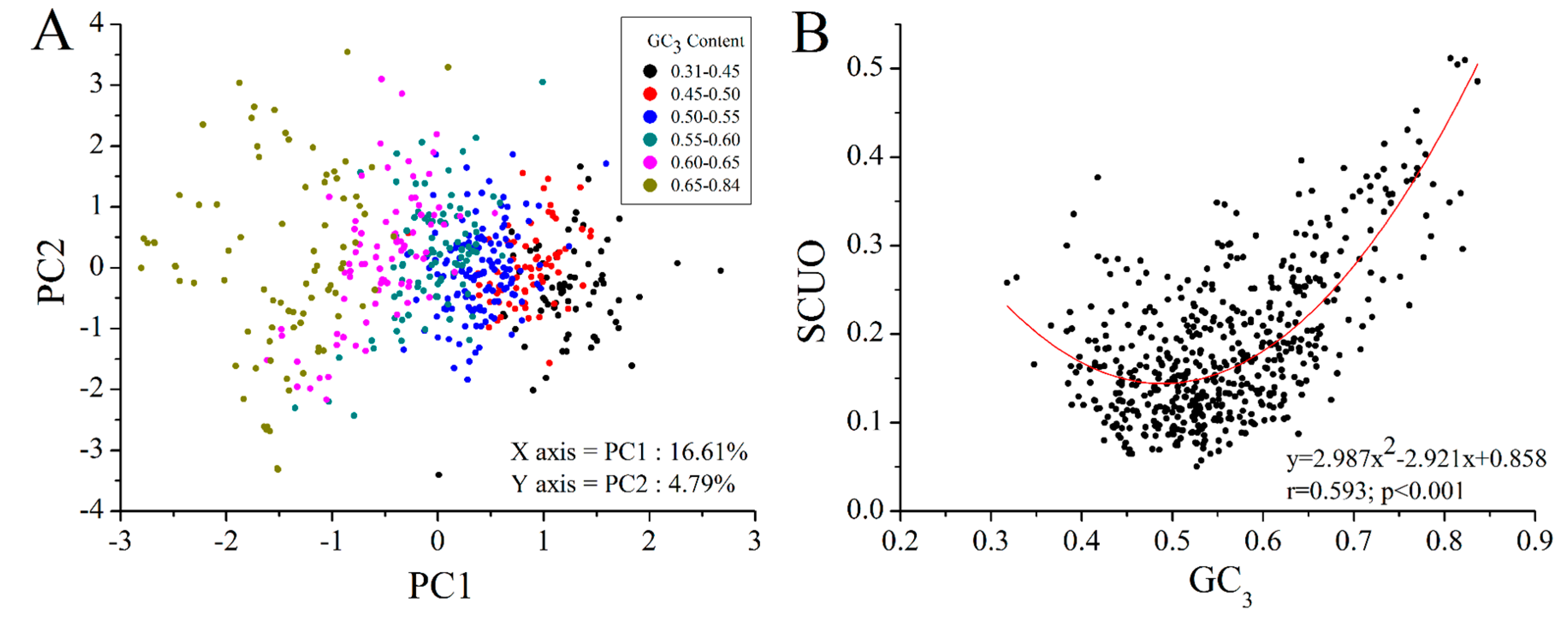
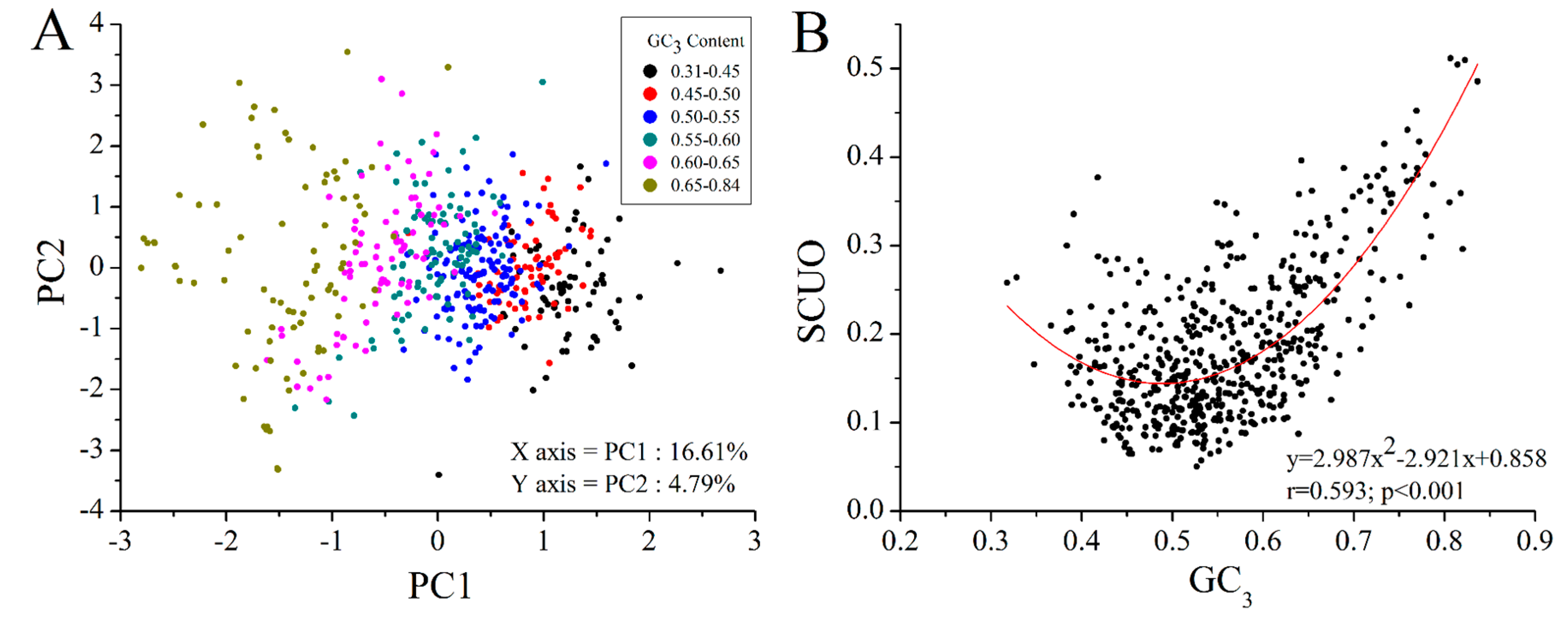
2.4. GC3 Bias in M. amblycephala
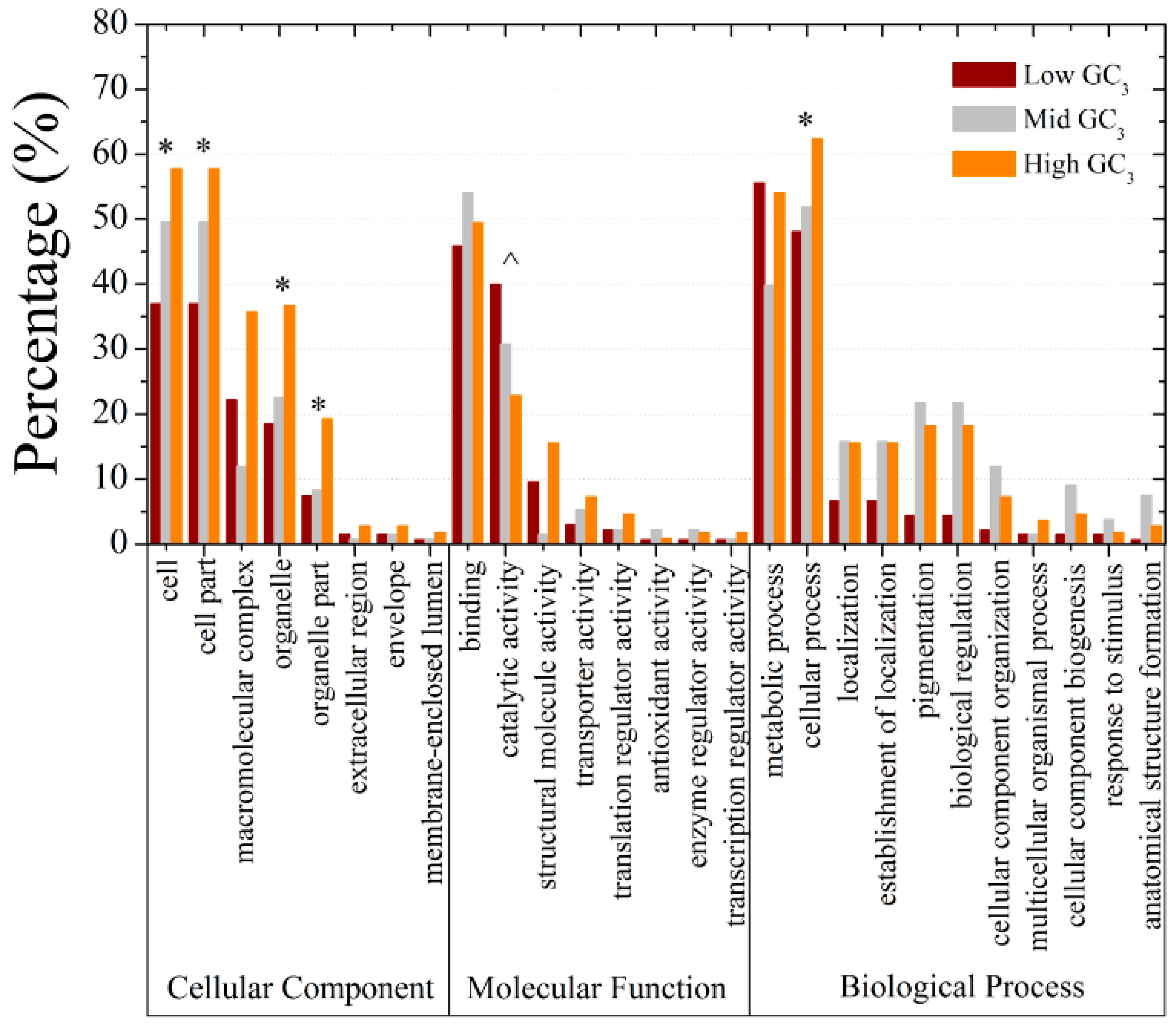
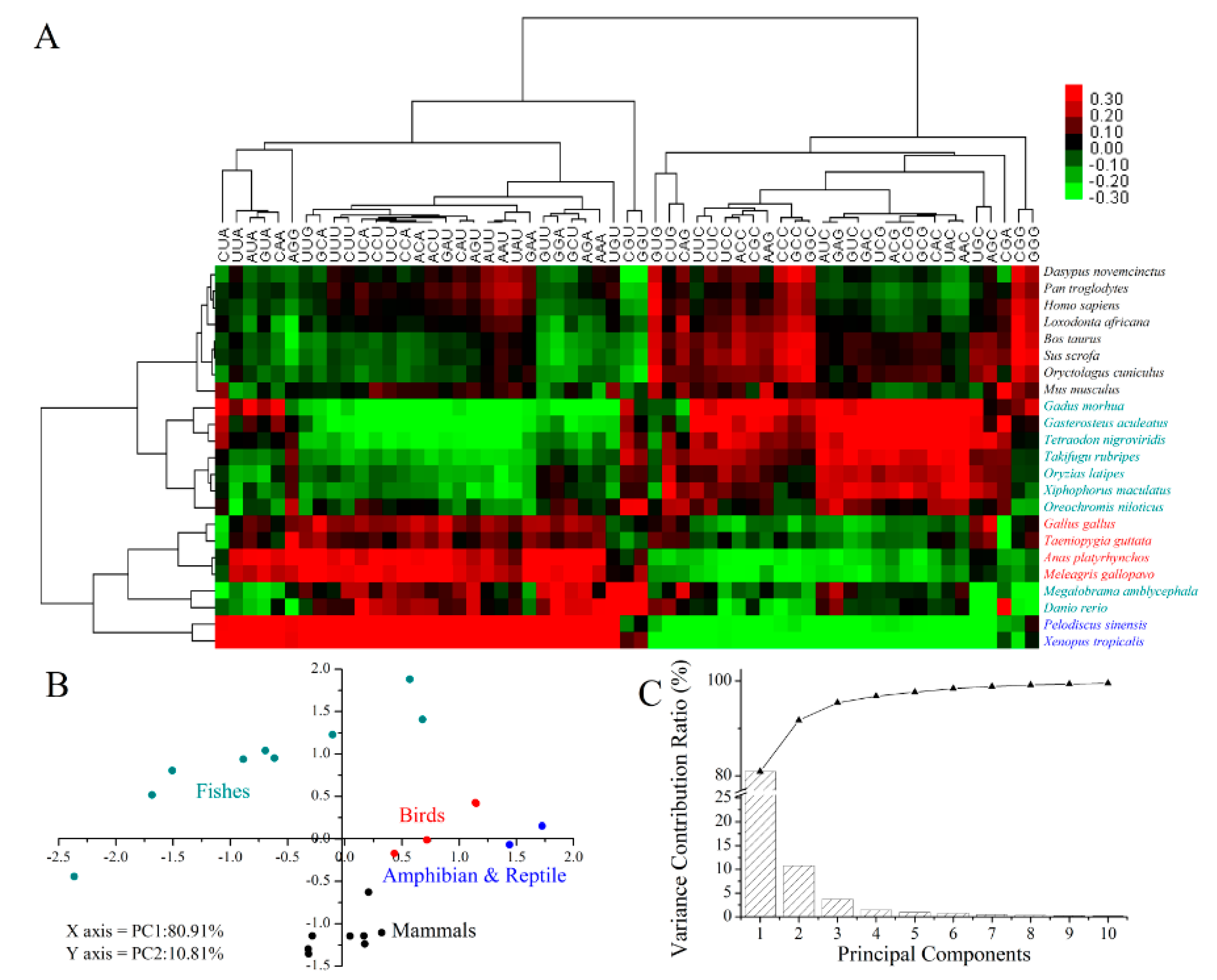
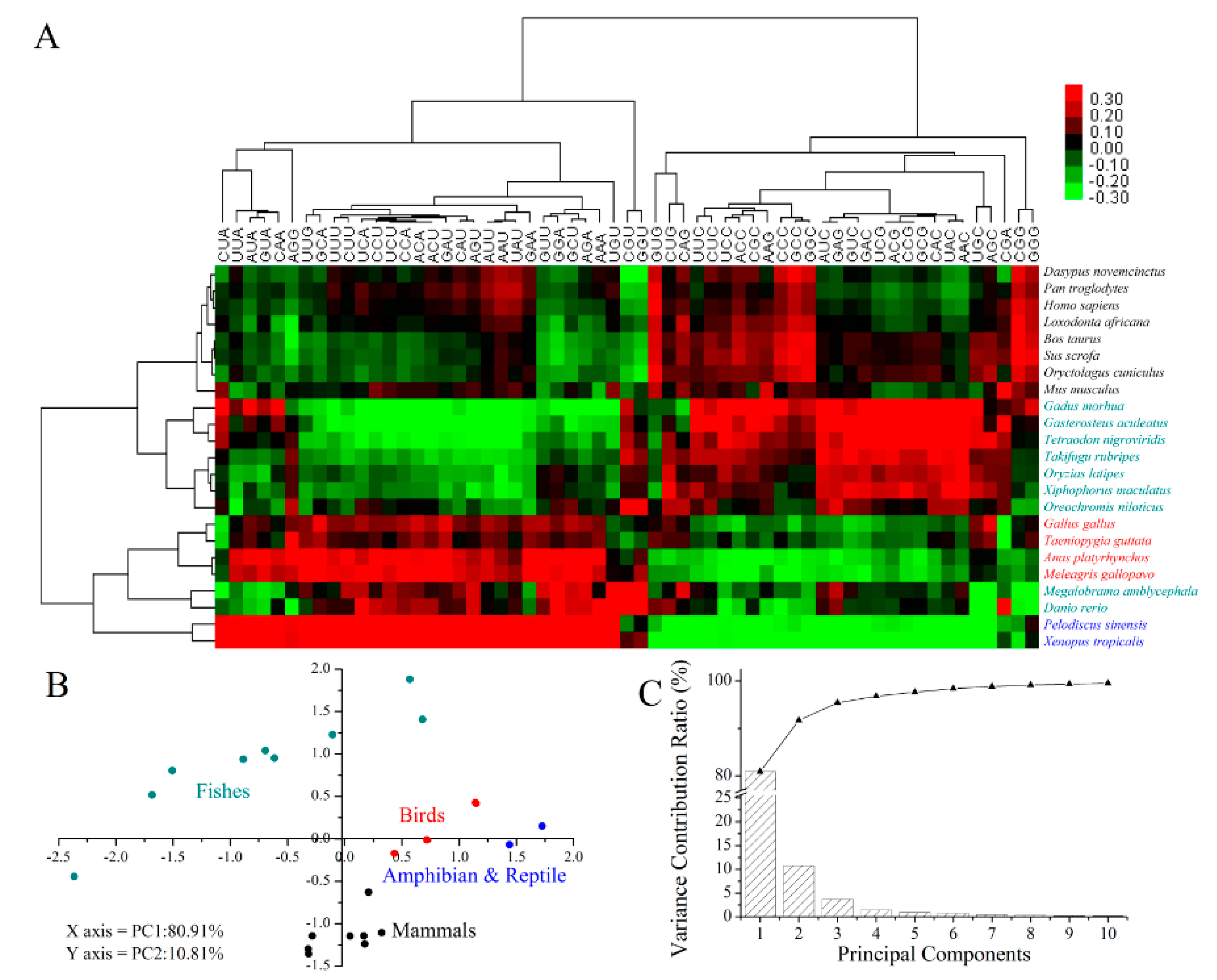
2.5. Codon Usage Patterns across the Vertebrates
3. Experimental Section
3.1. Sequence Data Collection, Filtering and Mining
3.2. Indices of Codon Usage
3.3. Null Models
3.4. Identification of High-Frequency, Preferred and Avoided Codons
3.5. Identification of High-Frequency, Preferred and Avoided Codon Pairs
3.6. Calculation of KLD Value and Logo Analyses
3.7. Calculation of Relative Abundance
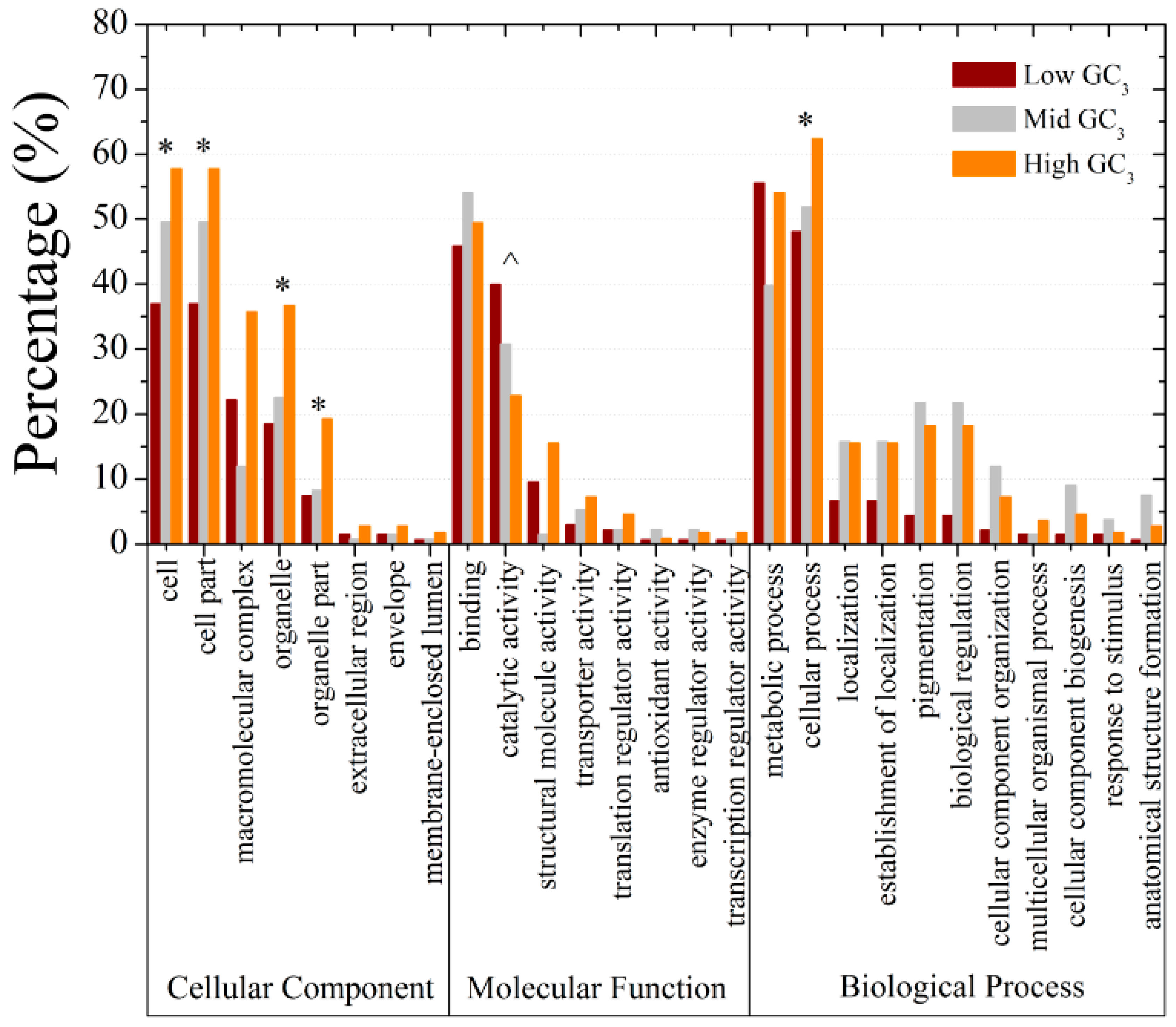
3.8. Gene Ontology Annotation
3.9. Clustering and Principal Component Analysis (PCA)
3.10. Codon Context Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Abbreviations
| AU3 | AU content at 3rd nucleotide in codon |
| C | Cytosine |
| G | Guanine |
| GC1 | GC content of 1st nucleotide in codon |
| GC2 | GC content of 2nd nucleotide in codon |
| GC3 | GC content of 3rd nucleotide in codon |
| GO | Gene ontology |
| KLD | Kullback–Leibler divergence |
| ORF | Open reading frame |
| PCA | Principal component analysis |
| RSCU | Relative synonymous codon usage |
| RSCPU | Relative synonymous codon pair usage |
| SSC | shuffled synonymous codons |
| SC | shuffled codons |
| SCUO | Synonymous codon usage order |
| ToL | The Tree of Life Web Project |
Conflicts of Interest
References
- Li, S.; Cai, W.; Zhou, B. Morphological and biochemical genetic variations among populations of blunt snout bream (Megalobrama amblycephala). J. Fish. China 1991, 15, 204–211. [Google Scholar]
- Ke, H. The artificial reproduction and culture experiment of Megalobrama amblycephala. Acta Hydrobiol. Sin. 1965, 5, 282–283. [Google Scholar]
- Wang, W. The aquaculture status of blunt snout bream (Megalobrama amblycephala). Sci. Fish Farming 2009, 4, 44–45. [Google Scholar]
- Gao, Z.; Luo, W.; Liu, H.; Zeng, C.; Liu, X.; Yi, S.; Wang, W. Transcriptome analysis and SSR/SNP markers information of the blunt snout bream (Megalobrama amblycephala). PLoS ONE 2012, 7, e42637. [Google Scholar] [CrossRef] [PubMed]
- Rao, H.; Deng, J.; Wang, W.; Gao, Z. An AFLP-based approach for the identification of sex-linked markers in blunt snout bream, Megalobrama amblycephala (Cyprinidae). Genet. Mol. Res. 2012, 11, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Gao, Z.; Zhao, H.; Zeng, C.; Luo, W.; Chen, B.; Wang, W. Identification and characterization of microRNAs involved in growth of blunt snout bream (Megalobrama amblycephala) by Solexa sequencing. BMC Genomics 2013, 14, 754. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Xu, C.; Wang, Y.; Liu, W.; Yin, X.; Li, X.; Chen, M.; Chen, K. Codon usage patterns in Chinese bayberry (Myrica rubra) based on RNA-Seq data. BMC Genomics 2013, 14, 732. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Doherty, A.; McInerney, J. Translational selection frequently overcomes genetic drift in shaping synonymous codon usage patterns in vertebrates. Mol. Biol. Evol. 2013, 30, 2263–2267. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Li, J.; Yang, Y.; Qi, L.; Chen, B.; Zhao, F.; Bao, Q.; Wu, J. Codon usage patterns and adaptive evolution of marine unicellular cyanobacteria Synechococcus and Prochlorococcus. Mol. Phylogenet. Evol. 2012, 62, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Duret, L.; Mouchiroud, D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA 1999, 96, 4482–4487. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Bergero, R.; Zeng, K.; Charlesworth, D. Patterns of codon usage bias in Silene latifolia. Mol. Biol. Evol. 2011, 28, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Behura, S.; Severson, D. Comparative analysis of codon usage bias and codon context patterns between dipteran and hymenopteran sequenced genomes. PLoS ONE 2012, 7, e43111. [Google Scholar] [CrossRef] [PubMed]
- Al-Saif, M.; Khabar, K. UU/UA dinucleotide frequency reduction in coding regions results in increased mRNA stability and protein expression. Mol. Ther. 2012, 20, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Sterky, F.; Bhalerao, R.; Unneberg, P.; Segerman, B.; Nilsson, P.; Brunner, A.; Charbonnel-Campaa, L.; Lindvall, J.; Tandre, K.; Strauss, S.; et al. A Populus EST resource for plant functional genomics. Proc. Natl. Acad. Sci. USA 2004, 101, 13951–13956. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ibeas, D.; Blanca, J.; Roig, C.; Gonzalez-To, M.; Pico, B.; Truniger, V.; Gomez, P.; Deleu, W.; Cano-Delgado, A.; Arus, P.; et al. MELOGEN: An EST database for melon functional genomics. BMC Genomics 2007, 8, 306. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Chen, M.; Xu, J.; Luo, J. Relationships among stop codon usage bias, its context, isochores, and gene expression level in various eukaryotes. J. Mol. Evol. 2005, 61, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Tats, A.; Tenson, T.; Remm, M. Preferred and avoided codon pairs in three domains of life. BMC Genomics 2008, 9, 463. [Google Scholar] [CrossRef] [PubMed]
- Irwin, B.; Heck, J.; Hatfield, G. Codon pair utilization biases influence translational elongation step times. J. Biol. Chem. 1995, 270, 22801–22806. [Google Scholar] [CrossRef] [PubMed]
- Moura, G.; Pinheiro, M.; Arrais, J.; Gomes, A.; Carreto, L. Large scale comparative codon-pair context analysis unveils general rules that fine-tune evolution of mRNA primary structure. PLoS ONE 2007, 2, e847. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Yang, J.; Pearson, N.; Maclean, C.; Zhang, J. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet. 2012, 8, e1002603. [Google Scholar] [CrossRef] [PubMed]
- Hockenberry, A.; Sirer, I.; Amaral, L.; Jewett, M. Quantifying position-dependent codon usage bias. Mol. Biol. Evol. 2014, 31, 1880–1893. [Google Scholar] [CrossRef] [PubMed]
- Bentele, K.; Saffert, P.; Rauscher, R.; Ignatova, Z.; Bluthgen, N. Efficient translation initiation dictates codon usage at gene start. Mol. Syst. Biol. 2013, 9, 675. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. The scanning model for translation: An update. J. Cell Biol. 1989, 108, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Noderer, W.; Flockhart, R.; Bhaduri, A.; Arce, A.; Zhang, J.; Khavari, P.; Wang, C. Quantitative analysis of mammalian translation initiation sites by FACS-Seq. Mol. Syst. Biol. 2014, 10, 748. [Google Scholar] [CrossRef] [PubMed]
- Grzegorski, S.; Chiari, E.; Robbins, A.; Kish, P.; Kahana, A. Natural variability of Kozak sequences correlates with function in a zebrafish model. PLoS ONE 2014, 9, e108475. [Google Scholar] [CrossRef] [PubMed]
- Mottagui-Tabar, S.; Isaksson, L. Only the last amino acids in the nascent peptide influence translation termination in Escherichia coli genes. FEBS Lett. 1997, 414, 165–170. [Google Scholar] [CrossRef]
- Bonetti, B.; Fu, L.; Moon, J.; Bedwell, D. The efficiency of translation termination is determined by a synergistic interplay between up-stream and down-stream sequences in saccharomyces cerevisiae. J. Mol. Biol. 1995, 251, 334–345. [Google Scholar] [CrossRef] [PubMed]
- McCaughan, K.; Brown, C.; Dalphin, M.; Berry, M.; Tate, W. Translational termination efficiency in mammals is influenced by the base following the stop codon. Proc. Natl. Acad. Sci. USA 1995, 6, 5431–5435. [Google Scholar] [CrossRef]
- Brown, C.; Stockwell, P.; Trotman, C.; Tate, W. Sequence analysis suggests that tetra-nucleotides signal the termination of protein synthesis in eukaryotes. Nucleic Acids Res. 1990, 18, 6339–6345. [Google Scholar] [CrossRef] [PubMed]
- Tatarinova, T.; Alexandrov, N.; Bouck, J.; Feldmann, K. GC3 biology in corn, rice, sorghum and other grasses. BMC Genomics 2010, 11, 308. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hickey, D. Rapid divergence of codon usage patterns within the rice genome. BMC Evol. Biol. 2007, 7, S6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.; Xu, D.; Kleinhofs, A.; Zhou, J. Quantitative relationship between synonymous codon usage bias and GC composition across unicellular genomes. BMC Evol. Biol. 2004, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeeberg, B. Shannon information theoretic computation of synonymous codon usage biases in coding regions of human and mouse genomes. Genome Res. 2002, 12, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Salinas, J.; Matassi, G.; Montero, L.; Bernardi, G. Compositional compartentalization and compositional patterns in the nuclear genomes of plants. Nucleic Acids Res. 1988, 16, 4269–4285. [Google Scholar] [PubMed]
- Palidwor, G.; Perkins, T.; Xia, X. A general model of codon bias due to GC mutational bias. PLoS ONE 2010, 5, e13431. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Urwin, N.; Jenkins, G.; Adams, R. Effect of CWG methylation on expression of plant genes. Biochem. J. 1999, 341, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Moura, G.; Pinheiro, M.; Silva, R.; Miranda, I.; Afreixo, V.; Dias, G.; Freitas, A.; Oliveira, J.; Santos, M. Comparative context analysis of codon pairs on an ORFeome scale. Genome Biol. 2005, 6, R28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, D.; Logue, M.; Butler, G.; Wolfe, K. Chromosomal G + C content evolution in yeasts: Systematic interspecies differences, and GC-poor troughs at centromeres. Genome Biol. Evol. 2010, 2, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.; Li, W. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Angellotti, M.; Bhuiyan, S.; Chen, G.; Wan, X. CodonO: Codon usage bias analysis within and across genomes. Nucleic Acids Res. 2007, 35, W132–W136. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Tong, C.; Shi, J. Analysis of codon usage between different poplar species. J. Genet. Genomics 2007, 34, 555–561. [Google Scholar] [CrossRef]
- Audic, S.; Claverie, J. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [PubMed]
- Feng, C.; Chen, M.; Xu, C.; Bai, L.; Yin, X.; Li, X.; Allan, A.; Ferguson, I.; Chen, K. Transcriptomic analysis of Chinese bayberry (Myrica rubra) fruit development and ripening using RNA-Seq. BMC Genomics 2012, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Roulston, M. Estimating the errors on measured entropy and mutual information. Phys. D 1999, 125, 285–294. [Google Scholar] [CrossRef]
- Crooks, G.; Hon, G.; Chandonia, J.-M.; Brenner, S. WebLogo: A sequence logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Karlin, S.; Mrazek, J. Compositional differences within and between eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 10227–10232. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- De Hoon, M.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, X.; Yi, S.; Guo, X.; Wang, W. A Comprehensive Analysis of Codon Usage Patterns in Blunt Snout Bream (Megalobrama amblycephala) Based on RNA-Seq Data. Int. J. Mol. Sci. 2015, 16, 11996-12013. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160611996
Duan X, Yi S, Guo X, Wang W. A Comprehensive Analysis of Codon Usage Patterns in Blunt Snout Bream (Megalobrama amblycephala) Based on RNA-Seq Data. International Journal of Molecular Sciences. 2015; 16(6):11996-12013. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160611996
Chicago/Turabian StyleDuan, Xiaoke, Shaokui Yi, Xianwu Guo, and Weimin Wang. 2015. "A Comprehensive Analysis of Codon Usage Patterns in Blunt Snout Bream (Megalobrama amblycephala) Based on RNA-Seq Data" International Journal of Molecular Sciences 16, no. 6: 11996-12013. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160611996