
Combinatorial Pharmacophore-Based 3D-QSAR Analysis and Virtual Screening of FGFR1 Inhibitors

Abstract
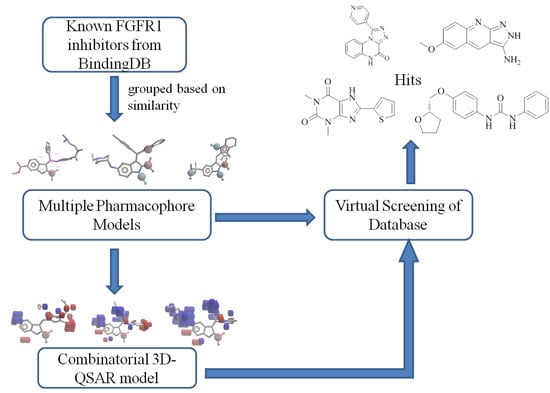
:
1. Introduction
2. Results and Discussion


{kind=link}
{kind=link}
{kind=link}
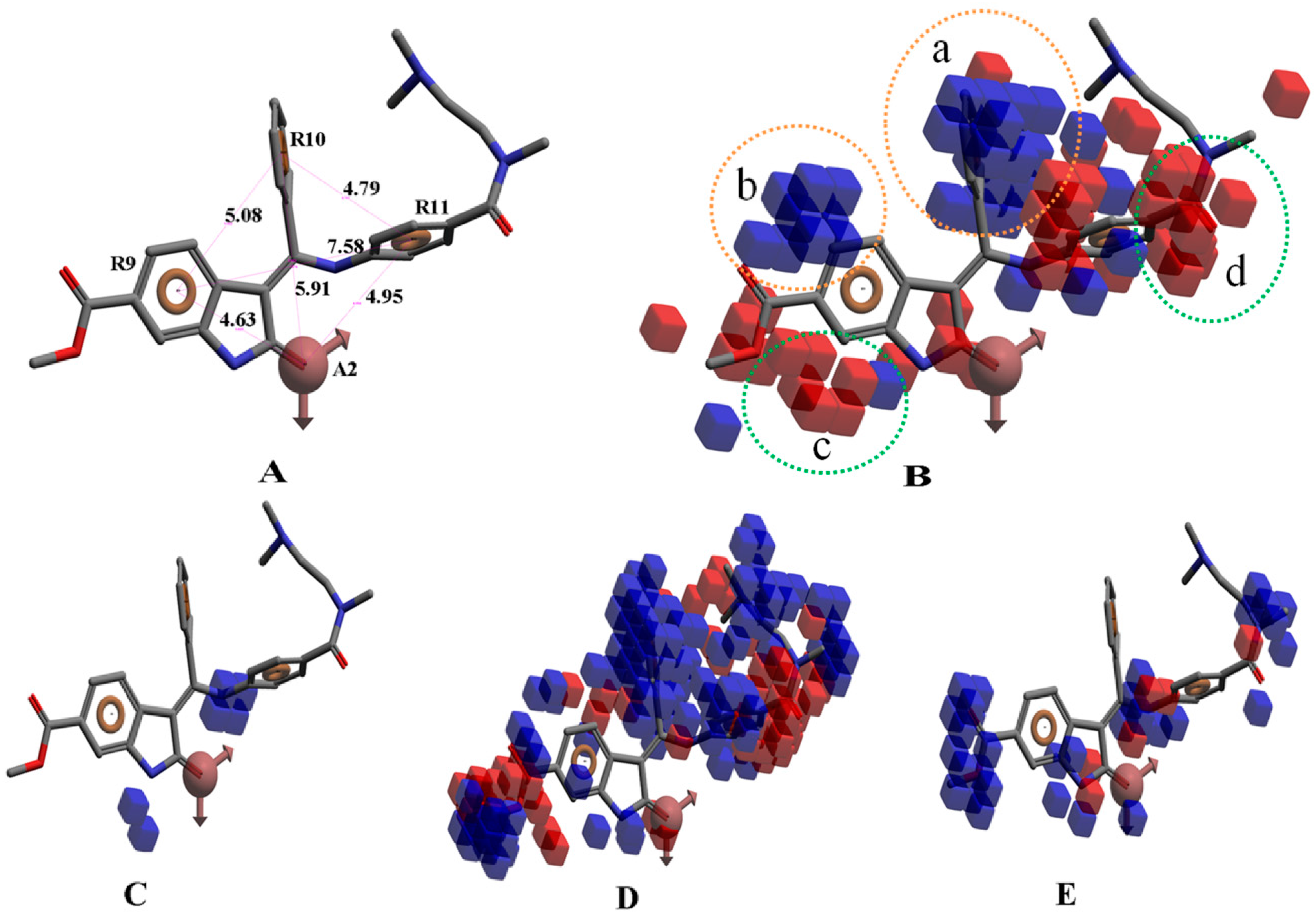{kind=link}
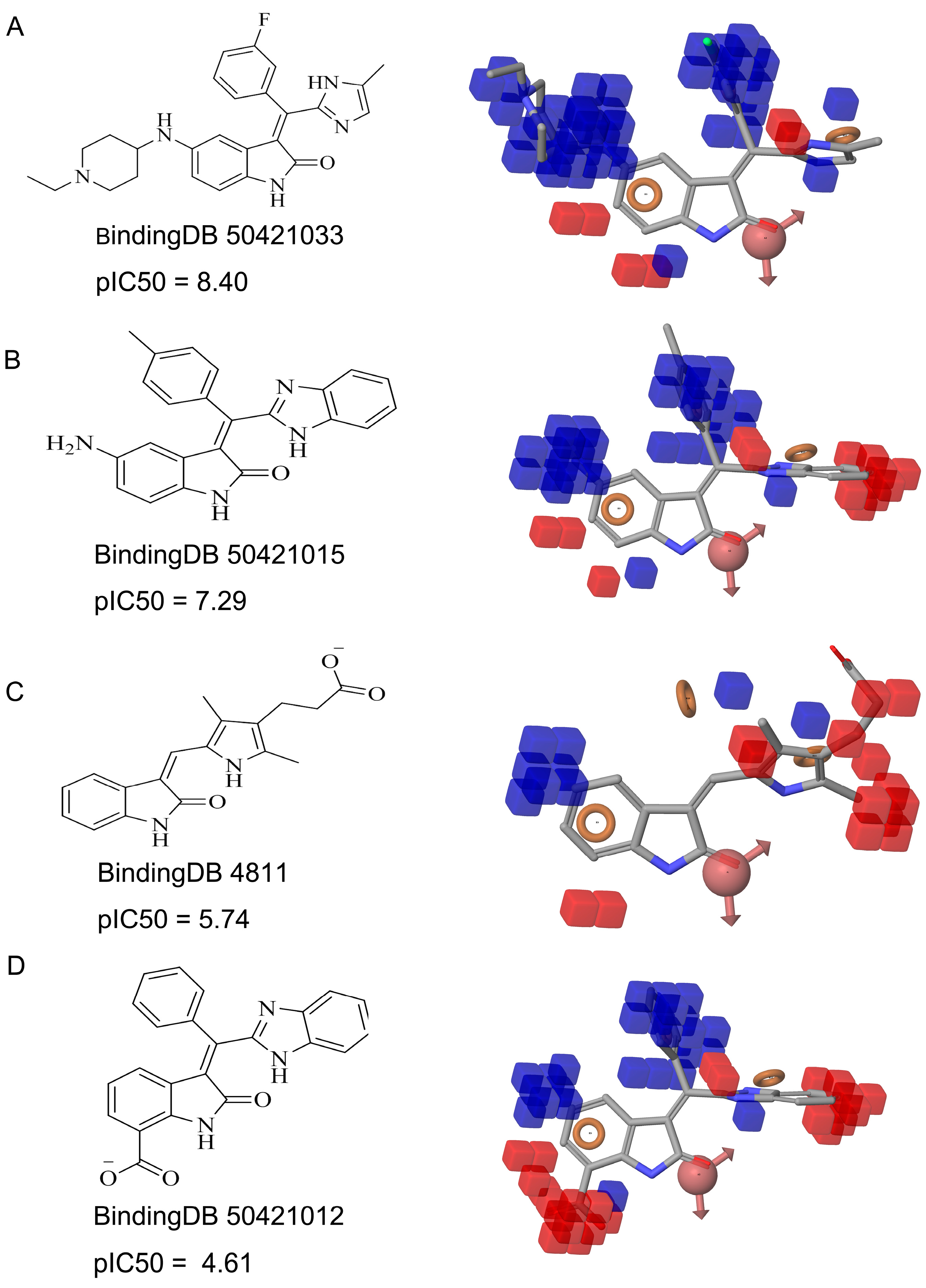{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Number of Compounds | R2 | SD | F | p | Stability |
|---|---|---|---|---|---|---|
| 1 (ARRR) | 83 | 0.81 | 0.41 | 114.50 | 7.35 × 10−29 | 0.51 |
| 2 (ADRRR) | 29 | 0.89 | 0.32 | 75.30 | 3.10 × 10−13 | 0.61 |
| 3 (AAAARR) | 62 | 0.86 | 0.32 | 121.70 | 5.14 × 10−25 | 0.74 |
| 4 (DDRRR) | 99 | 0.81 | 0.38 | 134.70 | 2.82 × 10−34 | 0.53 |
| 5 (ADHRR) | 18 | 0.99 | 0.09 | 362.60 | 1.67 × 10−13 | 0.53 |
| 6 (ADDH) | 25 | 0.96 | 0.14 | 170.90 | 6.58 × 10−15 | 0.89 |
| 7 (ADHR) | 20 | 0.98 | 0.15 | 237.80 | 1.78 × 10−13 | 0.56 |
| 8 (DRRRR) | 39 | 0.98 | 0.21 | 649.80 | 9.81 × 10−31 | 0.85 |

| Group | Test Set Grouped Based on Similarity | Total Test Set (232) | ||||
|---|---|---|---|---|---|---|
| Matched Hits | R2 | SD | Matched Hits | R2 | SD | |
| 1 (ARRR) | 51 | 0.53 | 0.64 | 229 | 0.00 * | 1.12 |
| 2 (ADRRR) | 16 | 0.37 | 0.71 | 231 | 0.00 * | 1.16 |
| 3 (AAAARR) | 29 | 0.41 | 0.68 | 227 | 0.01 | 1.09 |
| 4 (DDRRR) | 43 | 0.17 | 0.87 | 227 | 0.00 * | 1.12 |
| 5 (ADHRR) | 9 | 0.66 | 0.79 | 232 | 0.00 * | 1.09 |
| 6 (ADDH) | 16 | 0.54 | 0.68 | 184 | 0.00 * | 1.00 |
| 7 (ADHR) | 23 | 0.37 | 0.90 | 216 | 0.03 | 1.10 |
| 8 (DRRRR) | 42 | 0.87 | 0.33 | 229 | 0.12 | 1.01 |
| Combinatorial QSAR Model | – | – | – | 229 | 0.53 | 0.75 |


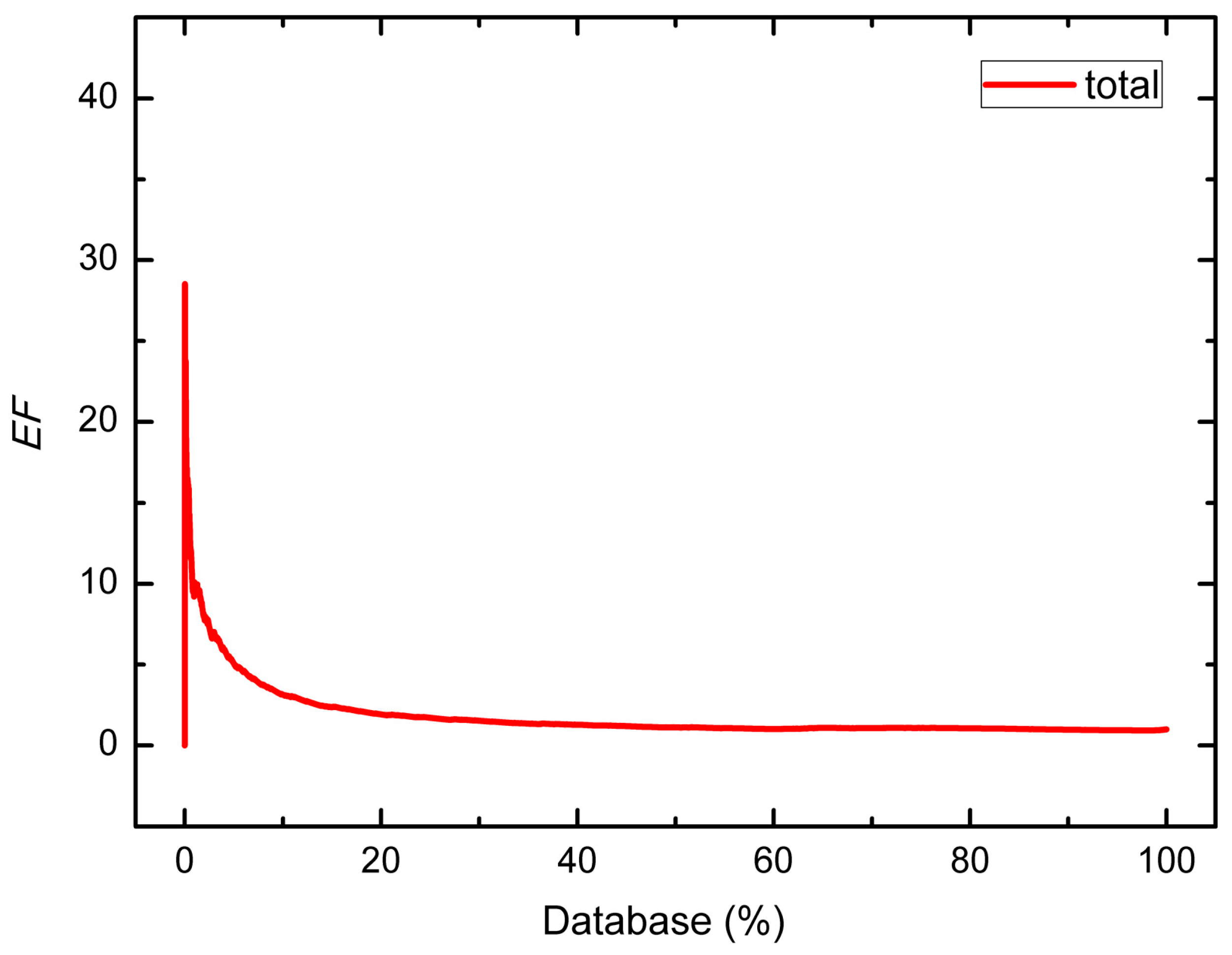
| Group | Number of Hits/Size of Dataset | EF | |||||
|---|---|---|---|---|---|---|---|
| Decoy Set | Decoys | Inhibitors | 1% | 2% | 5% | 10% | |
| 1 | 2063/2804 | 2012/2750 | 51/54 | 11.56 | 7.89 | 5.50 | 3.34 |
| 2 | 836/915 | 820/899 | 16/16 | 26.13 | 18.44 | 11.20 | 6.84 |
| 3 | 1048/1121 | 1019/1092 | 29/29 | 14.46 | 10.33 | 8.34 | 6.54 |
| 4 | 629/716 | 586/673 | 43/43 | 14.63 | 14.63 | 12.74 | 8.59 |
| 5 | 737/773 | 728/764 | 9/9 | 0.00 | 10.92 | 6.64 | 3.32 |
| 6 | 93/109 | 77/93 | 16/16 | 5.81 | 5.81 | 4.65 | 3.49 |
| 7 | 542/593 | 519/570 | 23/23 | 23.57 | 17.14 | 9.60 | 4.80 |
| 8 | 585/866 | 543/824 | 42/42 | 9.29 | 4.64 | 2.40 | 2.60 |
| Total | 6533/7897 | 6304/7665 | 229/232 | 10.09 | 7.84 | 5.06 | 3.15 |


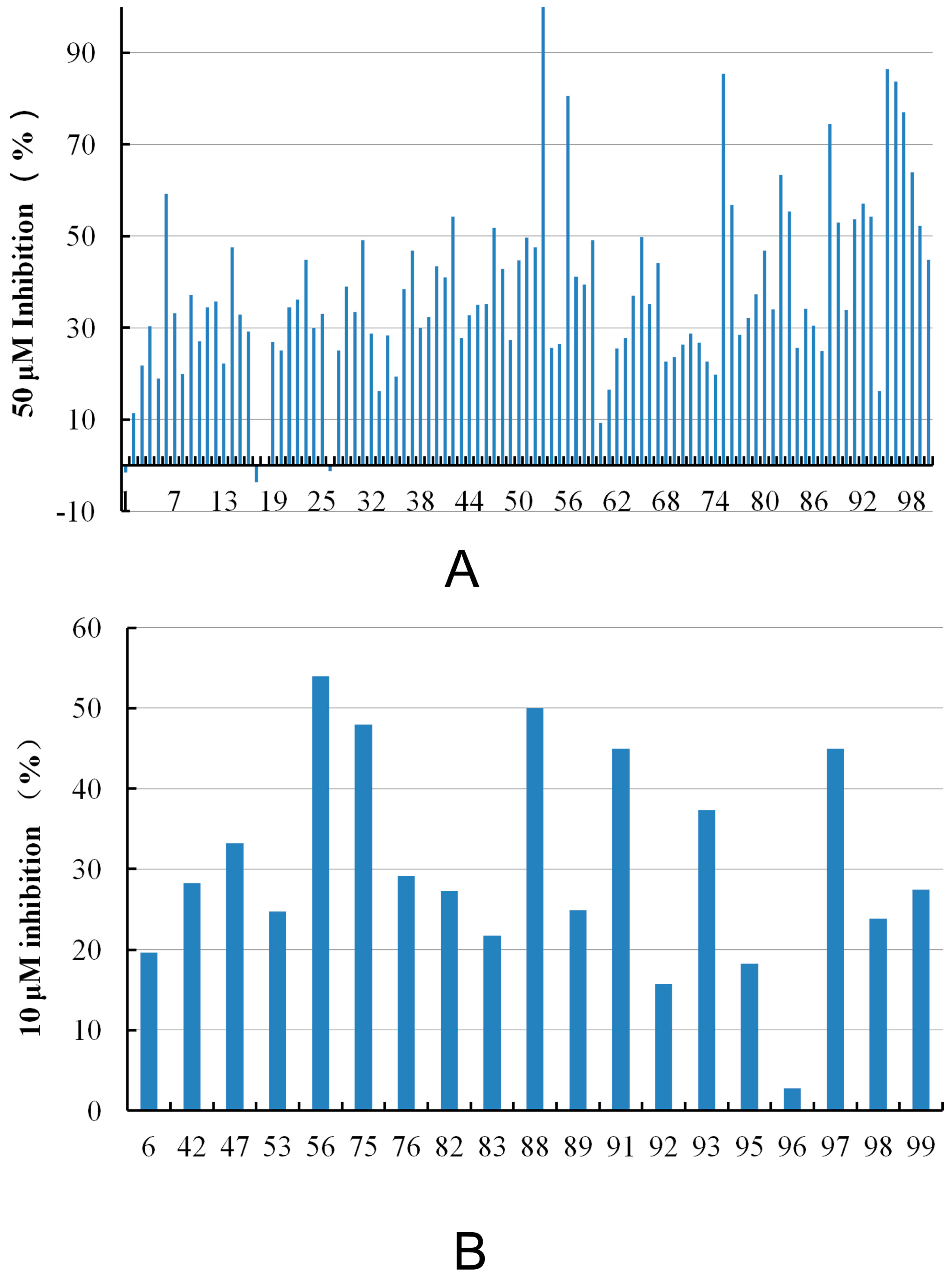
| Compound ID | Hit Compound Structure | Inhibition (%) a | Similar Structure in BindingDB | Similarity b | |
|---|---|---|---|---|---|
| 50 μM | 10 μM | ||||
| 6 |  | 59.20 | 19.65 |  | 0.25 |
| BindingDB4812 | |||||
| 42 |  | 54.20 | 28.25 |  | 0.27 |
| BindingDB50307880 | |||||
| 47 |  | 51.80 | 33.20 |  | 0.23 |
| BindingDB50421018 | |||||
| 53 |  | 104.30 | 24.75 |  | 0.37 |
| BindingDB50234144 | |||||
| 56 |  | 80.60 | 54.00 |  | 0.32 |
| BindingDB50420994 | |||||
| 75 |  | 85.40 | 48.00 |  | 0.26 |
| BindingDB50279238 | |||||
| 76 |  | 56.80 | 29.15 |  | 0.29 |
| BindingDB13533 | |||||
| 82 |  | 63.40 | 27.30 |  | 0.35 |
| BindingDB50121980 | |||||
| 83 |  | 55.40 | 21.80 |  | 0.32 |
| BindingDB3855 | |||||
| 88 |  | 74.50 | 50.00 |  | 0.35 |
| BindingDB3933 | |||||
| 89 |  | 53.00 | 24.95 |  | 0.30 |
| BindingDB50431812 | |||||
| 91 |  | 53.70 | 45.00 |  | 0.30 |
| BindingDB50420968 | |||||
| 92 |  | 57.10 | 15.75 |  | 0.25 |
| BindingDB50185172 | |||||
| 93 |  | 54.30 | 37.35 |  | 0.38 |
| BindingDB50185180 | |||||
| 95 |  | 86.40 | 18.30 |  | 0.28 |
| BindingDB3051 | |||||
| 96 |  | 83.70 | 2.80 |  | 0.28 |
| BindingDB50279045 | |||||
| 97 |  | 77.10 | 45.00 |  | 0.28 |
| BindingDB11242 | |||||
| 98 |  | 64.00 | 23.85 |  | 0.28 |
| BindingDB50345445 | |||||
| 99 |  | 52.30 | 27.45 |  | 0.27 |
| BindingDB6619 | |||||
3. Experimental Section
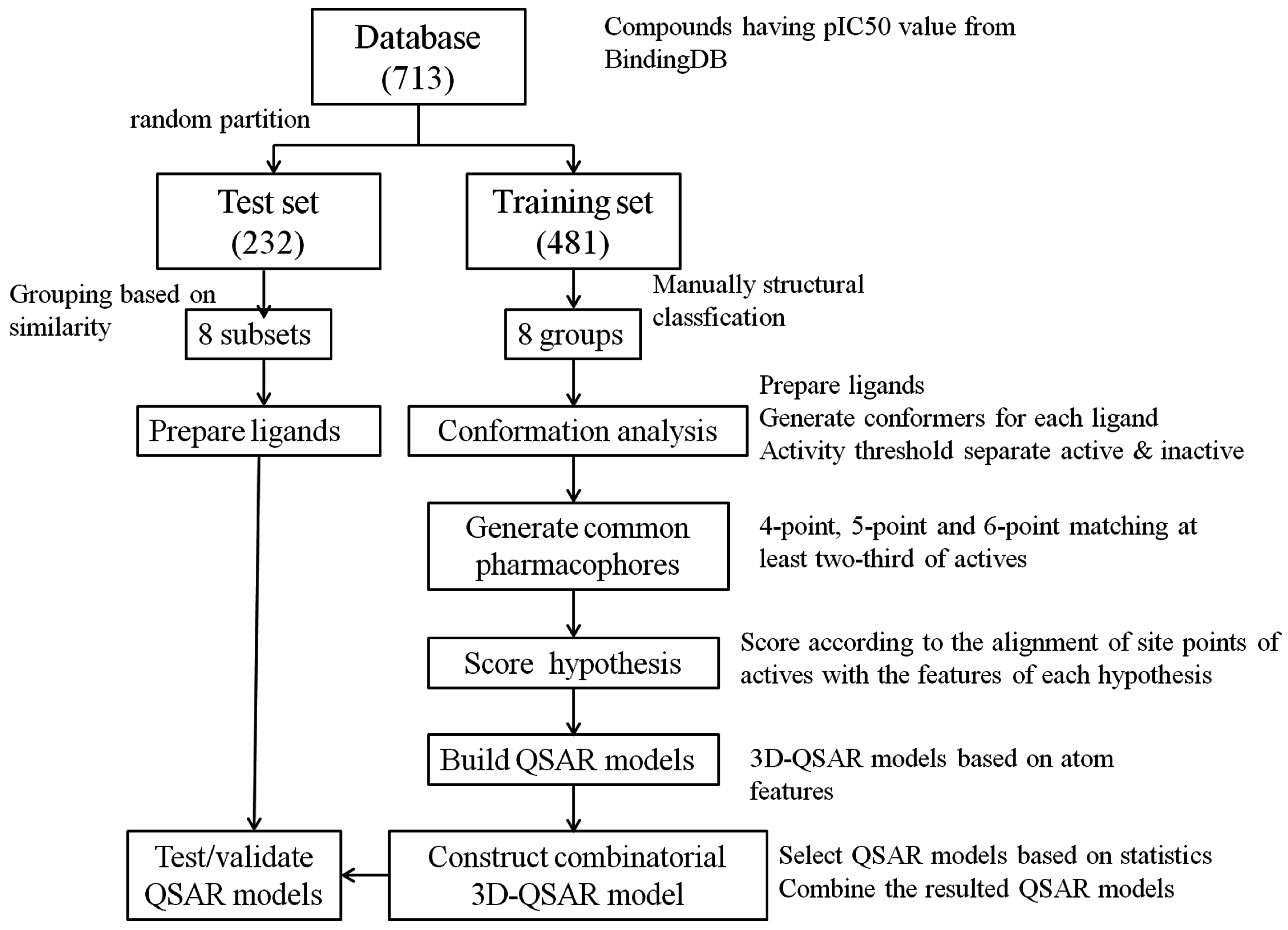
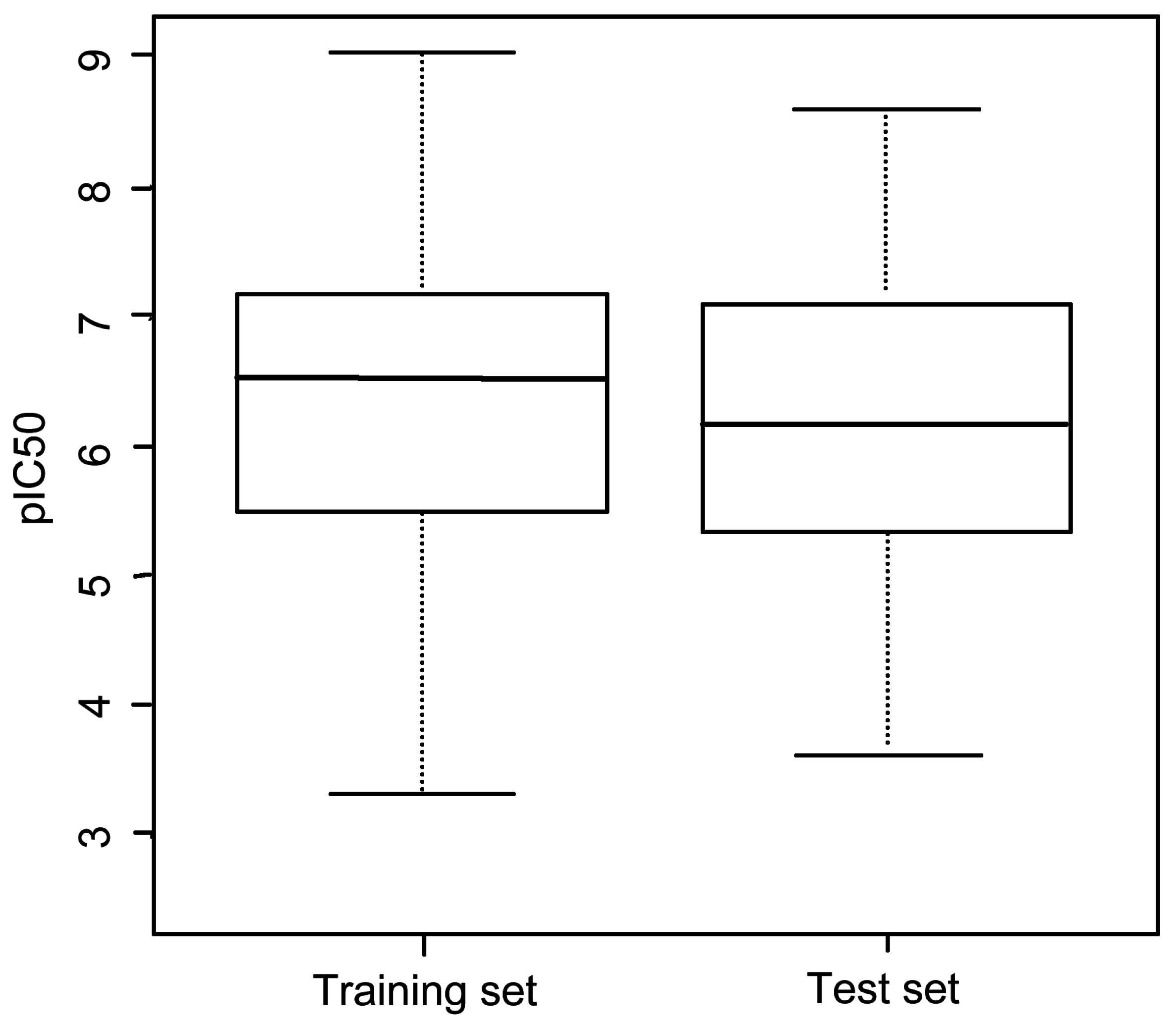
3.1. Dataset

| Group | Skeleton | Size of Training Set | Size of Test Set |
|---|---|---|---|
| 1 |  | 83 | 54 |
| 2 |  | 29 | 16 |
| 3 |  | 62 | 29 |
| 4 |  | 99 | 43 |
| 5 |  | 18 | 9 |
| 6 |  | 25 | 16 |
| 7 |  | 20 | 23 |
| 8 |  | 39 | 42 |
3.2. Pharmacophore Hypothesis Generation
3.2.1. Conformation Analysis
3.2.2. Generate Common Pharmacophore Hypothesis (CPH)
3.2.3. Score Hypotheses
3.3. Build QSAR Models
3.4. Construct Combinatorial 3D-QSAR Model
3.5. Model Validation
3.6. Pharmacophore-Based Virtual Screening and 3D-QSAR Analysis
3.7. Enzyme Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dieci, M.V.; Arnedos, M.; Andre, F.; Soria, J.C. Fibroblast growth factor receptor inhibitors as a cancer treatment: From a biologic rationale to medical perspectives. Cancer Discov. 2013, 3, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Easty, D.; Gray, S.; O’Byrne, K.; O’Donnell, D.; Bennett, D. Receptor tyrosine kinases and their activation in melanoma. Pigment Cell Melanoma Res. 2011, 24, 446–461. [Google Scholar] [CrossRef] [PubMed]
- Eswarakumar, V.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Rodon, J.; Prat, A.; Perez-Garcia, J.; Adamo, B.; Felip, E.; Cortes, J.; Iafrate, A.J.; Nuciforo, P.; Tabernero, J. Genomic aberrations in the FGFR pathway: Opportunities for targeted therapies in solid tumors. Ann. Oncol. 2014, 25, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Le Corre, L.; Girard, A.L.; Aubertin, J.; Radvanyi, F.; Benoist-Lasselin, C.; Jonquoy, A.; Mugniery, E.; Legeai-Mallet, L.; Busca, P.; le Merrer, Y. Synthesis and biological evaluation of a triazole-based library of pyrido [2,3-d] pyrimidines as FGFR3 tyrosine kinase inhibitors. Org. Biomol. Chem. 2010, 8, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- McBride, C.M.; Renhowe, P.A.; Heise, C.; Jansen, J.M.; Lapointe, G.; Ma, S.; Pineda, R.; Vora, J.; Wiesmann, M.; Shafer, C.M. Design and structure-activity relationship of 3-benzimidazol-2-yl-1H-indazoles as inhibitors of receptor tyrosine kinases. Bioorg. Med. Chem. Lett. 2006, 16, 3595–3599. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Tran, N.; Liang, C.X.; Tang, F.; Rice, A.; Schreck, R.; Waltz, K.; Shawver, L.K.; McMahon, G.; Tang, C. Design, synthesis, and evaluations of substituted 3-(3- or 4-carboxyethylpyrrol-2-yl)methylidenyl indolin-2-ones as inhibitors of VEGF, FGF, and PDGF receptor tyrosine kinases. J. Med. Chem. 1999, 42, 5120–5130. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.M.; Delaney, A.M.; Hamby, D.M.; Schroeder, M.C.; Spoon, T.A.; Crean, S.M.; Showalter, H.D.H.; Denny, W.A. Synthesis and structure-activity relationships of soluble 7-substituted 3-(3,5-dimethoxyphenyl)-1,6-naphthyridin-2-amines and related ureas as dual inhibitors of the fibroblast growth factor receptor-1 and vascular endothelial growth factor receptor-2 tyrosine kinases. J. Med. Chem. 2005, 48, 4628–4653. [Google Scholar] [PubMed]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2011, 365, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.; Ward, G.; Saxty, G.; Berdini, V.; Cleasby, A.; King, P.; Angibaud, P.; Perera, T.; Fazal, L.; Ross, D.; et al. Potent, selective inhibitors of fibroblast growth factor receptor define fibroblast growth factor dependence in preclinical cancer models. Mol. Cancer Ther. 2011, 10, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Molife, R.; Evans, T.R.J.; Hardie, M.; Marriott, C.; Butzberger-Zimmerli, P.; Morrison, R.; Fox, J.A.; Heise, C.; Louie, S.; et al. A phase I pharmacokinetic and pharmacodynamic study of TKI258, an oral, multitargeted receptor tyrosine kinase inhibitor in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Bello, E.; Colella, G.; Scarlato, V.; Oliva, P.; Berndt, A.; Valbusa, G.; Serra, S.C.; D’Incalci, M.; Cavalletti, E.; Giavazzi, R.; et al. E-3810 is a potent dual inhibitor of VEGFR and FGFR that exerts antitumor activity in multiple preclinical models. Cancer Res. 2011, 71, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H. Discovery of 3-(2, 6-dichloro-3, 5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Chen, G.; Wei, X.; Zhao, Y.; Li, X. Small molecule inhibition of fibroblast growth factor receptors in cancer. Cytokine Growth Factor Rev. 2013, 24, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Liu, Z.; Wu, J.; Cai, Y.; Li, X. Anticancer molecules targeting fibroblast growth factor receptors. Trends Pharmacol. Sci. 2012, 33, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.K.; Yeo, A.H.L.; Kang, T.S.; Chua, B.T. Current strategies for inhibiting FGFR activities in clinical applications: Opportunities, challenges and toxicological considerations. Drug Discov. Today 2014, 19, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.; Pan, Y.; Li, L.; Ferguson, S.S.; Xia, M.; Swaan, P.W.; Wang, H. Identification of novel activators of constitutive androstane receptor from FDA-approved drugs by integrated computational and biological approaches. Pharm. Res. 2013, 30, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.M.; Wang, Y.L.; Bryant, S.H. Pharmacophore and 3D-QSAR characterization of 6-arylquinazolin-4-amines as Cdc2-like kinase 4 (Clk4) and dual specificity tyrosine-phosphorylation-regulated kinase 1A (Dyrk1A) inhibitors. J. Chem. Inf. Model. 2013, 53, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Acharya, C.; Coop, A.; Polli, J.E.; Mackerell, A.D., Jr. Recent advances in ligand-based drug design: Relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Aided Drug Des. 2011, 7, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.A. The role of quantitative structure—Activity relationships (QSAR) in biomolecular discovery. Brief. Bioinform. 2002, 3, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, M. Current state and perspectives of 3D-QSAR. Curr. Top. Med. Chem. 2002, 2, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Moonsamy, S.; Dash, R.C.; Soliman, M.E. Integrated computational tools for identification of CCR5 antagonists as potential HIV-1 entry inhibitors: Homology modeling, virtual screening, molecular dynamics simulations and 3D QSAR analysis. Molecules 2014, 19, 5243–5265. [Google Scholar] [CrossRef] [PubMed]
- Shih, K.; Lin, C.; Zhou, J.; Chi, H.; Chen, T.; Wang, C.; Tseng, H.; Tang, C. Development of novel 3D-QSAR combination approach for screening and optimizing B-Raf inhibitors in silico. J. Chem. Inf. Model. 2011, 51, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.V.; Agarwal, S.; Kesharwani, R.K.; Misra, K. 3D QSAR and pharmacophore study of curcuminoids and curcumin analogs: Interaction with thioredoxin reductase. Interdiscip. Sci. 2013, 5, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Kristam, R.; Parmar, V.; Viswanadhan, V.N. 3D-QSAR analysis of TRPV1 inhibitors reveals a pharmacophore applicable to diverse scaffolds and clinical candidates. J. Mol. Graph. Model. 2013, 45, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; McMahon, G.; Sun, L.; Tang, C.; Hirth, P.; Yeh, B.K.; Hubbard, S.R.; Schlessinger, J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 1997, 276, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Tran, N.; Liang, C.X.; Hubbard, S.; Tang, F.; Lipson, K.; Schreck, R.; Zhou, Y.; McMahon, G.; Tang, C. Identification of substituted 3-(4,5,6,7-tetrahydro-1H-indol-2-yl)methylene-1,3-dihydroindol-2-ones as growth factor receptor inhibitors for VEGF-R2 (Flk-1/KDR), FGF-R1, and PDGF-Rβ tyrosine kinases. J. Med. Chem. 2000, 43, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (pains) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Mohan, C.G. Dual binding site and selective acetylcholinesterase inhibitors derived from integrated pharmacophore models and sequential virtual screening. Biomed. Res. Int. 2014, 2014, 291214. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.Q.; Lin, Y.M.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. Phase: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massague, A.; Guasch, L.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallve, S. DecoyFinder: An easy-to-use python GUI application for building target-specific decoy sets. Bioinformatics 2012, 28, 1661–1662. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Jin, H.; Liu, Z.; Zhang, L.; Wang, X.S. An unbiased method to build benchmarking sets for ligand-based virtual screening and its application to GPCRs. J. Chem. Inf. Model. 2014, 54, 1433–1450. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Tilahun, E.L.; Reid, T.-E.; Zhang, L.; Wang, X.S. Benchmarking methods and data sets for ligand enrichment assessment in virtual screening. Methods 2015, 71, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, H.L.; Zhang, Q.J.; Bao, X.G.; Yu, K.Q.; Luo, X.M.; Zhu, W.L.; Jiang, H.L. Pharmacophore-based virtual screening versus docking-based virtual screening: A benchmark comparison against eight targets. Acta Pharmacol. Sin. 2009, 30, 1694–1708. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, N.; Xu, Y.; Liu, X.; Wang, Y.; Peng, J.; Luo, X.; Zheng, M.; Chen, K.; Jiang, H. Combinatorial Pharmacophore-Based 3D-QSAR Analysis and Virtual Screening of FGFR1 Inhibitors. Int. J. Mol. Sci. 2015, 16, 13407-13426. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160613407
Zhou N, Xu Y, Liu X, Wang Y, Peng J, Luo X, Zheng M, Chen K, Jiang H. Combinatorial Pharmacophore-Based 3D-QSAR Analysis and Virtual Screening of FGFR1 Inhibitors. International Journal of Molecular Sciences. 2015; 16(6):13407-13426. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160613407
Chicago/Turabian StyleZhou, Nannan, Yuan Xu, Xian Liu, Yulan Wang, Jianlong Peng, Xiaomin Luo, Mingyue Zheng, Kaixian Chen, and Hualiang Jiang. 2015. "Combinatorial Pharmacophore-Based 3D-QSAR Analysis and Virtual Screening of FGFR1 Inhibitors" International Journal of Molecular Sciences 16, no. 6: 13407-13426. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160613407