
Cloning, Expression and Characterization of a Thermostable Esterase HydS14 from Actinomadura sp. Strain S14 in Pichia pastoris

Abstract
:1. Introduction
2. Results and Discussion
2.1. Cloning of a Novel Esterase Gene from Actinomadura sp. S14
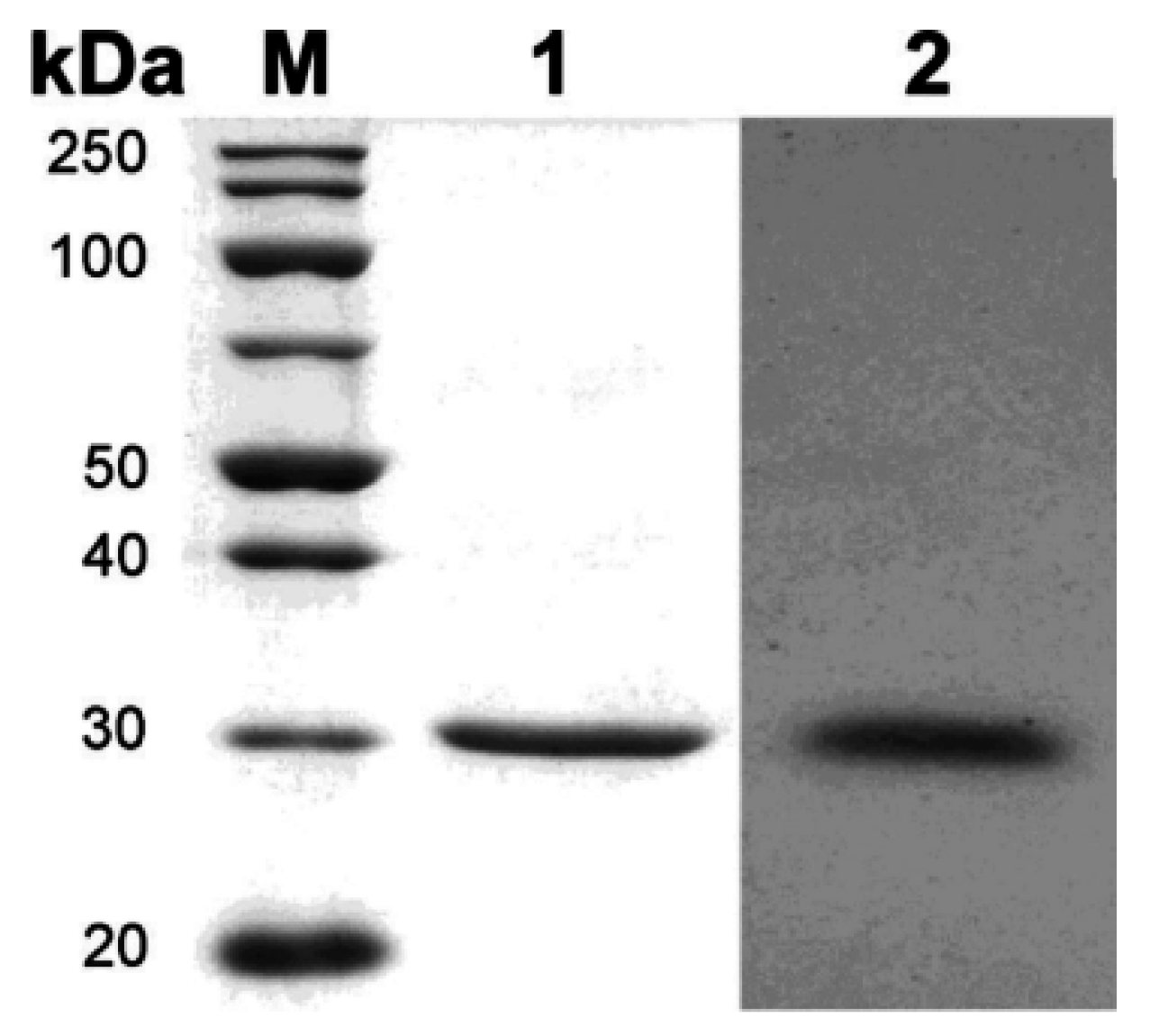
2.2. Expression and Purification of Recombinant HydS14





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Purification Step | Total Activity (U) | Total Protein (mg) | Specific Activity (U/mg) | Purification (fold) | Yield (%) |
|---|---|---|---|---|---|
| Culture supernatant | 2830 ± 110 | 60 | 47.2 ± 1.8 | 1 | 100 |
| Concentrated supernatant | 1270 ± 26 | 2.2 | 576 ± 12 | 12 | 45 |
| Ni-Sepharose column | 390 ± 9.8 | 0.39 | 999 ± 25 | 21 | 14 |
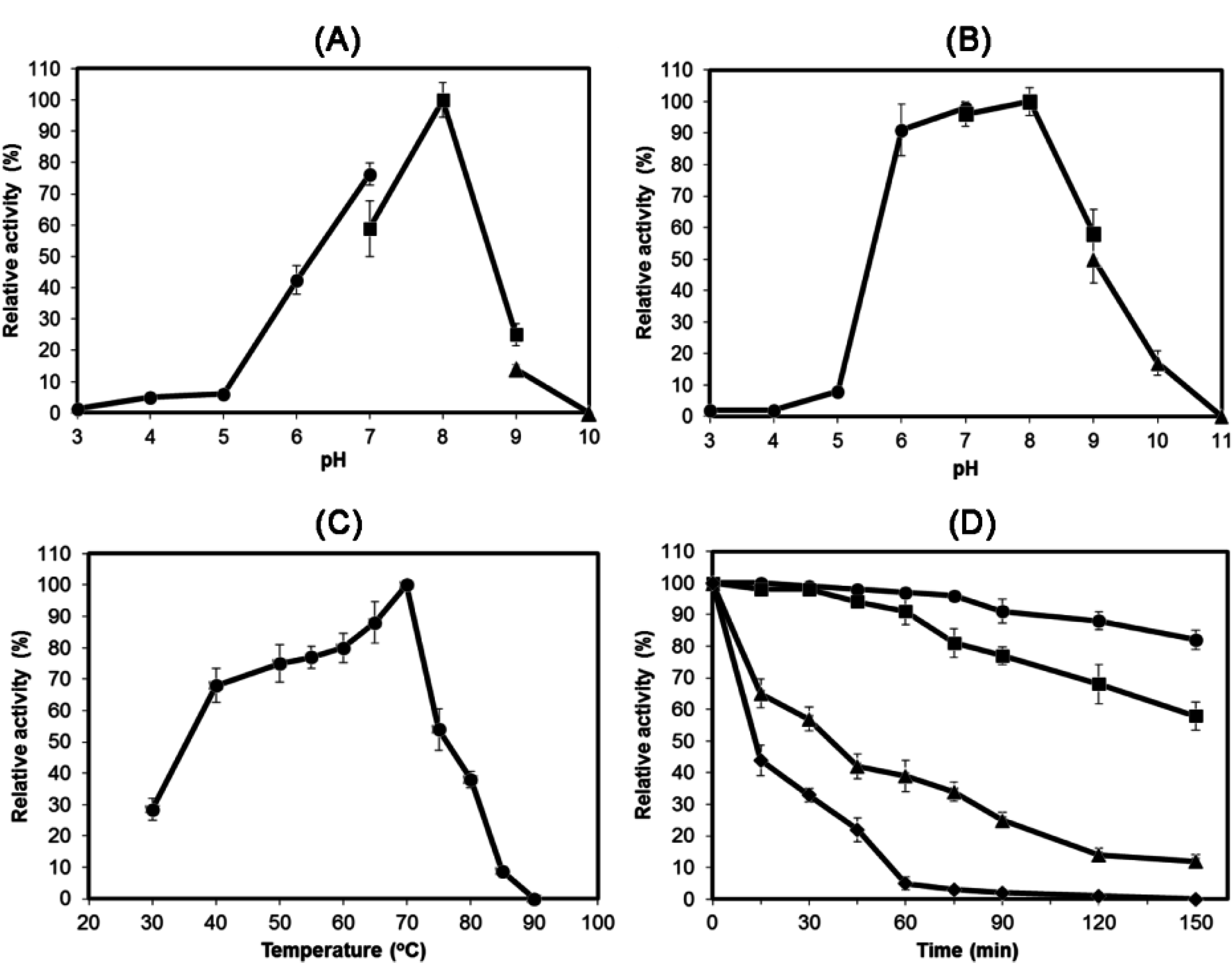
2.3. Characterization of Recombinant HydS14

| Substrates | Relative Activity (%) |
|---|---|
| pNP-acyl esters | |
| pNP-acetate (C2) | 91 ± 3.5 |
| pNP-butyrate (C4) | 100 |
| pNP-hexanoate (C6) | 26 ± 1.2 |
| pNP-caprylate (C8) | 17 ± 0.8 |
| Triglycerides | |
| Triacetin | 64 ± 2.8 |
| Tributyrin | 100 |
| Tricaproin | 53 ± 2.5 |
| Tricaprylin | 39 ± 1.8 |
| Substrates | Vmax (µmol/min/mg) | Km (mM) | Kcat (S−1) | Kcat/Km (mM−1·S−1) |
|---|---|---|---|---|
| pNP-acetate (C2) | 35.31 ± 1.19 | 0.36 ± 0.01 | 2.37 ± 0.06 | 6.59 ± 0.04 |
| pNP-butyrate (C4) | 37.07 ± 1.04 | 0.21 ± 0.02 | 2.47 ± 0.07 | 11.74 ± 0.78 |
| pNP-caprylate (C6) | 42.91 ± 0.77 | 1.84 ± 0.13 | 2.86 ± 0.05 | 1.56 ± 0.08 |
| Detergent or Solvent | Concentration (% (v/v)) | Relative Activity (%) |
|---|---|---|
| None | – | 100 |
| Tween 80 | 5 | 85 ± 3.5 |
| 10 | 73 ± 3.5 | |
| Triton X-100 | 5 | 100 |
| 10 | 68 ± 3.2 | |
| Methanol | 5 | 92 ± 4.5 |
| 10 | 87 ± 3.9 | |
| Ethanol | 5 | 86 ± 4.2 |
| 10 | 80 ± 3.8 | |
| 2-Propanol | 5 | 83 ± 4.0 |
| 10 | 75 ± 3.6 | |
| 1-Butanol | 5 | 77 ± 3.6 |
| 10 | 28 ± 1.3 | |
| Acetone | 5 | 96 ± 4.6 |
| 10 | 77 ± 3.6 | |
| DMSO | 5 | 91 ± 4.4 |
| 10 | 55 ± 2.6 |
| Metal or Inhibitor | Relative Activity (%) |
|---|---|
| None | 100 |
| Li+ | 73 ± 3.4 |
| K+ | 115 ± 5.1 |
| Rb+ | 82 ± 4.0 |
| Co2+ | 77 ± 3.6 |
| Mg2+ | 101 ± 4.9 |
| Ca2+ | 105 ± 5.0 |
| Cu2+ | 57 ± 2.4 |
| Ni2+ | 80 ± 3.9 |
| Zn2+ | 64 ± 3.1 |
| Fe2+ | 36 ± 1.7 |
| Mn2+ | 63 ± 3.0 |
| Hg2+ | 27 ± 1.2 |
| o-Phenanthroline | 91 ± 3.9 |
| p-Chloromercuribenzoic acid | 87 ± 4.2 |
| EDTA | 73 ± 3.6 |
| PMSF | 28 ± 1.3 |
| 2-Mercaptoethanol | 92 ± 4.4 |
3. Experimental Section
3.1. Bacterial Strains, Plasmids, Cultivation, and Chemicals
3.2. Construction and Screening of a Genomic Library for Esterase Activity
3.3. Cloning, Expression and Purification
3.4. SDS-PAGE and Zymography
3.5. Determination of Esterase Activity
3.6. Characterization of Recombinant HydS14
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arpigny, J.L.; Jaeger, K.E. Bacterial lipolytic enzymes: Classification and properties. Biochem. J. 1999, 343, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Handrick, R.; Reinhardt, S.; Focarete, M.T.; Scandola, M.; Adamus, G.; Kowalczuk, M.; Jendrossek, D. A new type of thermoalkalophilic hydrolase of Paucimonas lemoignei with high specificity for amorphous polyesters of short chain-length hydroxyalkanoic acids. J. Biol. Chem. 2001, 276, 36215–36224. [Google Scholar] [CrossRef] [PubMed]
- Levisson, M.; Oost, J.; Kengen, S.W.M. Characterization and structural modeling of a new type of thermostable esterase from Thermotoga maritime. FEBS J. 2007, 27, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Lee, C.H.; Oh, T.K.; Song, J.K.; Yoon, J.H. Isolation and characterization of a novel lipase from a metagenomic library of tidal flat sediments: Evidence for a new family of bacterial lipases. Appl. Environ. Microbiol. 2006, 72, 7406–7409. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Oh, K.H.; Lee, M.H.; Kang, C.H.; Oh, T.K.; Yoon, J.H. Novel cold-adapted alkaline lipase from an intertidal flat metagenome and proposal for a new family of bacterial lipases. Appl. Environ. Microbiol. 2009, 75, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Montoro-Garcia, S.; Martinez-Martinez, I.; Navarro-Fernandez, J.; Takami, H.; Garcia-Carmona, F.; Sanchez-Ferrer, A. A characterization of a novel thermostable carboxylesterase from Geobacillus kaustophilus HTA426 shows the existence of a new carboxylesterase family. J. Bacteriol. 2009, 191, 3076–3085. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Xue, Y.; Zhou, C.; Tao, J.; Li, G.; Lu, J.; Ma, Y.A. Thermostable esterase from Thermoanaerobacter tengcongensis opening up a new family of bacterial lipolytic enzymes. Biochim. Biophys. Acta 2011, 1814, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U.T. Microbial carboxyl esterases: Classification, properties and application in biocatalysis. FEMS Microbiol. Rev. 2002, 26, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Pio, T.F.; Macedo, G.A. Cutinases: Properties and industrial applications. Adv. Appl. Microbiol. 2009, 66, 77–95. [Google Scholar] [PubMed]
- Ollis, D.L.; Cheahm, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken, S.M.; Harel, M.; Remington, S.J.; Silman, I.; Schrag, J.; et al. The α/β hydrolase fold. Protein Eng. 1992, 5, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Pleiss, J.; Fischer, M.; Peiker, M.; Thiele, C.; Schmid, R.D. Lipase engineering database: Understanding and exploiting sequence-structure-function relationships. J. Mol. Catal. B-Enzym. 2000, 10, 491–508. [Google Scholar] [CrossRef]
- Akoh, C.C.; Lee, G.-C.; Liaw, Y.C.; Huang, T.H.; Shaw, J.-F. GDSL family of serine esterases/lipases. Prog. Lipid Res. 2004, 43, 534–552. [Google Scholar] [CrossRef]
- Haki, G.D.; Rakshit, S.K. Developments in industrially important thermostable enzymes: A review. Bioresour. Technol. 2003, 89, 17–34. [Google Scholar] [CrossRef]
- Sriyapai, T.; Somyoonsap, P.; Matsui, K.; Kawai, F.; Chansiri, K. Cloning of a thermostable xylanase from Actinomadura sp. S14 and its expression in Escherichia coli and Pichia pastoris. J. Biosci. Bioeng. 2011, 111, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Hong, K.S.; Malhotra, S.; Park, J.H.; Hwang, E.C.; Choi, H.K.; Kim, Y.S.; Tao, W.; Lee, S.W. A new esterase EstD2 isolated from plant rhizosphere soil metagenome. Appl. Microbiol. Biotechnol. 2010, 88, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Leiros, H.K.; de Pascale, D.; Johnson, K.A.; Blencke, H.M.; Landfald, B. Functional and structural studies of a novel cold-adapted esterase from an Arctic intertidal metagenomic library. Appl. Microbiol. Biotechnol. 2013, 97, 3965–3978. [Google Scholar] [CrossRef] [PubMed]
- Bains, J.; Kaufman, L.; Farnelln, B.; Boulanger, M.J. A product analog bound form of 3-oxoadipate-enol-lactonase (PcaD) reveals a multifunctional role for the divergent cap domain. J. Mol. Biol. 2011, 406, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Holmquist, M. α/β-Hydrolase fold enzymes: Structures, functions and mechanisms. Curr. Protein Pept. Sci. 2000, 1, 209–235. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.X.; Zhang, W.Y.; Ding, J.P.; Gao, P.J. Sequence pattern for the occurrence of N-glycosylation in proteins. J. Protein Chem. 1999, 18, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, S.B. Thermostable esterase from a thermoacidophilic archaeon: purification and characterization for enzymatic resolution of a chiral compound. Biosci. Biotechnol. Biochem. 2004, 68, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Benjamin, S.; Soccol, C.R.; Nigam, P.; Krieger, M.; Soccol, V.T. The realm of microbial lipases in biotechnology. Biotechnol. Appl. Biochem. 1999, 29, 119–131. [Google Scholar] [PubMed]
- Tirawongsaroj, P.; Sriprang, R.; Harnpicharnchai, P.; Thongaram, T.; Champreda, V.; Tanapongpipat, S.; Pootanakit, K.; Eurwilaichitr, L. Novel thermophilic and thermostable lipolytic enzymes from a Thailand hot spring metagenomic library. J. Biotechnol. 2008, 133, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Kieser, T.; Bibb, M.J.; Buttner, M.J.; Chater, K.F.; Hopwood, D.A. Practical Streptomyces Genetics; The John Innes Foundation: Norwich, UK, 2000; pp. 162–208. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Wakabayashi, K.; Nakai, R.; Aono, R.; Horikoshi, K. Purification and some properties of an alkaline xylanase from alkaliphilic Bacillus sp. strain 41 M-1. Appl. Environ. Microbiol. 1993, 59, 2311–2316. [Google Scholar] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Pinsirodom, P.; Parkin, K.L. Current Protocols in Food Analytical Chemistry; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sriyapai, P.; Kawai, F.; Siripoke, S.; Chansiri, K.; Sriyapai, T. Cloning, Expression and Characterization of a Thermostable Esterase HydS14 from Actinomadura sp. Strain S14 in Pichia pastoris. Int. J. Mol. Sci. 2015, 16, 13579-13594. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160613579
Sriyapai P, Kawai F, Siripoke S, Chansiri K, Sriyapai T. Cloning, Expression and Characterization of a Thermostable Esterase HydS14 from Actinomadura sp. Strain S14 in Pichia pastoris. International Journal of Molecular Sciences. 2015; 16(6):13579-13594. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160613579
Chicago/Turabian StyleSriyapai, Pichapak, Fusako Kawai, Somjai Siripoke, Kosum Chansiri, and Thayat Sriyapai. 2015. "Cloning, Expression and Characterization of a Thermostable Esterase HydS14 from Actinomadura sp. Strain S14 in Pichia pastoris" International Journal of Molecular Sciences 16, no. 6: 13579-13594. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160613579