
Effect of pH on the Aggregation of α-syn12 Dimer in Explicit Water by Replica-Exchange Molecular Dynamics Simulation


Abstract
:1. Introduction
2. Results and Discussion
2.1. Effect of pH on the Inter-Molecular States of (α-syn12)2
2.1.1. Free Energy Surface Obtained from PCA of Inter-Chain Side-Chain Inverse Distances


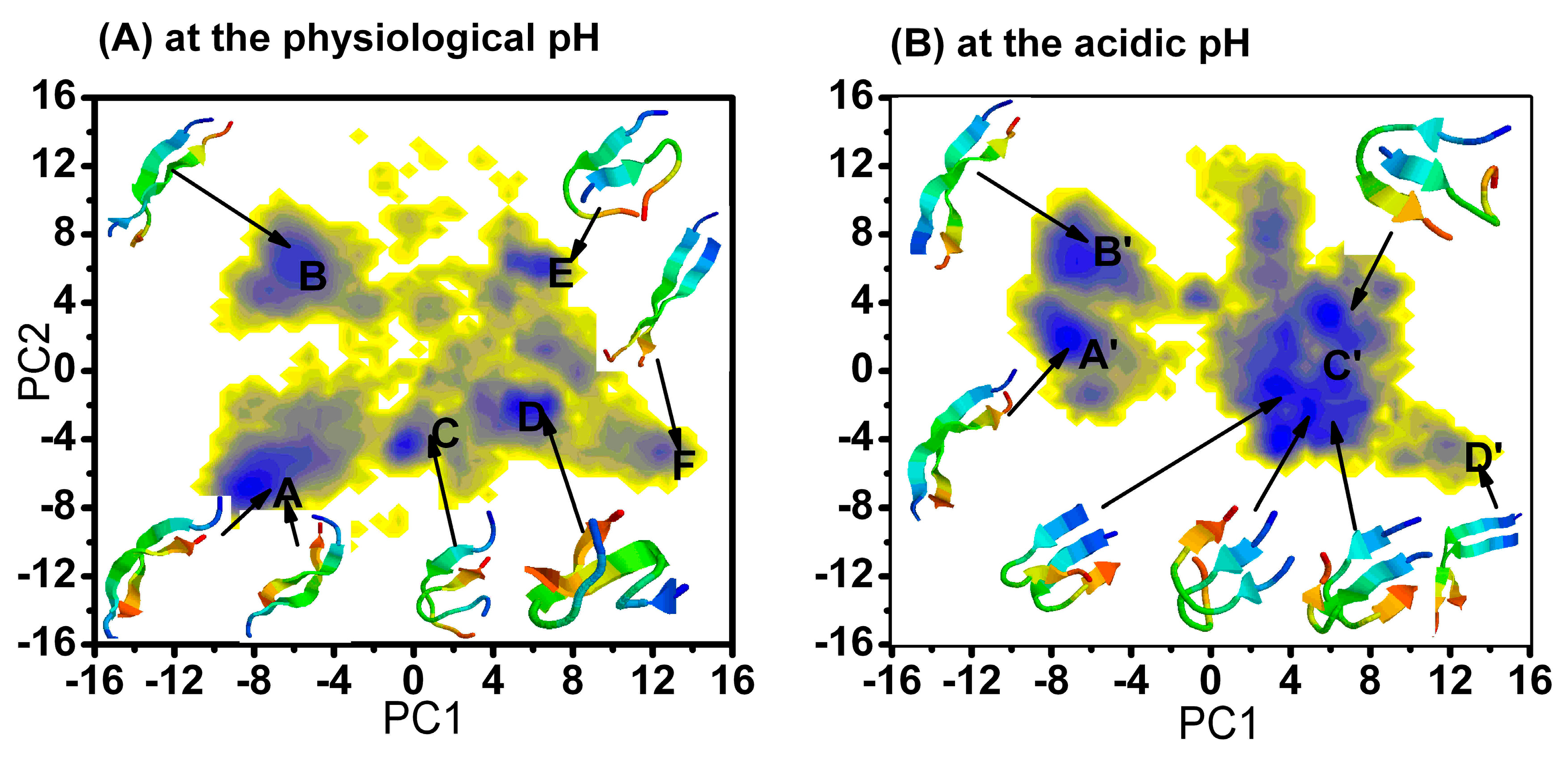{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Different Regions | The Relative Depths (kJ mol−1) of the Minima at Physiology pH |
|---|---|
| 201–500 ns | |
| A | 0 |
| B | 2.5 |
| C | 3.6 |
| D | 0.1 |
| E | 1.8 |
| F | 4.1 |
| The Different Regions | The Relative Depths (kJ mol−1) of the Minima at Acidic pH |
|---|---|
| 201–500 ns | |
| A' | 0 |
| B' | 1.1 |
| C' | −0.6 |
| D' | 6.1 |
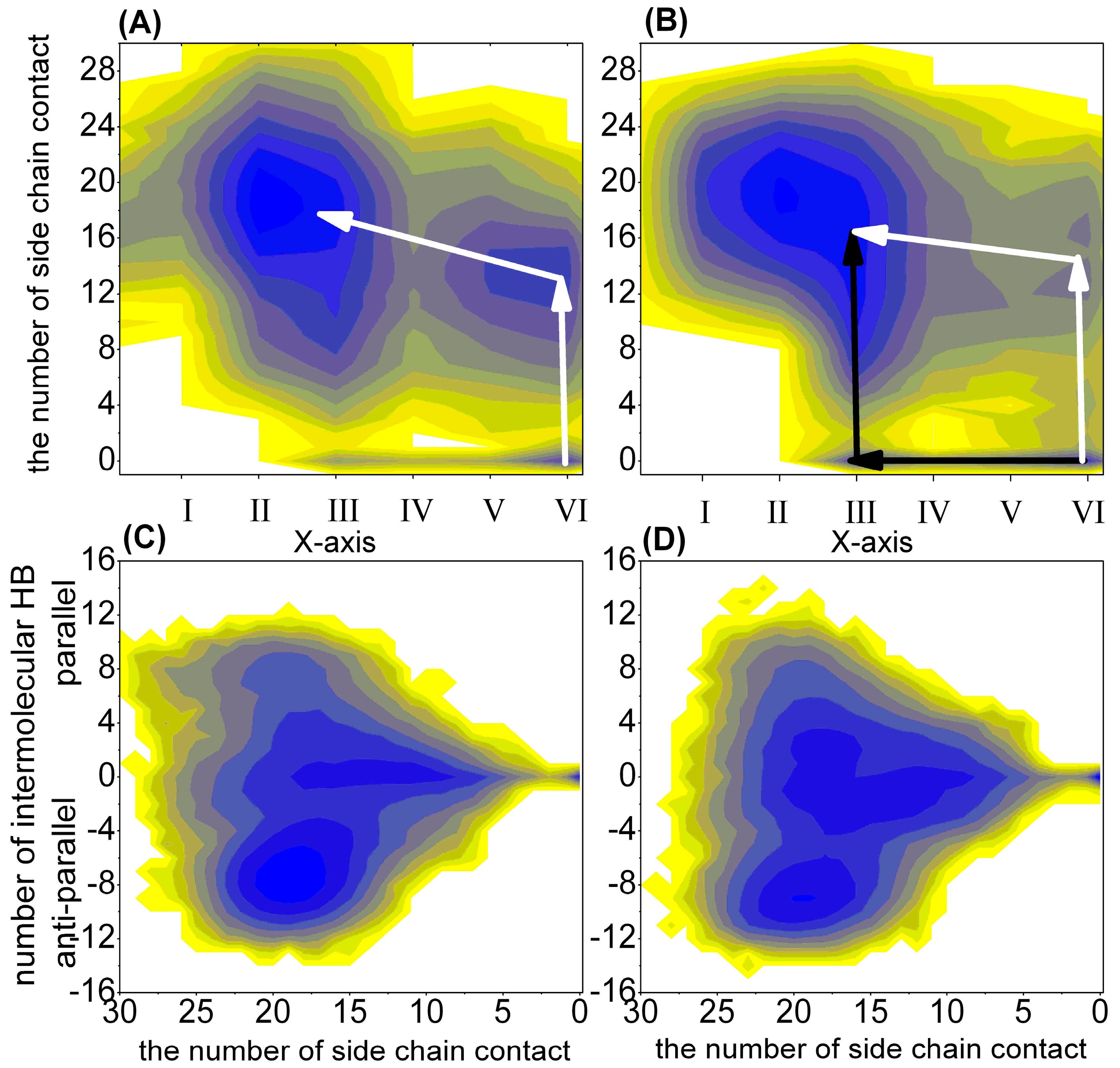
2.1.2. Free Energy Surface Based on the Other Representative Reaction Coordinates

| The Different Regions | 1–100 ns | 101–200 ns | 201–300 ns | 301–400 ns | 401–500 ns |
|---|---|---|---|---|---|
| NUMintraHB > 0, Lhelix > 0, Lsheet = 0; Cited as VI | 0.23 (0.11) | 0.04 (0) | 0 (0) | 0 (0) | 0 (0) |
| NUMintraHB > 0, Lhelix > 0, Lsheet > 0; Cited as V | 0.17 (0.04) | 0.04 (0) | 0 (0) | 0 (0) | 0 (0) |
| NUMintraHB > 0, Lhelix = 0, Lsheet = 0; Cited as IV | 0.04 (0.04) | 0.01 (0.01) | 0 (0.03) | 0 (0) | 0 (0.01) |
| NUMintraHB > 0, Lhelix = 0, Lsheet > 0; Cited as III | 0.48 (0.69) | 0.37 (0.54) | 0.32 (0.39) | 0.23 (0.41) | 0.16 (0.41) |
| NUMintraHB = 0, Lhelix > 0, Lsheet = 0 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| NUMintraHB = 0, Lhelix > 0, Lsheet > 0 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| NUMintraHB = 0, Lhelix = 0, Lsheet > 0; Cited as II | 0.07 (0.10) | 0.53 (0.40) | 0.67 (0.48) | 0.75 (0.49) | 0.82 (0.49) |
| NUMintraHB = 0, Lhelix = 0, Lsheet = 0; Cited as I | 0 (0.02) | 0 (0.04) | 0 (0.09) | 0 (0.09) | 0.01 (0.08) |
| The Different Regions | 1–100 ns | 101–200 ns | 201–300 ns | 301–400 ns | 401–500 ns |
|---|---|---|---|---|---|
| VI and NUMcon > 0 | 0.20 (0.06) | 0.04 (0) | 0 (0) | 0 (0) | 0 (0) |
| VI and NUMcon = 0 | 0.04 (0.05) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| V and NUMcon > 0 | 0.17 (0.03) | 0.04 (0) | 0 (0) | 0 (0) | 0 (0) |
| V and NUMcon = 0 | 0 (0.01) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| IV and NUMcon > 0 | 0.04 (0.04) | 0.01 (0.01) | 0 (0.03) | 0 (0) | 0 (0.01) |
| IV and NUMcon = 0 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| III and NUMcon > 0 | 0.48 (0.64) | 0.37 (0.54) | 0.32 (0.39) | 0.23 (0.41) | 0.16 (0.41) |
| III and NUMcon = 0 | 0 (0.05) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| II and NUMcon > 0 | 0.07 (0.10) | 0.53 (0.40) | 0.67 (0.48) | 0.75 (0.49) | 0.82 (0.49) |
| II and NUMcon = 0 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| I and NUMcon > 0 | 0 (0.02) | 0 (0.04) | 0 (0.09) | 0 (0.09) | 0.01 (0.08) |
| I and NUMcon = 0 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| The Different Regions | 1–100 ns | 101–200 ns | 201–300 ns | 301–400 ns | 401–500 ns |
|---|---|---|---|---|---|
| NUMinterHB = 0 | 0.18 (0.14) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| NUMinterHB > 0, 0° ≤ θ ≤ 50° | 0.28 (0.03) | 0.15 (0) | 0.10 (0) | 0.10 (0) | 0.07 (0) |
| NUMinterHB > 0, 50° ≤ θ ≤ 90° | 0.27 (0.24) | 0.14 (0.22) | 0.17 (0.25) | 0.10 (0.29) | 0.09 (0.20) |
| NUMinterHB > 0, 90° < θ < 130° | 0.15 (0.23) | 0.18 (0.20) | 0.13 (0.21) | 0.18 (0.22) | 0.12 (0.25) |
| NUMinterHB > 0, 130° ≤ θ ≤ 180° | 0.12 (0.23) | 0.53 (0.42) | 0.59 (0.41) | 0.62 (0.38) | 0.72 (0.45) |
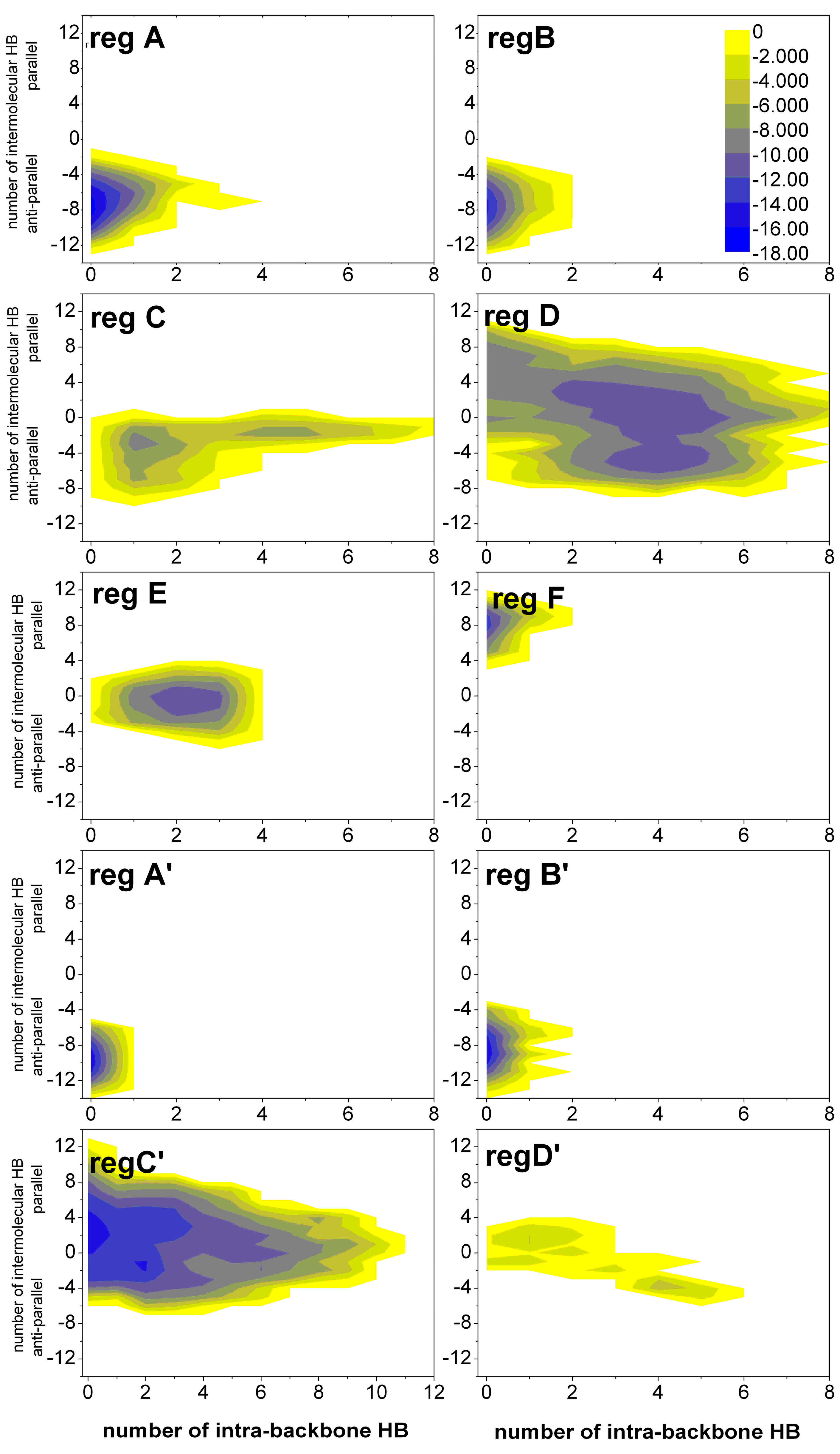
2.1.3. Free Energy Surfaces for Different States

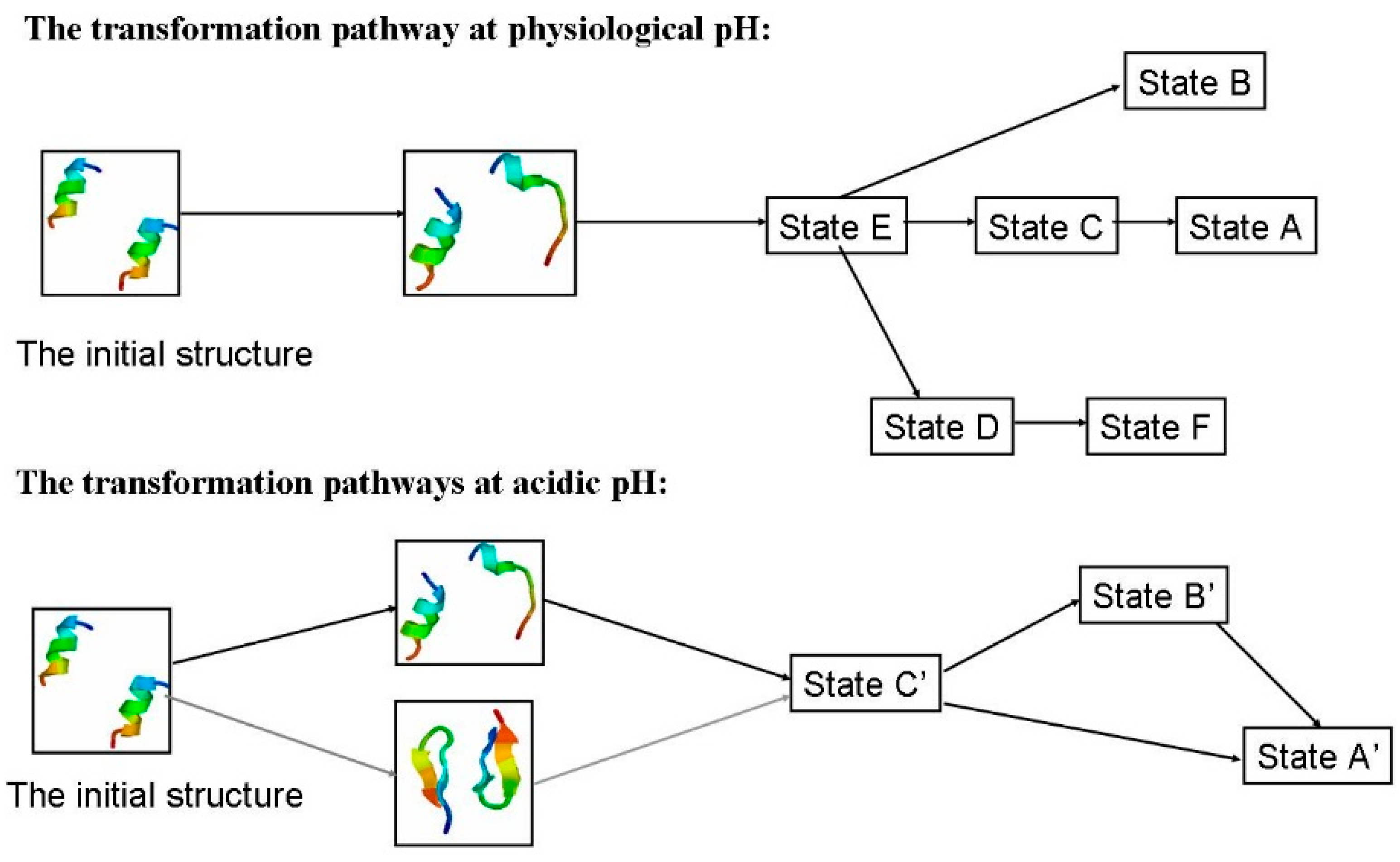
2.1.4. Possible Transformation Pathways

2.2. Effect of pH on the Intra-Molecular States of (α-syn12)2
3. Method

4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yu, J.; Lyubchenko, Y.L. Early stages for Parkinson’s development: α-Synuclein misfolding and aggregation. J. Neuroimmune Pharmacol. 2009, 4, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Gallea, J.I.; Celej, M.S. Structural insights into amyloid oligomers of the Parkinson disease-related protein α-synuclein. J. Biol. Chem. 2014, 289, 26733–26742. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.O.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.X.; Wood, N.W.; Latchman, D.S. Molecular basis of Parkinson’s disease. Neuroreport 2009, 20, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Volles, M.J.; Lee, S.J.; Rochet, J.C.; Shtilerman, M.D.; Ding, T.T.; Kessler, J.C.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar α-synuclein: Implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 2001, 40, 7812–7819. [Google Scholar] [CrossRef] [PubMed]
- Roostaee, A.; Beaudoin, S.; Staskevicius, A.; Roucou, X. Aggregation and neurotoxicity of recombinant α-synuclein aggregates initiated by dimerization. Mol. Neurodegener. 2013, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, E.; Pacifico, J.; Smith, D.P.; Hung, L.W.; Masters, C.L.; Cappai, R.; Wade, J.D.; Barnham, K.J. Dimeric structures of α-synuclein bind preferentially to lipid membranes. Biochim. Biophys. Acta 2008, 1778, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Yoshiki, Y.; Masami, M.; Hiroaki, S.; Takashi, N.; Shinya, H.; Shin-ichi, H.; Koichi, K.; Masato, H. Characterization of inhibitor-bound α-synuclein dimer: Role of α-synuclein N-terminal region in dimerization and inhibitor binding. J. Mol. Biol. 2010, 395, 445–456. [Google Scholar]
- Lorenzen, N.; Lemminger, L.; Pedersen, J.N.; Nielsen, S.B.; Otzen, D.E. The N-terminus of α-synuclein is essential for both monomeric and oligomeric interactions with membranes. FEBS Lett. 2014, 588, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.P.; Weinstock, D.S.; Narayanan, C.; Levy, R.M.; Baum, J. Structural reorganization of α-synuclein at low pH observed by NMR and REMD simulations. J. Mol. Biol. 2009, 391, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.L.; Chen, C.J.; Okumura, H.; Hu, C.K. Transformation between α-helix and β-sheet structures of one and two polyglutamine peptides in explicit water molecules by replica-exchange molecular dynamics simulations. J. Comput. Chem. 2014, 35, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Shea, J.E.; Urbanc, B. Insights into Aβ aggregation: A molecular dynamics perspective. Curr. Top. Med. Chem. 2012, 12, 2596–2610. [Google Scholar] [CrossRef] [PubMed]
- Barz, B.; Urbanc, B. Dimer formation enhances structural differences between amyloid β-protein (1–40) and (1–42): An explicit-solvent molecular dynamics study. PLoS ONE 2012, 7, e34345. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Bora, R.P.; Barman, A.; Singh, R.; Prabhakar, R. Dimerization of the full-length Alzheimer amyloid β-peptide (Aβ42) in explicit aqueous solution: A molecular dynamics study. J. Phys. Chem. 2012, 116, 4405–4416. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.H.; Ham, S. Atomic-level investigations on the amyloid-β dimerization process and its driving forces in water. Phys. Chem. Chem. Phys. 2012, 14, 1573–1575. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Srinivasa Rao, J.; Cruz, L. Spontaneous dimer states of the Aβ(21–30) decapeptide. Phys. Chem. Chem. Phys. 2014, 16, 13069–13073. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Li, M.S.; Derreumaux, P. Effects of all-atom force fields on amyloid oligomerization: Replica exchange molecular dynamics simulations of the Aβ(16–22) dimer and trimer. Phys. Chem. Chem. Phys. 2011, 13, 9778–9788. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Jewett, A.I.; Shea, J.E. Structural diversity of dimers of the Alzheimer amyloid-β(25–35) peptide and polymorphism of the resulting fibrils. Phys. Chem. Chem. Phys. 2010, 12, 3622–3629. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Luo, Y.; Ma, B.; Nussinov, R.; Wei, G. Conformational distribution and α-helix to β-sheet transition of human amylin fragment dimer. Biomacromolecules 2014, 15, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhang, Y.; Ning, L.; Jiao, P.; Liu, H.; Yao, X. Stabilities and structures of islet amyloid polypeptide (IAPP22–28) oligomers: From dimer to 16-mer. Biochim. Biophys. Acta 2014, 1840, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Eugene, C.; Laghaei, R.; Mousseau, N. Early oligomerization stages for the non-amyloid component of α-synuclein amyloid. J. Chem. Phys. 2014, 141, 135103. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cao, Z. Turn-directed α-β conformational transition of α-syn12 peptide at different pH revealed by unbiased molecular dynamics simulations. Int. J. Mol. Sci. 2013, 14, 10896–10907. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Liu, L.; Wu, P.; Wang, J. Structural and thermodynamics characters of isolated α-syn12 peptide: Long-time temperature replica-exchange molecular dynamics in aqueous solution. Acta Biochim. Biophys. Sin. 2011, 43, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Liu, L.; Wang, J. Effects of pH and temperature on the structural and thermodynamic character of α-syn12 peptide in aqueous solution. J. Biomol. Struct. Dyn. 2010, 28, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Li, M.S.; Derreumaux, P. Amyloid oligomer structure characterization from simulations: A general method. J. Chem. Phys. 2014, 140, 094105. [Google Scholar] [CrossRef] [PubMed]
- Viet, M.H.; Nguyen, P.H.; Ngo, S.T.; Li, M.S.; Derreumaux, P. Effect of the Tottori familial disease mutation (D7N) on the monomers and dimers of Aβ40 and Aβ42. ACS Chem. Neurosci. 2013, 4, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Viet, M.H.; Nguyen, P.H.; Derreumaux, P.; Li, M.S. Effect of the English familial disease mutation (H6R) on the monomers and dimers of Aβ40 and Aβ42. ACS Chem. Neurosci. 2014, 5, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Mao, A.H.; Crick, S.L.; Vitalis, A.; Chicoine, C.L.; Pappu, R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8183–8188. [Google Scholar] [CrossRef] [PubMed]
- Van der Spoel, D.; van Drunen, R.; Berendsen, H.J.C. GROningenMAchine for Chemical Simulation; Department of Biophysical Chemistry, BIOSON Research Institute, Nijenborgh 4 NL-9717 AG Groningen: Groningen, The Netherlands, 1994. [Google Scholar]
- Van Gunsteren, W.F.; Billeter, S.R.; Eising, A.A.; Hunenberger, P.H.; Krüger, P.K.; Mark, A.E.; Scott, W.R.P.; Tironi, I.G. Biomolecular Simulation: The GROMOS96 Manual and User Guide; Vdf Hochschulverlag AG and der ETH Zurich: Zurich, Switzerland, 1996. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.V.; Hermans, J. Interaction models for water in relation to protein hydration. Intermol. Forces 1981, 14, 331–342. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.; Darden, L.T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.V.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theor. Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Patriksson, A.; van der Spoel, D. A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys. 2008, 10, 2073–2077. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Nandel, F.S.; Hansmann, U.H. The Alzheimer β-amyloid (Aβ(1–39)) dimer in an implicit solvent. J. Chem. Phys. 2008, 129, 195102. [Google Scholar] [CrossRef] [PubMed]
- Heinig, M.; Frishman, D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Elstner, M.; Hermans, J. Comparison of a QM/MM force field and molecular mechanics force fields in simulations of alanine and glycine “dipeptides” (Ace–Ala–Nme and Ace–Gly–Nme) in water in relation to the problem of modeling the unfolded peptide backbone in solution. Proteins 2003, 50, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.E. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 1992, 68, 2696–2699. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Z.; Zhang, X.; Liu, L.; Zhao, L.; Li, H.; Wang, J. Effect of pH on the Aggregation of α-syn12 Dimer in Explicit Water by Replica-Exchange Molecular Dynamics Simulation. Int. J. Mol. Sci. 2015, 16, 14291-14304. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160714291
Cao Z, Zhang X, Liu L, Zhao L, Li H, Wang J. Effect of pH on the Aggregation of α-syn12 Dimer in Explicit Water by Replica-Exchange Molecular Dynamics Simulation. International Journal of Molecular Sciences. 2015; 16(7):14291-14304. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160714291
Chicago/Turabian StyleCao, Zanxia, Xiumei Zhang, Lei Liu, Liling Zhao, Haiyan Li, and Jihua Wang. 2015. "Effect of pH on the Aggregation of α-syn12 Dimer in Explicit Water by Replica-Exchange Molecular Dynamics Simulation" International Journal of Molecular Sciences 16, no. 7: 14291-14304. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160714291