
Combination of Aβ Secretion and Oxidative Stress in an Alzheimer-Like Cell Line Leads to the Over-Expression of the Nucleotide Excision Repair Proteins DDB2 and XPC

Abstract
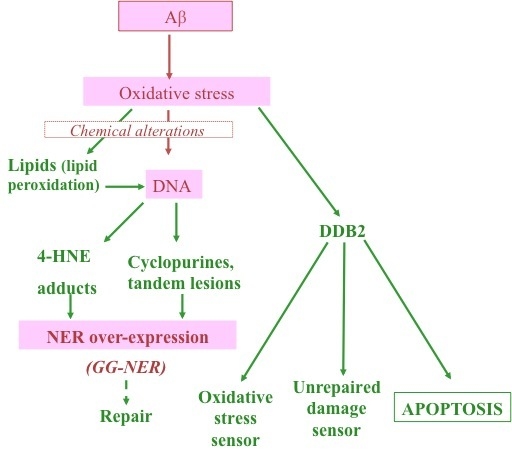
:
1. Introduction
2. Results
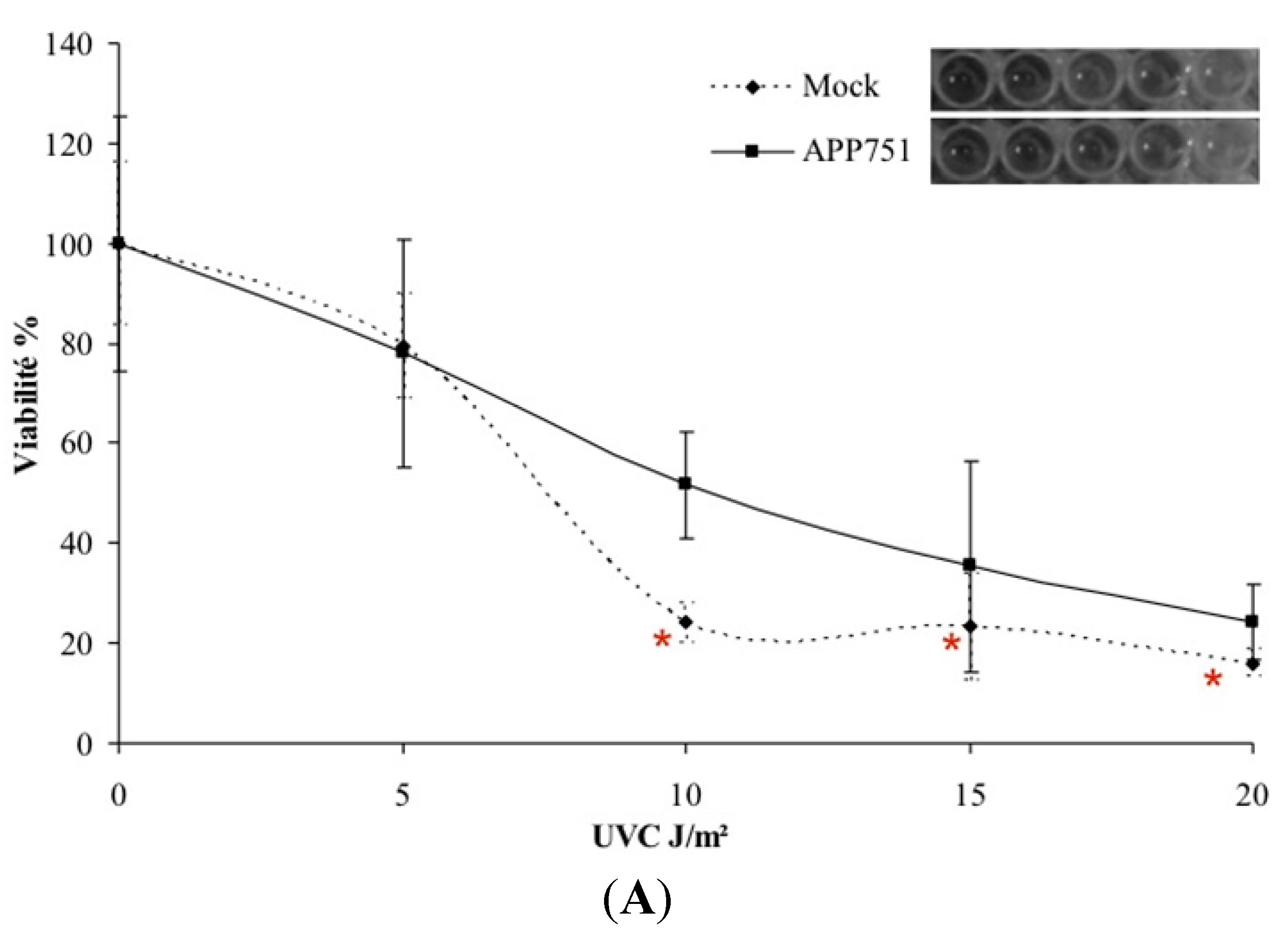
2.1. UVC-Induced Cytotoxicity Is Similar in Mock and APP751-Expressing Cells at Low Doses


2.2. Effect of Aβ, H2O2 Treatment, and UVC Radiation on the NER-Associated Gene Expression in Mock and APP751-Expressing Cells
2.2.1. Basal NER-Associated Gene Expression in the Mock and APP751-Expression Cells


2.2.2. Effect of H2O2 Treatment on NER-Associated Gene Expression in Mock and APP751 Cell Lines
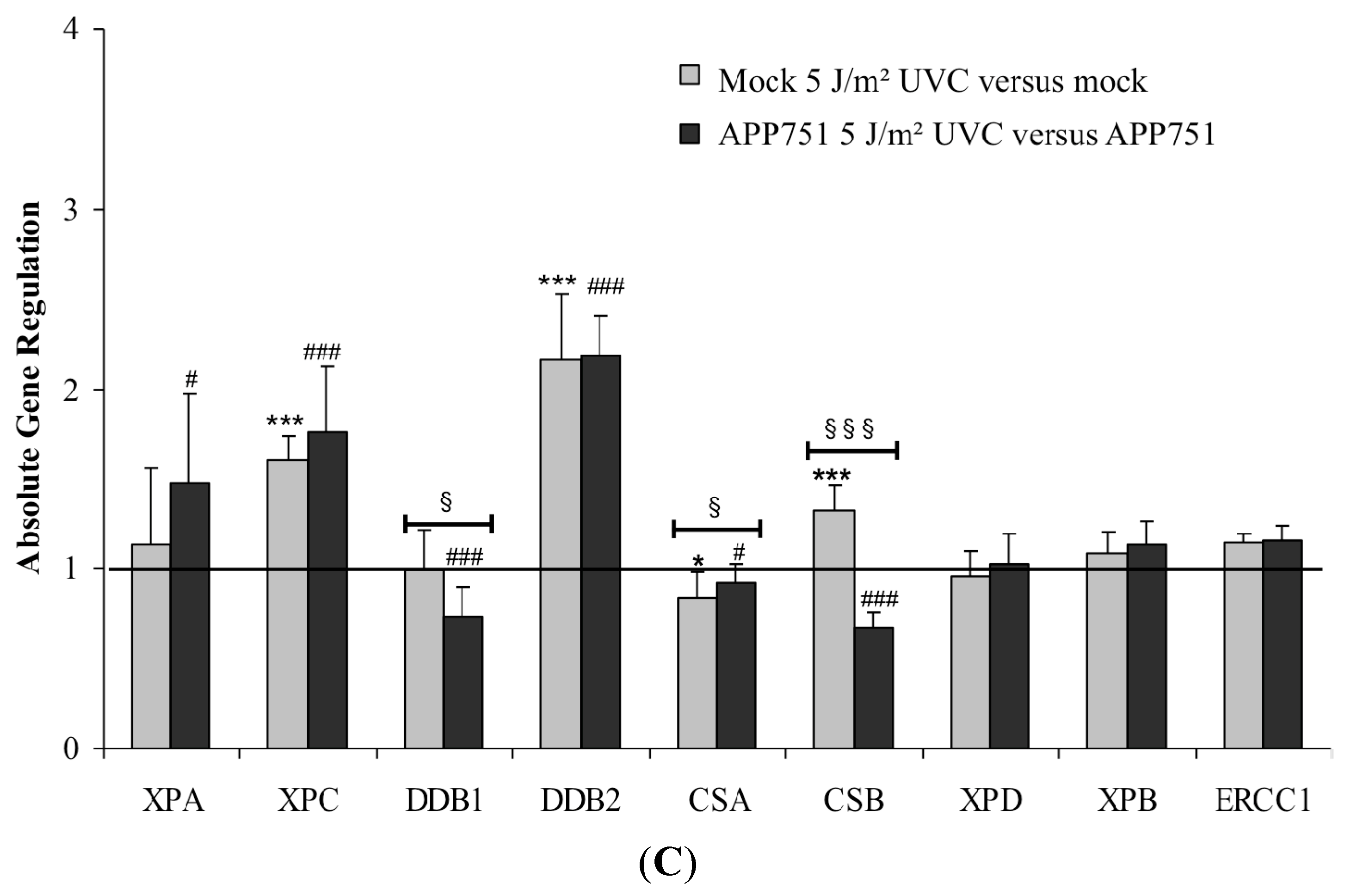
2.2.3. Effect of UVC Radiation on the NER-Associated Gene Expression in Mock and APP751-Expressing Cells
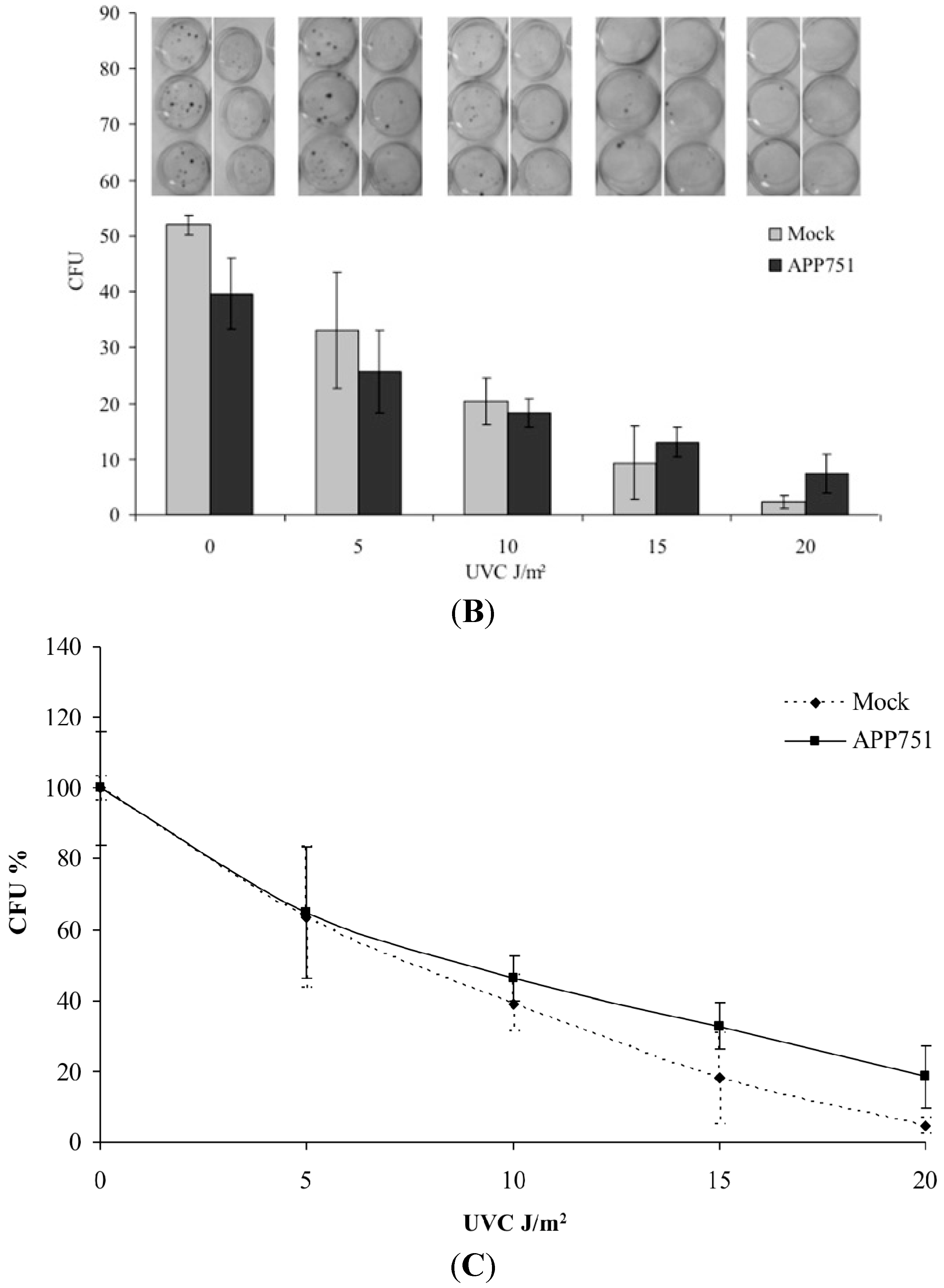
2.3. H2O2 Treatment Specifically Enhances Excision Capacity of UVC Lesions by the APP751 Cell Line

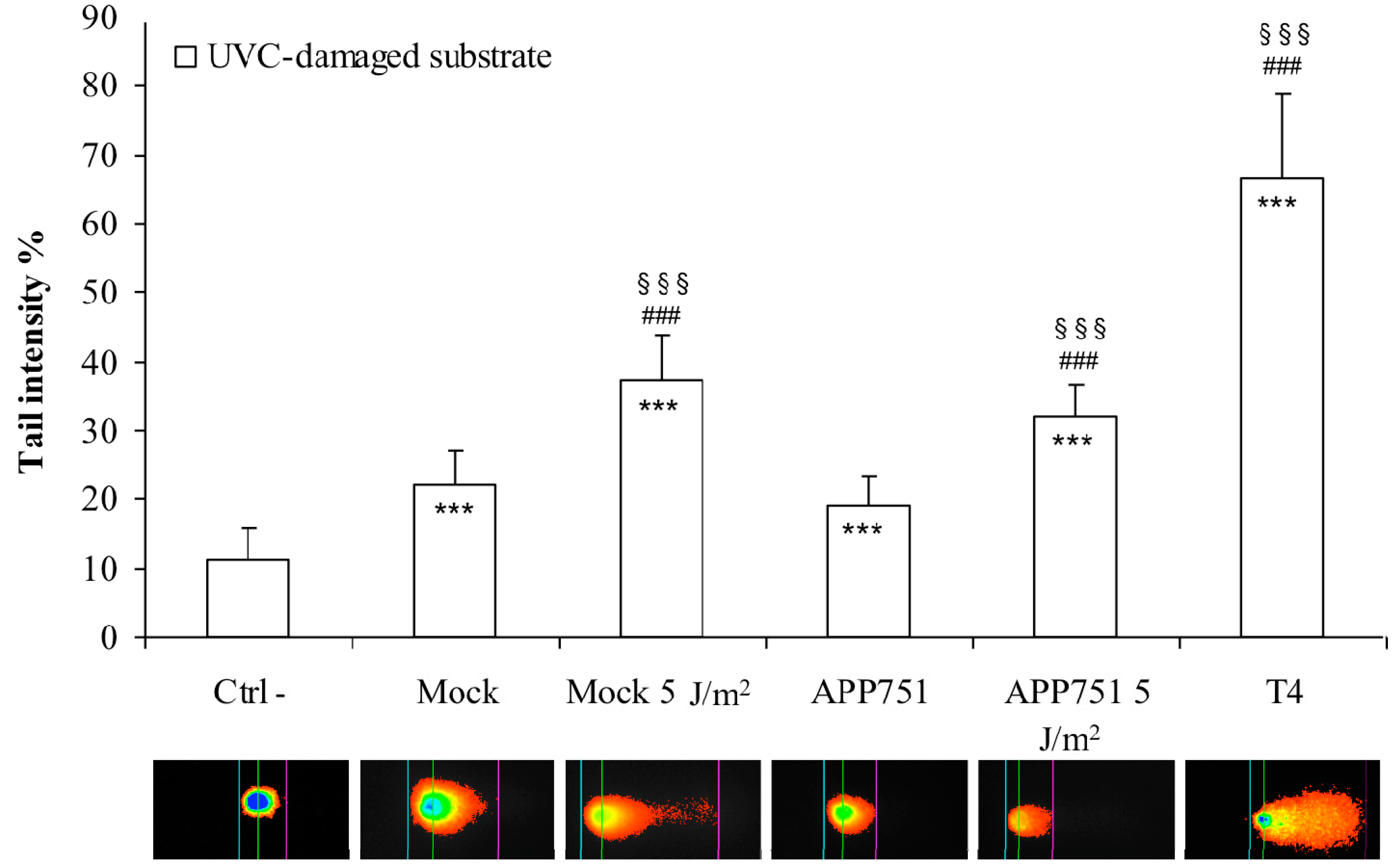
2.4. UVC Irradiation Induces a Similar Increase of Excision Capacity of UVC Lesions in Both Mock and APP751 Cell Lines

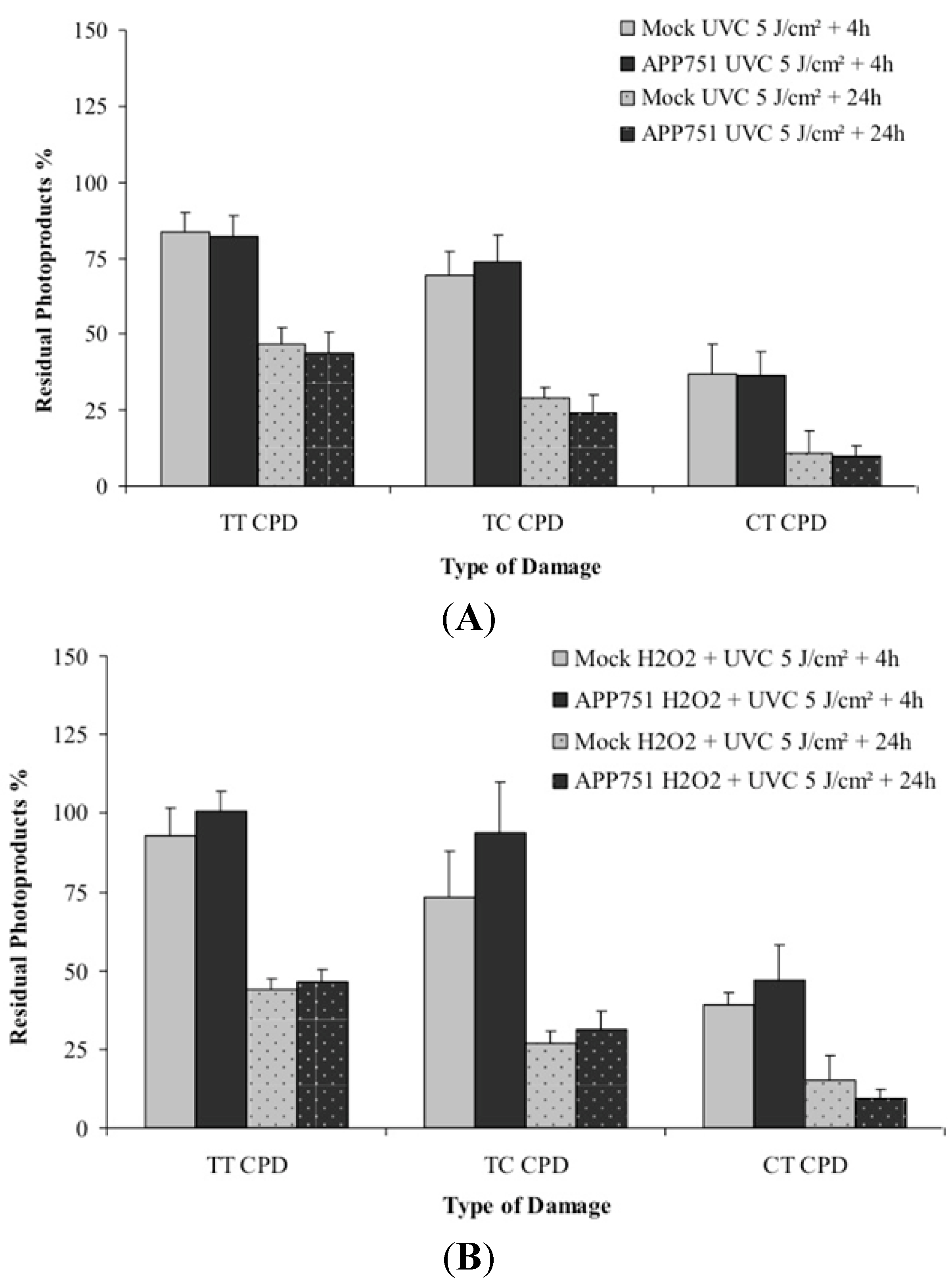
2.5. Assessments of the Photoproducts Repair Kinetics in the Mock and APP751-Expressing Cells with or without H2O2 Pre-Treatment

3. Discussion
4. Experimental Section
4.1. Cell Lines and Culture Conditions
4.2. UVC-Induced Cytotoxicity Measurements
4.3. Long-Term Toxicity of UVC
4.4. Preparation of Frozen Pellets
4.5. DNA Extraction and Digestion
4.6. Analysis of DNA Lesions (HPLC-MS/MS)
4.7. Gene Expression Analysis
4.7.1. mRNA Extraction and Quality Analysis
4.7.2. Reverse Transcription
4.7.3. Real Time Quantitative PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | GeneBank Entry | Forward Primer Sequence | Tm (°C) | Reverse Primer Sequence | Tm (°C) | Product Size (bp) |
|---|---|---|---|---|---|---|
| XPA | NM_000380.3 | gcagccccaaagataattga | 60.04 | tggcaaatcaaagtggttca | 60.09 | 183 |
| XPC | NM_004628.4 | ccatgaggacacacacaagg | 60 | tccaatgaaccacttcacca | 59.94 | 187 |
| DDB1 | NM_001923.3 | aacagagtggcgagagcatt | 60.02 | tcaatgacatgcagctcctc | 59.95 | 223 |
| DDB2 | NM_000107.2 | tcaaggacaaacccaccttc | 59.94 | gtgaccaccattcggctact | 60 | 226 |
| XPD | NM_000400.3 | gctggacatctaccccaaga | 60.07 | ccggatcacagcaatatcct | 59.92 | 166 |
| XPB | NM_000122.1 | gcggcagagattcttggtag | 59.98 | ggccccagacatagaactca | 60.07 | 235 |
| ERCC1 | NM_202001.1 | ttgtccaggtggatgtgaaa | 59.94 | gctggtttctgctcataggc | 59.98 | 151 |
| PCNA | NM_002592.2 | ggctctagcctgacaaatgc | 59.98 | gcctccaacaccttcttgag | 59.84 | 224 |
4.7.4. Validation of Stably Expressed Housekeeping Genes
4.7.5. mRNA Quantification and Statistical Analyses
4.8. DNA Repair Assays
4.9. Cell Nuclear Extracts
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mattson, M.P. Pathways towards and away from Alzheimerʼs disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, A. DNA damage, neuronal and glial cell death and neurodegeneration. Apoptosis 2010, 15, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E.; Beach, T.; Shen, Y.; Li, R.; Chang, Y. Deficiency of the Mre11 DNA repair complex in Alzheimerʼs disease brains. Brain Res. Mol. Brain Res. 2004, 128, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimerʼs disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Xiong, S.; Li, G.M.; Gu, L.; Mao, G.; Markesbery, W.R.; Lovell, M.A. Altered 8-oxoguanine glycosylase in mild cognitive impairment and late-stage Alzheimerʼs disease brain. Free Radic. Biol. Med. 2008, 45, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Weissman, L.; Jo, D.G.; Sorensen, M.M.; de Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimerʼs disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007, 35, 5545–5555. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Shamanna, R.A.; Croteau, D.L.; Bohr, V.A. Base excision DNA repair levels in mitochondrial lysates of Alzheimerʼs disease. Neurobiol. Aging 2014, 35, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Lillenes, M.S.; Stoen, M.; Gomez-Munoz, M.; Torp, R.; Gunther, C.C.; Nilsson, L.N.; Tonjum, T. Transient OGG1, APE1, PARP1 and Polbeta expression in an Alzheimerʼs disease mouse model. Mech. Ageing Dev. 2013, 134, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Bucholtz, N.; Demuth, I. DNA-repair in mild cognitive impairment and Alzheimerʼs disease. DNA Repair 2013, 12, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Leandro, G.S.; Lobo, R.R.; Oliveira, D.V.; Moriguti, J.C.; Sakamoto-Hojo, E.T. Lymphocytes of patients with Alzheimerʼs disease display different DNA damage repair kinetics and expression profiles of DNA repair and stress response genes. Int. J. Mol. Sci. 2013, 14, 12380–12400. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Misiak, M.; Wang, Y.; Ghosh, S.; Leandro, G.S.; Liu, D.; Tian, J.; Baptiste, B.A.; Cong, W.N.; Brenerman, B.M.; et al. DNA polymerase beta deficiency leads to neurodegeneration and exacerbates Alzheimer disease phenotypes. Nucleic Acids Res. 2015, 43, 943–959. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Nilsen, H.; Skorpen, F.; Otterlei, M.; Slupphaug, G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000, 476, 73–77. [Google Scholar] [CrossRef]
- Wood, R.D. Nucleotide excision repair in mammalian cells. J. Biol. Chem. 1997, 272, 23465–23468. [Google Scholar] [CrossRef] [PubMed]
- Fousteri, M.; Mullenders, L.H. Transcription-coupled nucleotide excision repair in mammalian cells: Molecular mechanisms and biological effects. Cell Res. 2008, 18, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, M.; de Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, K.H.; Patronas, N.J.; Schiffmann, R.; Brooks, B.P.; Tamura, D.; di Giovanna, J.J. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype-phenotype relationship. Neuroscience 2007, 145, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Rass, U.; Ahel, I.; West, S.C. Defective DNA repair and neurodegenerative disease. Cell 2007, 130, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Nardo, T.; Oneda, R.; Spivak, G.; Vaz, B.; Mortier, L.; Thomas, P.; Orioli, D.; Laugel, V.; Stary, A.; Hanawalt, P.C.; et al. A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc. Natl Acad. Sci. USA 2009, 106, 6209–6214. [Google Scholar] [CrossRef] [PubMed]
- Nouspikel, T. Nucleotide excision repair and neurological diseases. DNA Repair 2008, 7, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Forestier, A.; Douki, T.; Sauvaigo, S.; Rosa, V.D.; Demeilliers, C.; Rachidi, W. Alzheimerʼs disease-associated neurotoxic Peptide amyloid-beta impairs base excision repair in human neuroblastoma cells. Int. J. Mol. Sci. 2012, 13, 14766–14787. [Google Scholar] [CrossRef] [PubMed]
- Staropoli, J.F. Tumorigenesis and neurodegeneration: Two sides of the same coin? Bioessays 2008, 30, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R. The role of oxidative stress in Alzheimer disease. Arch. Neurol. 1999, 56, 1449–1452. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Nunomura, A.; Nakamura, M.; Takeda, A.; Shenk, J.C.; Aliev, G.; Smith, M.A.; Perry, G. Nucleic acid oxidation in Alzheimer disease. Free Radic. Biol. Med. 2008, 44, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimerʼs disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Dusinska, M.; Dzupinkova, Z.; Wsolova, L.; Harrington, V.; Collins, A.R. Possible involvement of XPA in repair of oxidative DNA damage deduced from analysis of damage, repair and genotype in a human population study. Mutagenesis 2006, 21, 205–211. [Google Scholar] [CrossRef] [PubMed]
- DʼErrico, M.; Parlanti, E.; Teson, M.; de Jesus, B.M.; Degan, P.; Calcagnile, A.; Jaruga, P.; Bjoras, M.; Crescenzi, M.; Pedrini, A.M.; et al. New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 2006, 25, 4305–4315. [Google Scholar] [CrossRef] [PubMed]
- Reardon, J.T.; Bessho, T.; Kung, H.C.; Bolton, P.H.; Sancar, A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: Possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc. Natl Acad. Sci. USA 1997, 94, 9463–9468. [Google Scholar] [CrossRef] [PubMed]
- Yang, W. Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair 2006, 5, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Bernardes de Jesus, B.M.; Bjoras, M.; Coin, F.; Egly, J.M. Dissection of the molecular defects caused by pathogenic mutations in the DNA repair factor XPC. Mol. Cell. Biol. 2008, 28, 7225–7235. [Google Scholar] [CrossRef] [PubMed]
- Low, G.K.; Fok, E.D.; Ting, A.P.; Hande, M.P. Oxidative damage induced genotoxic effects in human fibroblasts from Xeroderma Pigmentosum group A patients. Int. J. Biochem. Cell Biol. 2008, 40, 2583–2595. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.; Bischoff, C.; Piotrowski, J.; Bohr, V.A. Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J. Biol. Chem. 1998, 273, 33811–33816. [Google Scholar] [CrossRef] [PubMed]
- De Waard, H.; de Wit, J.; Andressoo, J.O.; van Oostrom, C.T.; Riis, B.; Weimann, A.; Poulsen, H.E.; van Steeg, H.; Hoeijmakers, J.H.; van der Horst, G.T. Different effects of CSA and CSB deficiency on sensitivity to oxidative DNA damage. Mol. Cell. Biol. 2004, 24, 7941–7948. [Google Scholar] [CrossRef] [PubMed]
- DʼErrico, M.; Parlanti, E.; Teson, M.; Degan, P.; Lemma, T.; Calcagnile, A.; Iavarone, I.; Jaruga, P.; Ropolo, M.; Pedrini, A.M.; et al. The role of CSA in the response to oxidative DNA damage in human cells. Oncogene 2007, 26, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Rolig, R.L.; McKinnon, P.J. Linking DNA damage and neurodegeneration. Trends Neurosci. 2000, 23, 417–424. [Google Scholar] [CrossRef]
- Hermon, M.; Cairns, N.; Egly, J.M.; Fery, A.; Labudova, O.; Lubec, G. Expression of DNA excision-repair-cross-complementing proteins p80 and p89 in brain of patients with down Syndrome and Alzheimerʼs disease. Neurosci. Lett. 1998, 251, 45–48. [Google Scholar] [CrossRef]
- Wang, X.W.; Vermeulen, W.; Coursen, J.D.; Gibson, M.; Lupold, S.E.; Forrester, K.; Xu, G.; Elmore, L.; Yeh, H.; Hoeijmakers, J.H.; et al. The XPB and XPD DNA helicases are components of the p53-mediated apoptosis pathway. Genes Dev. 1996, 10, 1219–1232. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Bulky DNA lesions induced by reactive oxygen species. Chem. Res. Toxicol. 2008, 21, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.H.; Polinsky, R.J.; Moshell, A.N. Evidence that lack of deoxyribonucleic acid repair causes death of neurons in xeroderma pigmentosum. Ann. Neurol. 1983, 13, 682–684. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, F.; Auvre, F.; Radicella, J.P.; Ravanat, J.L. HO• radicals induce an unexpected high proportion of tandem base lesions refractory to repair by DNA glycosylases. Proc. Natl Acad. Sci. USA 2010, 107, 5528–5533. [Google Scholar] [CrossRef] [PubMed]
- Catala, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 2009, 157, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sepe, S.; Payan-Gomez, C.; Milanese, C.; Hoeijmakers, J.H.; Mastroberardino, P.G. Nucleotide excision repair in chronic neurodegenerative diseases. DNA Repair 2013, 12, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Chu, G. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair 2002, 1, 601–616. [Google Scholar] [CrossRef]
- Bagchi, S.; Raychaudhuri, P. Damaged-DNA binding protein-2 drives apoptosis following DNA damage. Cell Div. 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, T.; Roy, N.; Kopanja, D.; Bagchi, S.; Raychaudhuri, P. DDB2 decides cell fate following DNA damage. Proc. Natl Acad. Sci. USA 2009, 106, 10690–10695. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.A.; Catao, C.S.; Martins, N.M.; Curti, C.; Bianchi, M.L.; Santos, A.C. Cisplatin-induced nephrotoxicity is associated with oxidative stress, redox state unbalance, impairment of energetic metabolism and apoptosis in rat kidney mitochondria. Arch. Toxicol. 2007, 81, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Barakat, B.M.; Wang, Q.E.; Han, C.; Milum, K.; Yin, D.T.; Zhao, Q.; Wani, G.; Arafa el, S.A.; El-Mahdy, M.A.; Wani, A.A. Overexpression of DDB2 enhances the sensitivity of human ovarian cancer cells to cisplatin by augmenting cellular apoptosis. Int. J. Cancer 2010, 127, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Stergiou, L.; Eberhard, R.; Doukoumetzidis, K.; Hengartner, M.O. NER and HR pathways act sequentially to promote UV-C-induced germ cell apoptosis in Caenorhabditis elegans. Cell Death Differ. 2011, 18, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhang, X.-P.; Liu, F. Coordination between p21 and DDB2 in the Cellular Response to UV Radiation. PLoS ONE 2013, 8, e80111. [Google Scholar] [CrossRef] [PubMed]
- Nouspikel, T.; Hanawalt, P.C. When parsimony backfires: Neglecting DNA repair may doom neurons in Alzheimerʼs disease. Bioessays 2003, 25, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Brasnjevic, I.; Hof, P.R.; Steinbusch, H.W.; Schmitz, C. Accumulation of nuclear DNA damage or neuron loss: Molecular basis for a new approach to understanding selective neuronal vulnerability in neurodegenerative diseases. DNA Repair 2008, 7, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Hamdane, M.; Loyens, A.; Gele, P.; Drobeck, H.; Begard, S.; Galas, M.C.; Delacourte, A.; Beauvillain, J.C.; Buee, L.; et al. Alkalizing drugs induce accumulation of amyloid precursor protein by-products in luminal vesicles of multivesicular bodies. J. Biol. Chem. 2007, 282, 18197–18205. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Hamdane, M.; Gompel, M.; Begard, S.; Drobecq, H.; Ghestem, A.; Grosjean, M.E.; Kostanjevecki, V.; Grognet, P.; Vanmechelen, E.; et al. Phosphorylation of amyloid precursor carboxy-terminal fragments enhances their processing by a gamma-secretase-dependent mechanism. Neurobiol. Dis. 2005, 20, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Ravanat, J.L.; Douki, T.; Duez, P.; Gremaud, E.; Herbert, K.; Hofer, T.; Lasserre, L.; Saint-Pierre, C.; Favier, A.; Cadet, J. Cellular background level of 8-oxo-7,8-dihydro-2′-deoxyguanosine: An isotope based method to evaluate artefactual oxidation of DNA during its extraction and subsequent work-up. Carcinogenesis 2002, 23, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30. [Google Scholar] [CrossRef]
- Millau, J.F.; Raffin, A.L.; Caillat, S.; Claudet, C.; Arras, G.; Ugolin, N.; Douki, T.; Ravanat, J.L.; Breton, J.; Oddos, T.; et al. A microarray to measure repair of damaged plasmids by cell lysates. Lab Chip 2008, 8, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forestier, A.; Douki, T.; De Rosa, V.; Béal, D.; Rachidi, W. Combination of Aβ Secretion and Oxidative Stress in an Alzheimer-Like Cell Line Leads to the Over-Expression of the Nucleotide Excision Repair Proteins DDB2 and XPC. Int. J. Mol. Sci. 2015, 16, 17422-17444. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160817422
Forestier A, Douki T, De Rosa V, Béal D, Rachidi W. Combination of Aβ Secretion and Oxidative Stress in an Alzheimer-Like Cell Line Leads to the Over-Expression of the Nucleotide Excision Repair Proteins DDB2 and XPC. International Journal of Molecular Sciences. 2015; 16(8):17422-17444. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160817422
Chicago/Turabian StyleForestier, Anne, Thierry Douki, Viviana De Rosa, David Béal, and Walid Rachidi. 2015. "Combination of Aβ Secretion and Oxidative Stress in an Alzheimer-Like Cell Line Leads to the Over-Expression of the Nucleotide Excision Repair Proteins DDB2 and XPC" International Journal of Molecular Sciences 16, no. 8: 17422-17444. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160817422