
Formation of Chlorotriophenoxy Radicals from Complete Series Reactions of Chlorotriophenols with H and OH Radicals

Abstract
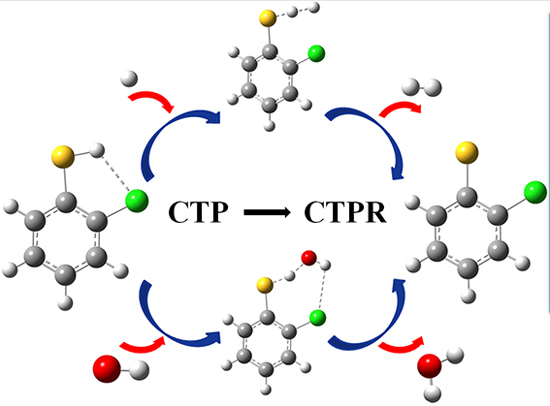
:
1. Introduction
2. Results and Discussion

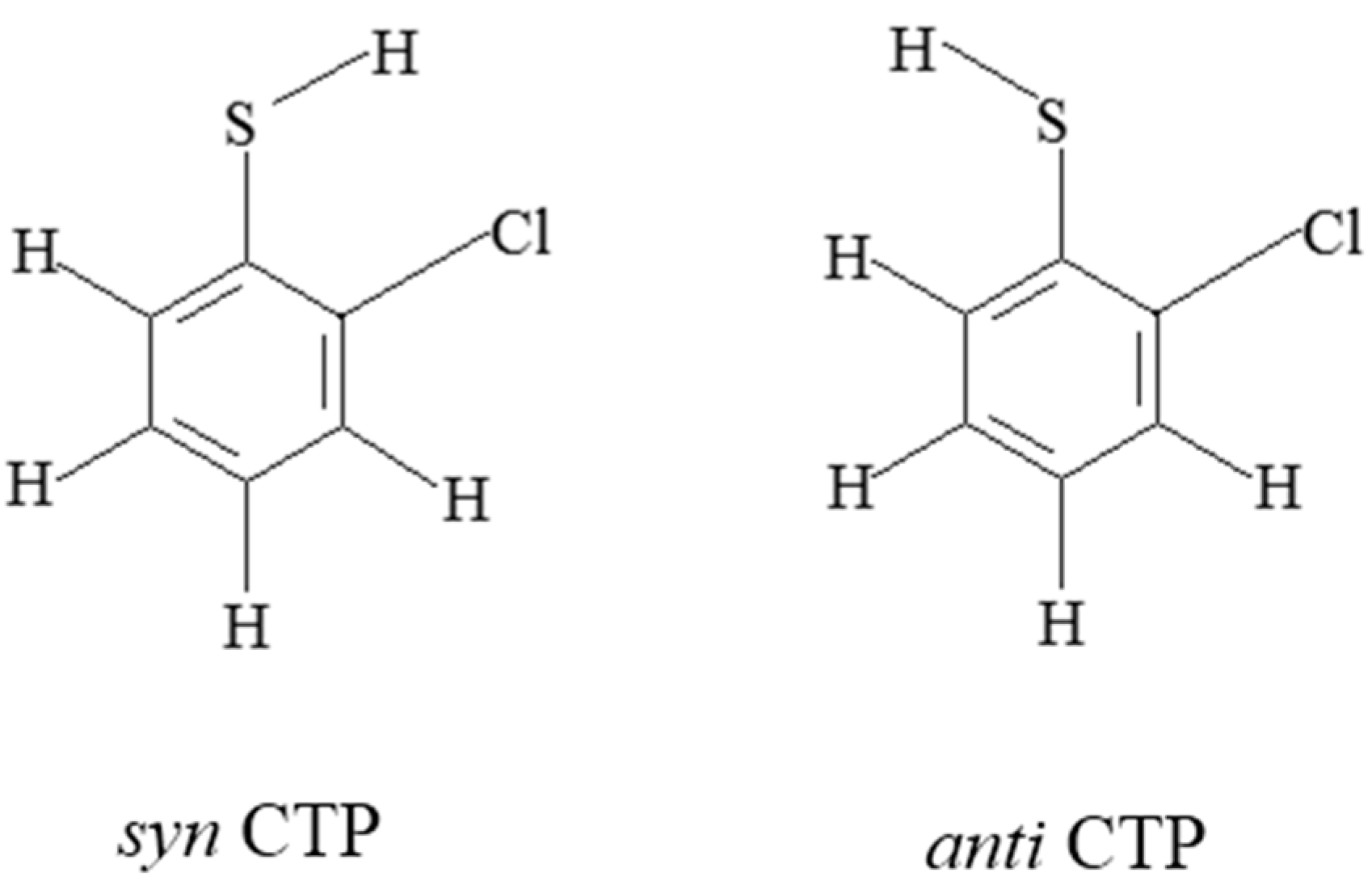
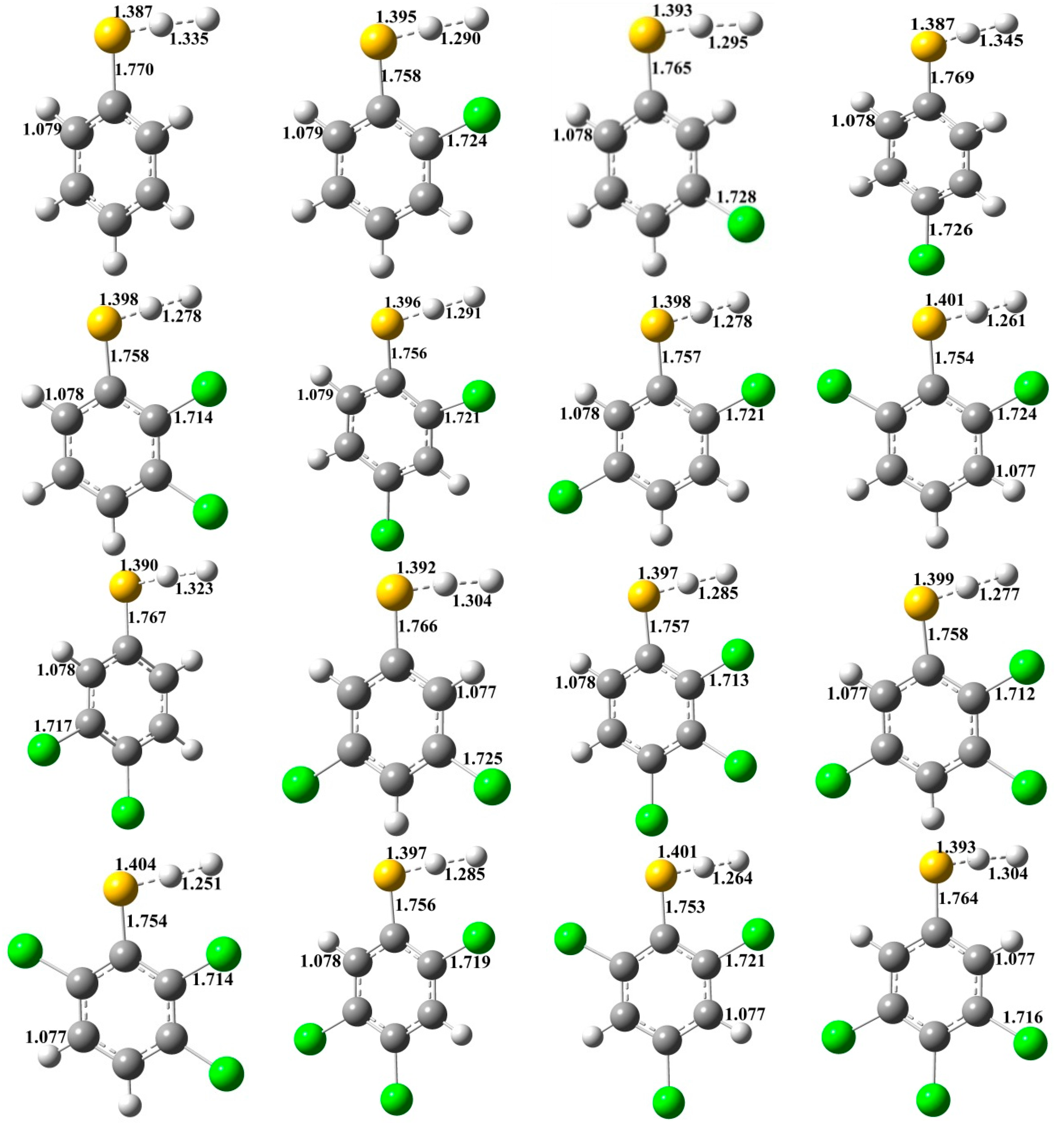
2.1. Reactions of CTPs with H



{kind=link}
{kind=link}
{kind=link}
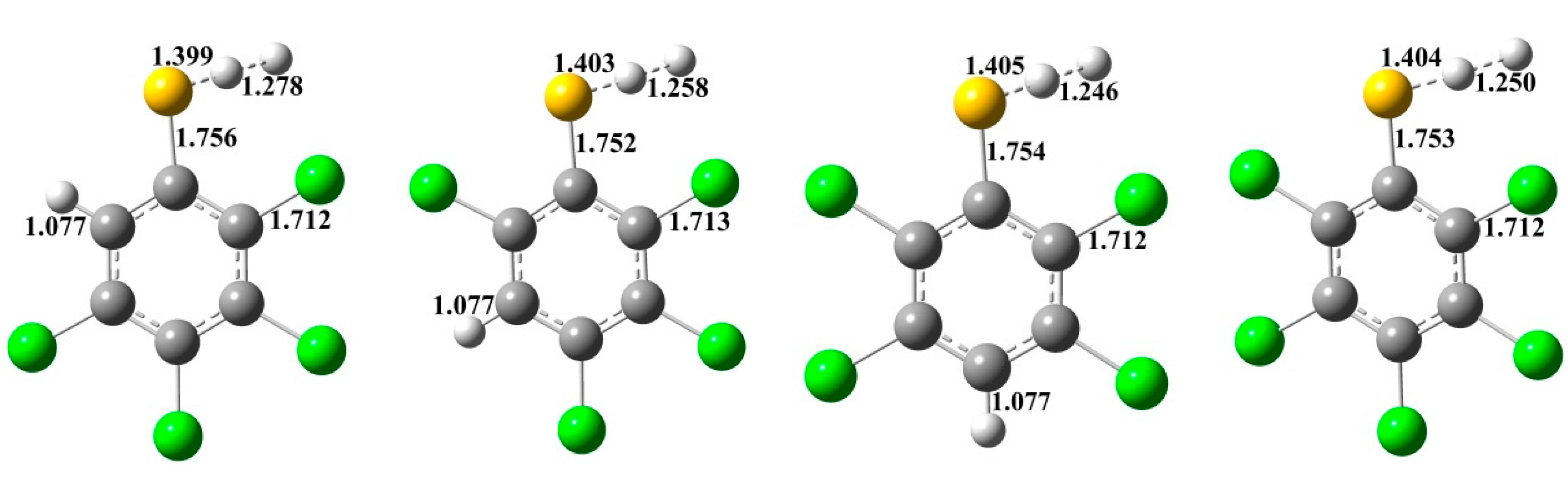{kind=link}
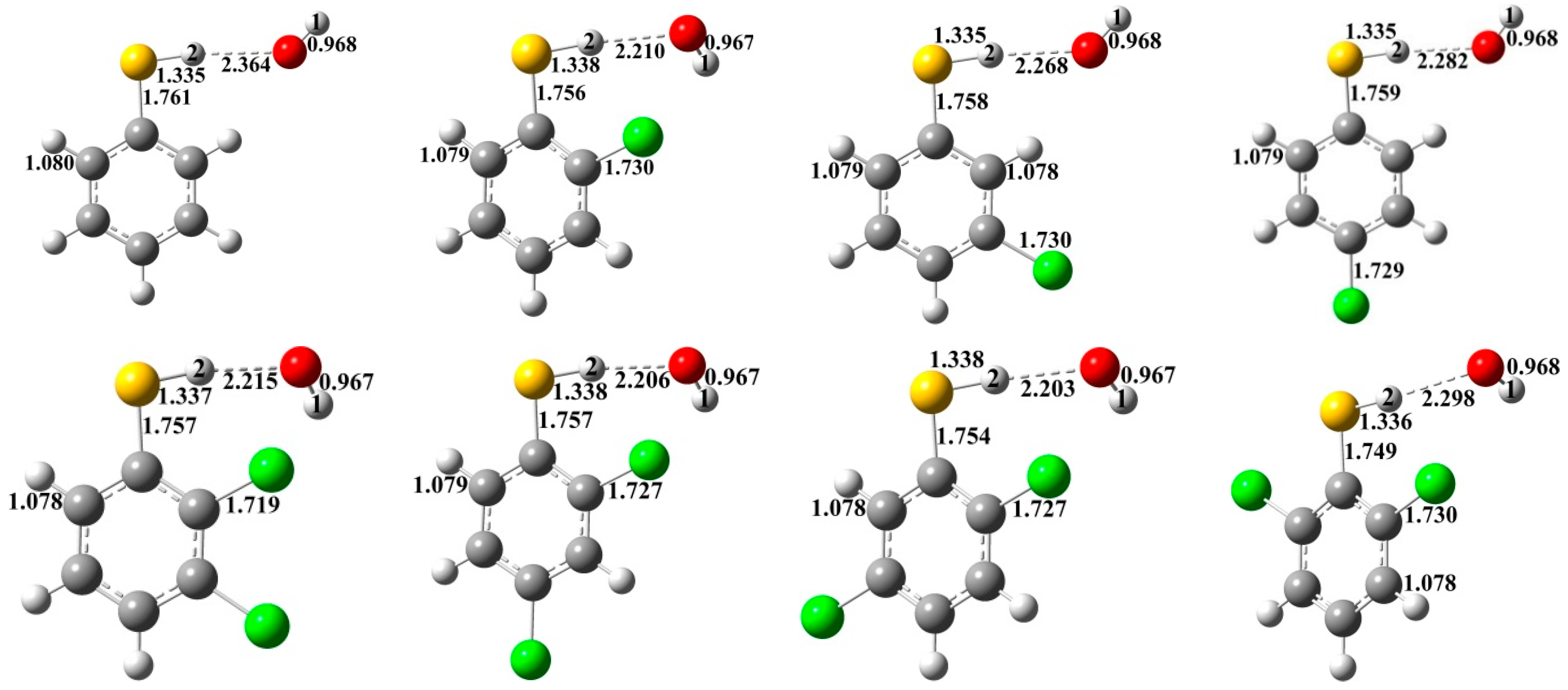{kind=link}
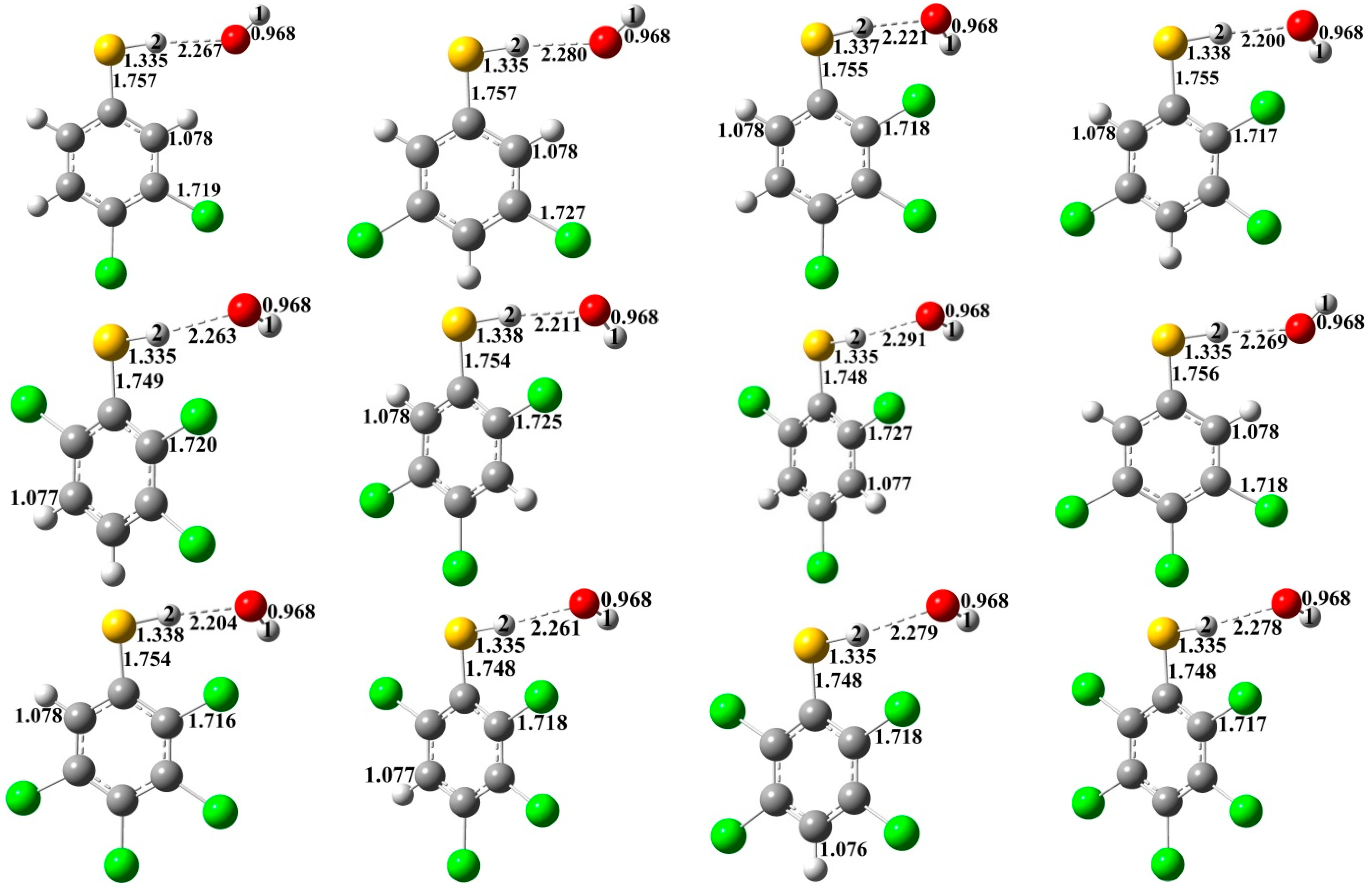{kind=link}
{kind=link}
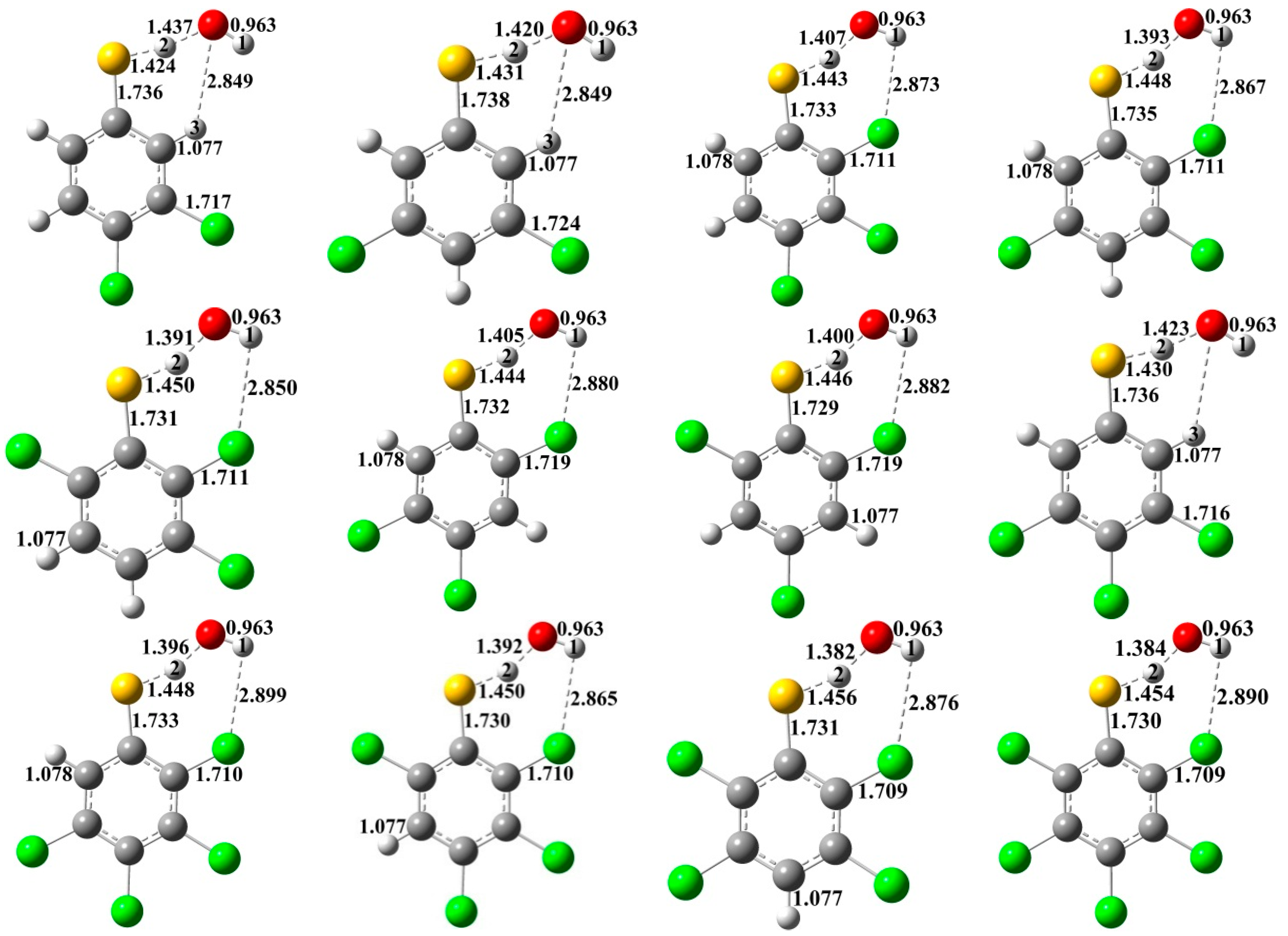{kind=link}
| CTP | ∆E | ∆H | ν | D0 (S-H) |
|---|---|---|---|---|
| Thiophenol | 2.50 | −14.16 | −789i | 86.51 |
| 2-CTP | 3.42 | −14.43 | −879i | 86.24 |
| 3-CTP | 2.76 | −13.94 | −871i | 86.73 |
| 4-CTP | 2.31 | −23.50 | −775i | 77.16 |
| 2,3-DCTP | 3.56 | −14.71 | −922i | 85.59 |
| 2,4-DCTP | 3.44 | −21.52 | −889i | 79.15 |
| 2,5-DCTP | 3.64 | −12.90 | −918i | 87.76 |
| 2,6-DCTP | 4.38 | −15.68 | −966i | 84.99 |
| 3,4-DCTP | 2.65 | −22.68 | −820i | 77.98 |
| 3,5-DCTP | 3.00 | −13.74 | −851i | 86.92 |
| 2,3,4-TCTP | 3.21 | −21.10 | −903i | 79.57 |
| 2,3,5-TCTP | 3.70 | −14.65 | −917i | 86.01 |
| 2,3,6-TCTP | 4.43 | −13.03 | −994i | 87.64 |
| 2,4,5-TCTP | 3.48 | −20.94 | −897i | 79.72 |
| 2,4,6-TCTP | 4.27 | −20.12 | −965i | 80.55 |
| 3,4,5-TCTP | 2.67 | −22.12 | −860i | 78.55 |
| 2,3,4,5-TeCTP | 3.17 | −20.57 | −921i | 80.10 |
| 2,3,4,6-TeCTP | 4.37 | −19.65 | −980i | 81.02 |
| 2,3,5,6-TeCTP | 4.52 | −16.48 | −1010i | 84.18 |
| PCTP | 4.62 | −18.98 | −1000i | 81.69 |
2.2. Reactions of CTPs with OH


| CTP | ΔEIM | ΔETS | ΔH | ν |
|---|---|---|---|---|
| Thiophenol | −1.28 | 7.03 | −27.69 | −2237i |
| 2-CTP | −0.90 | 8.67 | −27.96 | −2521i |
| 3-CTP | −1.12 | 7.64 | −27.47 | −2484i |
| 4-CTP | −1.38 | 6.99 | −37.03 | −2267i |
| 2,3-DCTP | −0.59 | 9.29 | −28.24 | −2588i |
| 2,4-DCTP | −0.73 | 8.80 | −35.05 | −2518i |
| 2,5-DCTP | −0.77 | 9.20 | −27.97 | −2633i |
| 2,6-DCTP | −0.99 | 10.27 | −29.21 | −2682i |
| 3,4-DCTP | −1.42 | 7.39 | −36.21 | −2484i |
| 3,5-DCTP | −1.48 | 8.13 | −27.27 | −2623i |
| 2,3,4-TCTP | −0.91 | 9.10 | −34.63 | −2584i |
| 2,3,5-TCTP | −0.64 | 9.48 | −28.18 | −2683i |
| 2,3,6-TCTP | −0.85 | 10.48 | −26.56 | −2753i |
| 2,4,5-TCTP | −0.78 | 8.98 | −34.47 | −2594i |
| 2,4,6-TCTP | −1.00 | 9.95 | −33.65 | −2680i |
| 3,4,5-TCTP | −1.65 | 7.66 | −35.65 | −2594i |
| 2,3,4,5-TeCTP | −1.00 | 9.29 | −34.10 | −2680i |
| 2,3,4,6-TeCTP | −1.04 | 10.18 | −33.18 | −2732i |
| 2,3,5,6-TeCTP | −1.40 | 10.85 | −30.01 | −2792i |
| PCTP | −1.71 | 10.55 | −32.51 | −2796i |


2.3. Rate Constant Calculations
| Reactions | Arrhenius Formulas |
|---|---|
| Thiophenol + H → C6H5O + H2 | k(T) = (2.43 × 10−1) exp (−1877/T) |
| 2-CTP + H → 2-CTPR + H2 | k(T) = (9.86 × 10−12) exp (−2069/T) |
| 3-CTP + H → 3-CTPR + H2 | k(T) = (1.45 × 10−11) exp (−1903/T) |
| 4-CTP + H → 4-CTPR + H2 | k(T) = (2.23 × 10−11) exp (−1753/T) |
| 2,3-DCTP + H → 2,3-DCTPR + H2 | k(T) = (2.43 × 10−11) exp (−2189/T) |
| 2,4-DCTP + H → 2,4-DCTPR + H2 | k(T) = (2.84 × 10−11) exp (−2229/T) |
| 2,5-DCTP + H → 2,5-DCTPR + H2 | k(T) = (1.12 × 10−11) exp (−2105/T) |
| 2,6-DCTP + H → 2,6-DCTPR + H2 | k(T) = (4.12 × 10−11) exp (−2707/T) |
| 3,4-DCTP + H → 3,4-DCTPR + H2 | k(T) = (4.02 × 10−11) exp (−1513/T) |
| 3,5-DCTP + H → 3,5-DCTPR + H2 | k(T) = (3.42 × 10−11) exp (−1566/T) |
| 2,3,4-TCTP + H → 2,3,4-TCTPR + H2 | k(T) = (3.42 × 10−11) exp (−1566/T) |
| 2,3,5-TCTP + H → 2,3,5-TCTPR + H2 | k(T) = (1.67 × 10−11) exp (−1866/T) |
| 2,3,6-TCTP + H → 2,3,6-TCTPR + H2 | k(T) = (1.47 × 10−11) exp (−2365/T) |
| 2,4,5-TCTP + H → 2,4,5-TCTPR + H2 | k(T) = (2.24 × 10−11) exp (−2689/T) |
| 2,4,6-TCTP + H → 2,4,6-TCTPR + H2 | k(T) = (1.43 × 10−11) exp (−2155/T) |
| 3,4,5-TCTP + H → 3,4,5-TCTPR + H2 | k(T) = (2.03 × 10−11) exp (−2476/T) |
| 2,3,4,5-TeCTP + H → 2,3,4,5-TeCTPR + H2 | k(T) = (2.41 × 10−11) exp (−1817/T) |
| 2,3,4,6-TeCTP + H → 2,3,4,6-TeCTPR + H2 | k(T) = (1.72 × 10−11) exp (−2710/T) |
| 2,3,5,6-TeCTP + H → 2,3,5,6-TeCTPR + H2 | k(T) = (4.70 × 10−11) exp (−2893/T) |
| PCTP + H → PCTPR + H2 | k(T) = (3.07 × 10−11) exp (−2865/T) |
| Reactions | Arrhenius Formulas |
|---|---|
| Thiophenol + OH → C6H5O + H2O | k(T) = (1.25 × 10−11) exp (−5356/T) |
| 2-CTP + OH → 2-CTPR + H2O | k(T) = (2.00 × 10−13) exp (−6304/T) |
| 3-CTP + OH → 3-CTPR + H2O | k(T) = (3.66 × 10−12) exp (−5632/T) |
| 4-CTP + OH → 4-CTPR + H2O | k(T) = (1.40 × 10−11) exp (−5025/T) |
| 2,3-DCTP + OH → 2,3-DCTPR + H2O | k(T) = (1.13 × 10−12) exp (−5934/T) |
| 2,4-DCTP + OH → 2,4-DCTPR + H2O | k(T) = (6.96 × 10−14) exp (−5861/T) |
| 2,5-DCTP + OH → 2,5-DCTPR + H2O | k(T) = (9.41 × 1013) exp (−6074/T) |
| 2,6-DCTP + OH → 2,6-DCTPR + H2O | k(T) = (9.90 × 10−14) exp (−6661/T) |
| 3,4-DCTP + OH → 3,4-DCTPR + H2O | k(T) = (7.20 × 10−12) exp (−5460/T) |
| 3,5-DCTP + OH → 3,5-DCTPR + H2O | k(T) = (1.99 × 10−12) exp (−5706/T) |
| 2,3,4-TCTP + OH → 2,3,4-TCTPR + H2O | k(T) = (1.32 × 10−12) exp (−6059 /T) |
| 2,3,5-TCTP + OH → 2,3,5-TCTPR + H2O | k(T) = (1.32 × 10−12) exp (−6839/T) |
| 2,3,6-TCTP + OH → 2,3,6-TCTPR + H2O | k(T) = (3.89 × 10−12) exp (−7354/T) |
| 2,4,5-TCTP + OH → 2,4,5-TCTPR + H2O | k(T) = (1.17 × 10−12) exp (−6143/T) |
| 2,4,6-TCTP + OH → 2,4,6-TCTPR + H2O | k(T) = (1.47 × 10−13) exp (−7076/T) |
| 3,4,5-TCTP + OH → 3,4,5-TCTPR + H2O | k(T) = (9.80 × 10−12) exp (−5620/T) |
| 2,3,4,5-TeCTP + OH → 2,3,4,5-TeCTPR + H2O | k(T) = (1.86 × 10−13) exp (−6366/T) |
| 2,3,4,6-TeCTP + OH → 2,3,4,6-TeCTPR + H2O | k(T) = (6.18 × 10-13) exp (−7325/T) |
| 2,3,5,6-TeCTP + OH → 2,3,5,6-TeCTPR + H2O | k(T) = (2.33 × 10−13) exp (−7239/T) |
| PCTP + OH → PCTPR + H2O | k(T) = (1.09 × 10−13) exp (−7015/T) |
3. Experimental Section
3.1. Density Functional Theory
3.2. Kinetic Calculation
3.3. Accuracy Verification
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, Y.; Zeng, X.L.; Chen, H.J.; Wang, H.J. Thermodynamic properties and relative stability of polychlorinated thianthrenes by density functional theory. J. Chem. Eng. Data 2007, 52, 1442–1448. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, X.L.; Chen, H.J.; Wang, H.J. Calculation of thermodynamic properties of polychlorinated phenoxathiins. J. Chem. Eng. Data 2008, 53, 513–519. [Google Scholar] [CrossRef]
- Chen, S.D.; Liu, H.X.; Wang, Z.Y. Study of structural and thermodynamic properties for polychlorinated dibenzothiophenes by density functional theory. J. Chem. Eng. Data 2007, 52, 1195–1202. [Google Scholar] [CrossRef]
- Nakai, S.; Kishita, S.; Nomura, Y.; Hosomi, M. Polychlorinated dibenzothiophenes in Japanese environmental samples and their photodegradability and dioxin-like endocrine-disruption potential. Chemosphere 2007, 67, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Sinkkonen, S. New types of persistent halogenated compounds. Springer 2000, 3, 289–314. [Google Scholar]
- Puzyn, T.; Rostkowski, P.; Swieczkowski, A.; Jeudrusiak, A.; Falandysz, J. Prediction of environmental partition coefficients and the Henry’s law constants for 135 congeners of chlorodibenzothiophene. Chemosphere 2006, 62, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Kopponen, P.; Sinkkonen, S.; Poso, A.; Gynther, J.; Karenlampi, S. Sulfur analogues of polychlorinated dibenzo-p-dioxins, dibenzofurans and diphenyl ethers as inducers of CYP1A1 in mouse hepatoma cellculture and structure activity relationships. Environ. Toxicol. Chem. 1994, 13, 1543–1548. [Google Scholar] [CrossRef]
- Weber, R.; Hagenmaler, H.; Schrenk, D. Elimination kinetics and toxicity of 2,3,7,8-tetrachlorothianthren, a thio analogue of 2,3,7,8-TCDD. Chemosphere 1998, 36, 2635–2641. [Google Scholar] [CrossRef]
- Nakai, S.; Nomura, Y.; Hosomi, M.; Espino, M.P. Detection of polychlorinated dibenzothiophenes (PCDTs) in environmental samples and investigation of their photodegradability and dioxin-like endocrine disruption potency. J. Environ. Chem. 2004, 14, 835–844. [Google Scholar] [CrossRef]
- Sinkkonen, S.; Paasivirta, J.; Lahtipera, M. Chlorinated and methylated dibenzothiophenes in sediment samples from a river contaminated by organochlorine wastes. J. Soils Sediments 2001, 1, 1–6. [Google Scholar] [CrossRef]
- Sinkkonen, S. Sources and environmental fate of PCDTs. Toxicol. Environ. Chem. 1998, 66, 105–112. [Google Scholar] [CrossRef]
- Sinkkonen, S. PCDTs in the environment. Chemosphere 1997, 34, 2585–2594. [Google Scholar] [CrossRef]
- Buser, H.R.; Rappe, C. Determination of polychlorodibenzothiophenes, the sulfur analogues of polychlorodibenzofurans, using various gas chromatographic/mass spectrometric techniques. Anal. Chem. 1991, 63, 1210–1217. [Google Scholar] [CrossRef]
- Sinkkonen, S.; Vattulainen, A.; Aittola, J.P.; Paasivirta, J.; Tarhanen, J.; Lahtipera, M. Mental reclamation produces sulfur analogues of toxic dioxins and furans. Chemosphere 1994, 28, 1279–1288. [Google Scholar] [CrossRef]
- Czerwinski, J. Pathways of polychlorinated dibenzothiophenes (PCDTs) in the environment. Arch. Environ. Prot. 2008, 34, 169–181. [Google Scholar]
- Buser, H.R. Identification and sources of dioxin-like compounds: I. Polychlorodibenzothiophenes and polychlorothianthrenes, the sulfur analogues of the polychlorodibenzofurans and polychlorodibenzodioxins. Chemosphere 1992, 25, 45–48. [Google Scholar] [CrossRef]
- Khachatryan, L.; Asatryan, R.; Dellinger, B. Development of expanded and core kinetic models for the gas phase formation of dioxins from chlorinated phenols. Chemosphere 2003, 52, 695–708. [Google Scholar] [CrossRef]
- Khachatryan, L.; Asatryan, R.; Dellinger, B. An elementary reaction kinetic model of the gas-phase formation of polychlorinated dibenzofurans from chlorinated phenols. J. Phys. Chem. A 2004, 108, 9567–9572. [Google Scholar] [CrossRef]
- Altarawneh, M.; Dlugogorski, B.Z.; Kennedy, E.M.; Mackie, J.C. Quantum chemical investigation of formation of polychlorodibenzo-p-dioxins and dibenzofurans from oxidation and pyrolysis of 2-chlorophenol. J. Phys. Chem. A 2007, 111, 2563–2573. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.Y.; Mulholland, J.A.; Kim, D.H.; Takeuchi, M. Homologue and isomer patterns of polychlorinated dibenzo-p-dioxins and dibenzofurans from phenol precursors: Comparison with municipal waste incinerator data. Environ. Sci. Technol. 2005, 39, 4398–4406. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.Y.; Mulholland, J.A.; Oh, J.E. Prediction of polychlorinated dibenzofuran congener distribution from gas-phase phenol condensation pathways. Chemosphere 2004, 55, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, J.A.; Akki, U.; Yang, Y.; Ryu, J.Y. Temperature dependence of PCDD/F isomer distributions from chlorophenol precursors. Chemosphere 2001, 42, 719–727. [Google Scholar] [CrossRef]
- Parette, R.; Pearson, W.N. 2,4,6,8-Tetrachlorodibenzothiophene in the Newark Bay Estuary: The likely source and reaction pathways. Chemosphere 2014, 111, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Dar, T.; Altarawneh, M.; Dlugogorski, B.Z. Theoretical study in the dimerization of 2-chlorothiophenol/2-chlorothiophenoxy: Precursors to PCDT/TA. Organohalogen Compd. 2012, 74, 657–660. [Google Scholar]
- Dar, T.; Altarawneh, M.; Dlugogorski, B.Z. Quantum chemical study on formation of PCDT/TA from 2-chlorothiophenol precursor. Environ. Sci. Technol. 2013, 47, 11040–11047. [Google Scholar] [CrossRef] [PubMed]
- Benz, T.; Hagenmaier, H.; Lindig, C.; She, J. Occurrence of the sulphur analogue of octachlorodibenzo-p-dioxin in the environment and investigations on its potential source. Fresenius′ J. Anal. Chem. 1992, 344, 286–291. [Google Scholar] [CrossRef]
- Navarro, R.; Bierbrauer, K.; Mijangos, C.; Goiti, E.; Reinecke, H. Modification of poly(vinyl chloride) with new aromatic thiol compounds Synthesis and characterization. Polym. Degrad. Stab. 2008, 93, 585–591. [Google Scholar] [CrossRef]
- Shi, J.Q.; Cheng, J.; Wanga, F.Y.; Flamm, A.; Wang, Z.Y.; Yang, X.; Gao, S.X. Acute toxicity and n-octanol/water partition coefficients of substituted thiophenols: Determination and QSAR analysis. Ecotoxicol. Environ. Saf. 2012, 78, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Yamada, K.; Takemoto, I.; Mizutani, T.; Saeki, K. Inhibition of human cytochrome P450 2E1 by halogenated anilines, phenols, and thiophenols. Biol. Pharm. Bull. 2005, 28, 1221–1223. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.S.; Dellinger, B. Mechanisms of dioxin formation from the high-temperature pyrolysis of 2-chlorophenol. Environ. Sci. Technol. 2003, 37, 1325–1330. [Google Scholar] [CrossRef]
- Evans, C.S.; Dellinger, B. Mechanisms of dioxin formation from the high-temperature oxidation of 2-chlorophenol. Environ. Sci. Technol. 2005, 39, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Z.; Li, S.Q.; Qu, X.H.; Shi, X.Y.; Wang, W.W. A Quantum mechanical study on the formation of PCDD/Fs from 2-chlorophenol as precursor. Environ. Sci. Technol. 2008, 42, 7301–7308. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.H.; Wang, H.; Zhang, Q.Z.; Shi, X.Y.; Xu, F.; Wang, W.X. Mechanistic and kinetic studies on the homogeneous gas-phase formation of PCDD/Fs from 2,4,5-trichlorophenol. Environ. Sci. Technol. 2009, 43, 4068–4075. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Z.; Qu, X.H.; Xu, F.; Shi, X.Y.; Wang, W.X. Mechanism and thermal rate constants for the complete series reactions of chlorophenols with H. Environ. Sci. Technol. 2009, 43, 4105–4112. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wang, H.; Zhang, Q.Z.; Zhang, R.X.; Qu, X.H.; Wang, W.X. Kinetic properties for the complete series reactions of chlorophenols with OH radicals—Relevance for dioxin formation. Environ. Sci. Technol. 2010, 44, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Khachatryan, L.; Burcat, A.; Dellinger, B. An elementary reaction-kinetic model for the gas-phase formation of 1,3,6,8- and 1,3,7,9-tetrachlorinated dibenzo-p-dioxins from 2,4,6-trichlorophenol. Combust. Flame 2003, 132, 406–421. [Google Scholar] [CrossRef]
- Larsen, N.W.; Schulz, L. Internal rotation and structure of thiophenol and 4-fluorothiophenol studied by microwave spectroscopy and quantum chemistry. J. Mol. Struct. 2009, 920, 30–39. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Hybrid meta density functional theory methods for therochemistry, thermochemical kinetics, and noncovalent interactions: The MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Baldridge, M.S.; Gordon, R.; Steckler, R.; Truhlar, D.G. Ab initio reaction paths and direct dynamics calculations. J. Phys. Chem. 1989, 93, 5107–5119. [Google Scholar] [CrossRef]
- Gonzalez-Lafont, A.; Truong, T.N.; Truhlar, D.G. Interpolated variational transition-state theory: Practical methods for estimating variational transition-state properties and tunneling contributions to chemical reaction rates from electronic structure calculations. J. Chem. Phys. 1991, 95, 8875–8894. [Google Scholar] [CrossRef]
- Garrett, B.C.; Truhlar, D.G. Generalized transition state theory Classical mechanical theory and applications to collinear reactions of hydrogen molecules. J. Phys. Chem. 1979, 83, 1052–1079. [Google Scholar] [CrossRef]
- Fernandez-Ramos, A.; Ellingson, B.A.; Garret, B.C.; Truhlar, D.G. Variational transition state theory with multidimensional tunneling. In Reviews in Computational Chemistry; Lipkowitz, K.B., Cundari, T.R., Eds.; Wiley-VCH: Hoboken, NJ, USA, 2007. [Google Scholar]
- Corchado, J.C.; Chuang, Y.Y.; Fast, P.L.; Villa, J.; Hu, W.P.; Liu, Y.P.; Lynch, G.C.; Nguyen, K.A.; Jackels, C.F.; Melissas, V.S.; et al. POLYRATE Version 9.7; University of Minnesota: Minneapolis, MN, USA, 2007. [Google Scholar]
- Larson, N.W.; Nicolaisen, F.M. Far-infrared gas spectra of phenol, 4-fluorophenol, thiophenol and some deuterated species: Barrier to internal rotation. J. Mol. Struct. 1974, 22, 29–43. [Google Scholar] [CrossRef]
- Nyquist, R.A.; Evans, J.C. The vibrational spectra of p-chlorobenzenethiol. Spectrochim. Acta 1961, 17, 795–801. [Google Scholar] [CrossRef]
- Luo, Y.R. Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, F.; Shi, X.; Zhang, Q.; Wang, W. Formation of Chlorotriophenoxy Radicals from Complete Series Reactions of Chlorotriophenols with H and OH Radicals. Int. J. Mol. Sci. 2015, 16, 18714-18731. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160818714
Xu F, Shi X, Zhang Q, Wang W. Formation of Chlorotriophenoxy Radicals from Complete Series Reactions of Chlorotriophenols with H and OH Radicals. International Journal of Molecular Sciences. 2015; 16(8):18714-18731. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160818714
Chicago/Turabian StyleXu, Fei, Xiangli Shi, Qingzhu Zhang, and Wenxing Wang. 2015. "Formation of Chlorotriophenoxy Radicals from Complete Series Reactions of Chlorotriophenols with H and OH Radicals" International Journal of Molecular Sciences 16, no. 8: 18714-18731. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160818714