
Quantitative Structure-Activity Relationships Study on the Rate Constants of Polychlorinated Dibenzo-p-Dioxins with OH Radical

Abstract
:1. Introduction
2. Results and Discussion
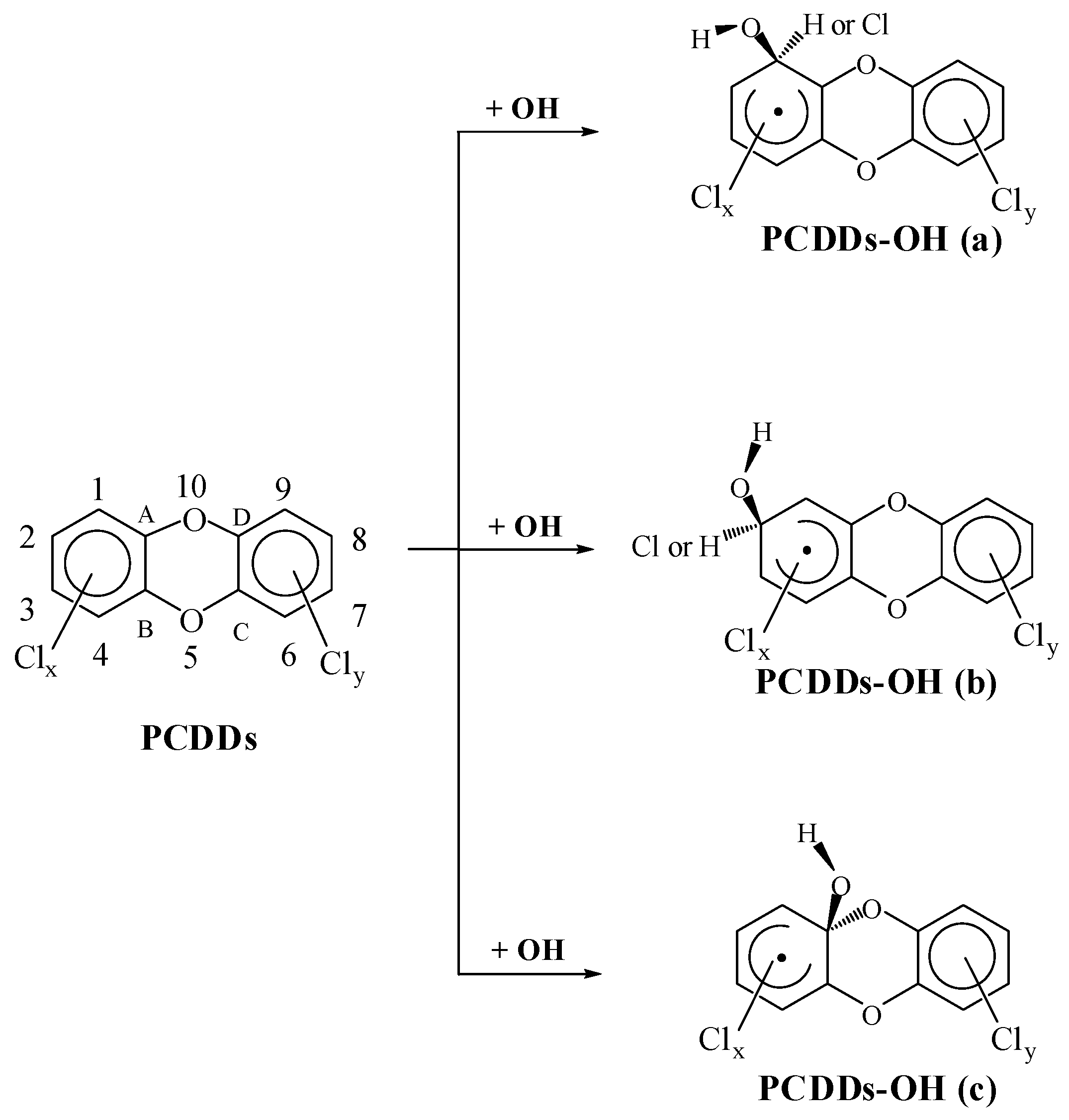
2.1. Reactions with OH Radicals

2.2. Relation between Rate Constants and the Configuration Parameters
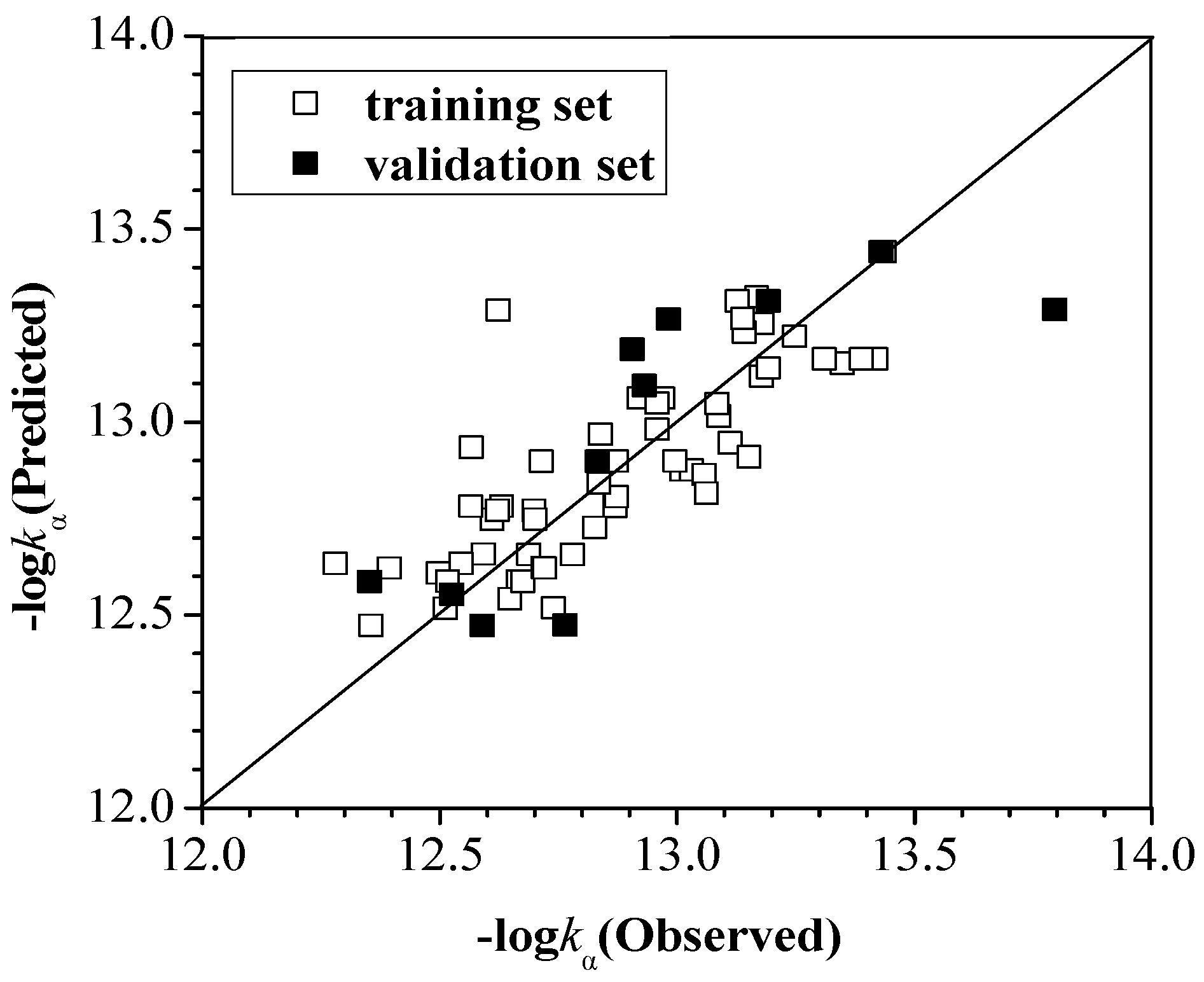
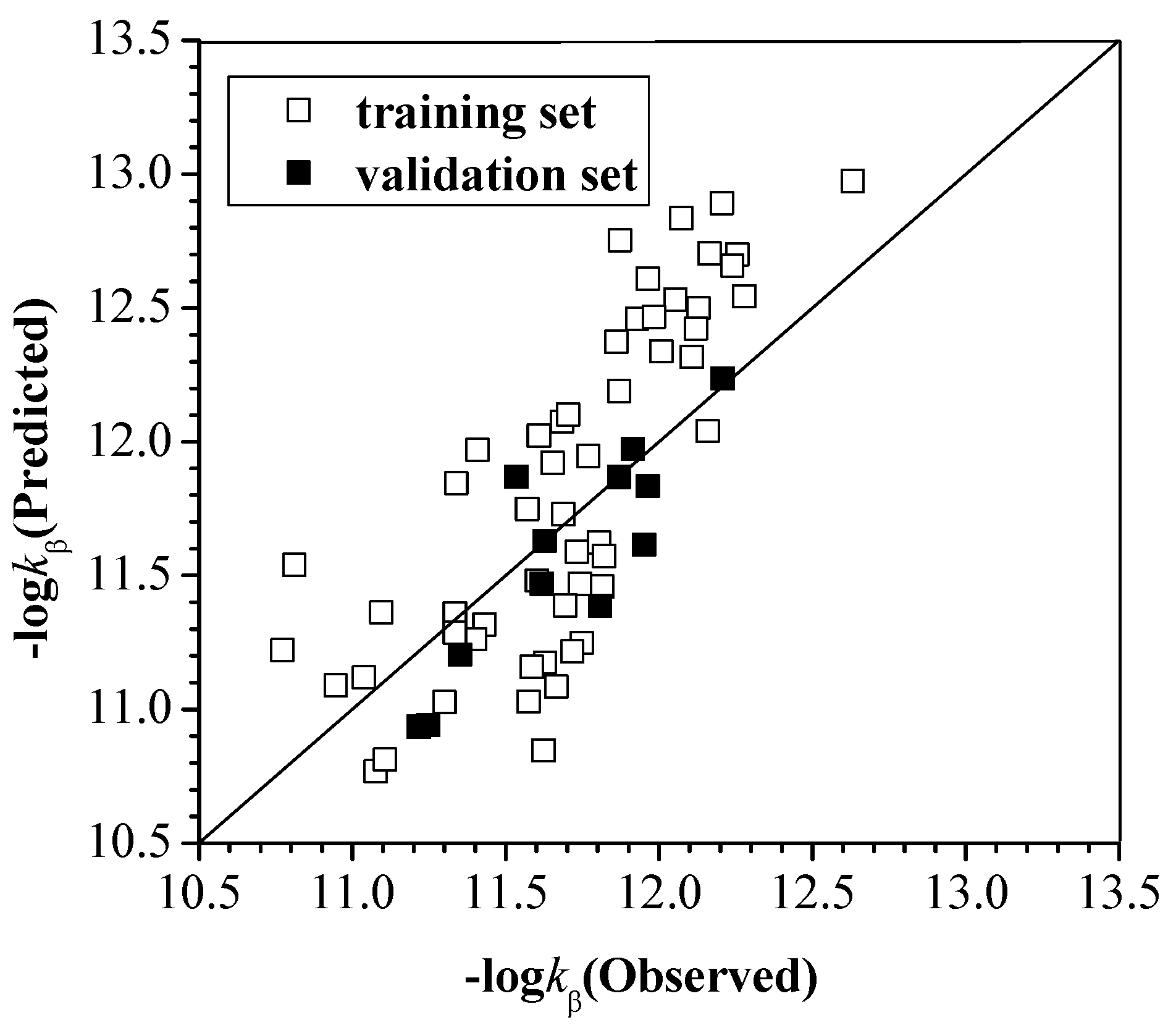
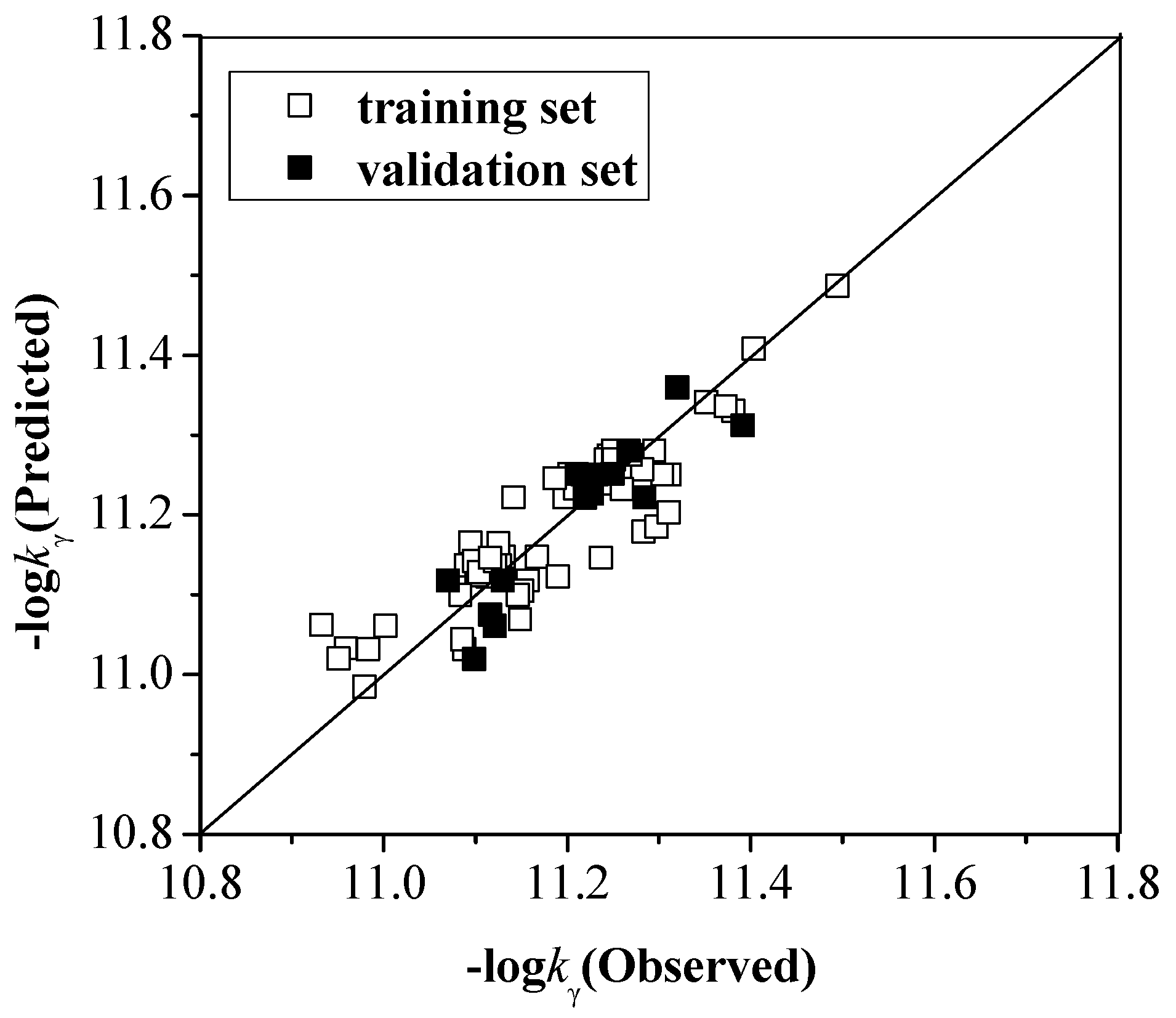
2.2.1. kα, kβ and kγ with the Configuration Parameters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | kα | kβ | kγ | Nα | Nβ | N1,9 | N2,8 | No | Nm | Np | N | EHOMO | ELUMO | ΔEHOMO-LUMO | D |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MCDDs | |||||||||||||||
| 1 | 1.35 × 10−13 | 9.15 × 10−12 | 7.40 × 10−12 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | −0.247 | −0.003 | 0.245 | 1.63 |
| 2 | 3.18 × 10−13 | 1.69 × 10−11 | 1.12 × 10−11 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | −0.246 | −0.003 | 0.243 | 2.07 |
| DCDDs | |||||||||||||||
| 1,2 | 8.66 × 10−14 | 3.98 × 10−12 | 8.21 × 10−12 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 2 | −0.251 | −0.010 | 0.242 | 2.86 |
| 1,3 | 4.44 × 10−13 | 5.66 × 10−12 | 7.97 × 10−12 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 2 | −0.253 | −0.011 | 0.242 | 2.29 |
| 1,4 | 8.23 × 10−14 | 1.13 × 10−11 | 5.50 × 10−12 | 2 | 0 | 1 | 0 | 0 | 0 | 1 | 2 | −0.255 | −0.011 | 0.244 | 0.88 |
| 1,6 | 5.25 × 10−13 | 1.54 × 10−11 | 5.62 × 10−12 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 2 | −0.255 | −0.010 | 0.245 | 0.00 |
| 1,7 | 2.34 × 10−13 | 8.03 × 10−12 | 6.81 × 10−12 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 2 | −0.253 | −0.011 | 0.243 | 1.52 |
| 1,8 | 2.72 × 10−13 | 7.81 × 10−12 | 8.25 × 10−12 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 2 | −0.253 | −0.011 | 0.242 | 2.79 |
| 1,9 | 2.85 × 10−13 | 8.39 × 10−12 | 5.57 × 10−12 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 2 | −0.255 | −0.010 | 0.245 | 2.99 |
| 2,3 | 3.07 × 10−13 | 4.58 × 10−12 | 7.92 × 10−12 | 0 | 2 | 0 | 1 | 1 | 0 | 0 | 2 | −0.251 | −0.009 | 0.242 | 3.16 |
| 2,7 | 2.45 × 10−13 | 3.90 × 10−12 | 1.10 × 10−11 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 2 | −0.252 | −0.012 | 0.241 | 0.00 |
| 2,8 | 1.99 × 10−13 | 4.63 × 10−12 | 1.05 × 10−11 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 2 | −0.252 | −0.012 | 0.240 | 1.79 |
| Tri-CDDs | |||||||||||||||
| 1,2,3 | 2.11 × 10−13 | 2.46 × 10−12 | 7.40 × 10−12 | 1 | 2 | 1 | 1 | 2 | 1 | 0 | 3 | −0.256 | −0.015 | 0.241 | 3.42 |
| 1,2,4 | 1.04 × 10−13 | 6.06 × 10−12 | 7.66 × 10−12 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 3 | −0.258 | −0.018 | 0.240 | 2.40 |
| 1,2,6 | 8.76 × 10−14 | 1.85 × 10−12 | 5.61 × 10−12 | 2 | 1 | 1 | 1 | 1 | 0 | 0 | 3 | −0.259 | −0.017 | 0.242 | 1.59 |
| 1,2,7 | 9.32 × 10−14 | 2.22 × 10−12 | 7.55 × 10−12 | 1 | 2 | 1 | 1 | 1 | 0 | 0 | 3 | −0.257 | −0.017 | 0.240 | 1.14 |
| 1,2,8 | 9.77 × 10−14 | 1.81 × 10−12 | 8.14 × 10−12 | 1 | 2 | 1 | 2 | 1 | 0 | 0 | 3 | −0.257 | −0.017 | 0.240 | 2.89 |
| 1,2,9 | 7.05 × 10−14 | 2.66 × 10−12 | 5.68 × 10−12 | 2 | 1 | 2 | 1 | 1 | 0 | 0 | 3 | −0.258 | −0.016 | 0.242 | 3.76 |
| 1,3,6 | 4.03 × 10−13 | 4.99 × 10−12 | 7.66 × 10−12 | 2 | 1 | 1 | 0 | 0 | 1 | 0 | 3 | −0.260 | −0.018 | 0.242 | 1.96 |
| 1,3,7 | 2.16 × 10−13 | 1.57 × 10−12 | 8.17 × 10−12 | 1 | 2 | 1 | 0 | 0 | 1 | 0 | 3 | −0.259 | −0.019 | 0.240 | 0.48 |
| 1,3,8 | 3.04 × 10−13 | 4.63 × 10−12 | 1.04 × 10−11 | 1 | 2 | 1 | 1 | 0 | 1 | 0 | 3 | −0.259 | −0.019 | 0.240 | 1.51 |
| 1,3,9 | 1.90 × 10−13 | 2.38 × 10−12 | 5.80 × 10−12 | 2 | 1 | 2 | 0 | 0 | 1 | 0 | 3 | −0.260 | −0.018 | 0.242 | 2.66 |
| 1,4,6 | 7.72 × 10−14 | 2.60 × 10−12 | 3.95 × 10−12 | 3 | 0 | 1 | 0 | 0 | 0 | 1 | 3 | −0.262 | −0.017 | 0.245 | 1.48 |
| 1,4,7 | 1.17 × 10−13 | 4.45 × 10−12 | 5.93 × 10−12 | 2 | 1 | 1 | 0 | 0 | 0 | 1 | 3 | −0.260 | −0.018 | 0.242 | 1.27 |
| 2,3,6 | 2.98 × 10−13 | 2.92 × 10−12 | 5.63 × 10−12 | 1 | 2 | 0 | 1 | 1 | 0 | 0 | 3 | −0.258 | −0.016 | 0.242 | 3.06 |
| 2,3,7 | 1.82 × 10−13 | 1.04 × 10−12 | 7.47 × 10−12 | 0 | 3 | 0 | 1 | 1 | 0 | 0 | 3 | −0.257 | −0.017 | 0.240 | 1.58 |
| Compound | kα | kβ | kγ | Nα | Nβ | N1,9 | N2,8 | No | Nm | Np | N | EHOMO | ELUMO | ΔEHOMO-LUMO | D |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TCDDs | |||||||||||||||
| 1,2,3,4 | 6.62 × 10−14 | 2.07 × 10−12 | 7.06 × 10−12 | 2 | 2 | 1 | 1 | 3 | 2 | 1 | 4 | −0.260 | −0.022 | 0.239 | 3.30 |
| 1,2,3,6 | 1.91 × 10−13 | 1.35 × 10−12 | 5.38 × 10−12 | 2 | 2 | 1 | 1 | 2 | 1 | 0 | 4 | −0.263 | −0.022 | 0.241 | 2.77 |
| 1,2,3,7 | 2.00 × 10−13 | 1.34 × 10−12 | 7.56 × 10−12 | 1 | 3 | 1 | 1 | 2 | 1 | 0 | 4 | −0.261 | −0.023 | 0.239 | 1.42 |
| 1,2,3,8 | 2.39 × 10−13 | 7.45 × 10−13 | 7.97 × 10−12 | 1 | 3 | 1 | 2 | 2 | 1 | 0 | 4 | −0.261 | −0.023 | 0.239 | 2.40 |
| 1,2,3,9 | 1.34 × 10−13 | 1.70 × 10−12 | 5.23 × 10−12 | 2 | 2 | 2 | 1 | 2 | 1 | 0 | 4 | −0.262 | −0.022 | 0.241 | 3.71 |
| 1,2,4,6 | 4.89 × 10−14 | 1.79 × 10−12 | 4.88 × 10−12 | 3 | 1 | 1 | 1 | 1 | 1 | 1 | 4 | −0.265 | −0.024 | 0.241 | 2.05 |
| 1,2,4,7 | 6.42 × 10−14 | 1.09 × 10−12 | 7.42 × 10−12 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 4 | −0.263 | −0.025 | 0.239 | 0.61 |
| 1,2,4,8 | 7.46 × 10−14 | 2.02 × 10−12 | 7.10 × 10−12 | 2 | 2 | 1 | 2 | 1 | 1 | 1 | 4 | −0.263 | −0.025 | 0.238 | 1.48 |
| 1,2,4,9 | 3.81 × 10−14 | 2.16 × 10−12 | 4.97 × 10−12 | 3 | 1 | 2 | 1 | 1 | 1 | 1 | 4 | −0.265 | −0.024 | 0.241 | 2.68 |
| 1,2,6,7 | 6.42 × 10−14 | 1.18 × 10−12 | 6.06 × 10−12 | 2 | 2 | 1 | 1 | 2 | 0 | 0 | 4 | −0.262 | −0.023 | 0.239 | 0.00 |
| 1,2,6,8 | 1.47 × 10−13 | 2.36 × 10−12 | 8.51 × 10−12 | 2 | 2 | 1 | 2 | 1 | 1 | 0 | 4 | −0.263 | −0.024 | 0.239 | 1.41 |
| 1,2,6,9 | 5.65 × 10−14 | 1.92 × 10−12 | 4.16 × 10−12 | 3 | 1 | 2 | 1 | 1 | 0 | 1 | 4 | −0.265 | −0.023 | 0.241 | 2.17 |
| 1,2,7,8 | 1.45 × 10−13 | 8.84 × 10−13 | 6.35 × 10−12 | 1 | 3 | 1 | 2 | 2 | 0 | 0 | 4 | −0.261 | −0.023 | 0.239 | 2.28 |
| 1,2,7,9 | 1.93 × 10−13 | 1.51 × 10−12 | 8.04 × 10−12 | 2 | 2 | 2 | 1 | 1 | 1 | 0 | 4 | −0.263 | −0.024 | 0.240 | 2.49 |
| 1,2,8,9 | 6.79 × 10−14 | 1.53 × 10−12 | 7.22 × 10−12 | 2 | 2 | 2 | 2 | 2 | 0 | 0 | 4 | −0.261 | −0.023 | 0.239 | 3.88 |
| 1,3,6,8 | 4.41 × 10−13 | 2.69 × 10−12 | 9.96 × 10−12 | 2 | 2 | 1 | 1 | 0 | 2 | 0 | 4 | −0.265 | −0.025 | 0.240 | 0.00 |
| 1,3,6,9 | 2.71 × 10−13 | 2.35 × 10−12 | 5.68 × 10−12 | 3 | 1 | 2 | 0 | 0 | 1 | 1 | 4 | −0.267 | −0.025 | 0.242 | 1.39 |
| 1,3,7,8 | 2.25 × 10−13 | 1.38 × 10−12 | 6.97 × 10−12 | 1 | 3 | 1 | 1 | 1 | 1 | 0 | 4 | −0.263 | −0.024 | 0.239 | 1.08 |
| 1,3,7,9 | 2.57 × 10−13 | 2.41 × 10−12 | 7.57 × 10−12 | 2 | 2 | 2 | 0 | 0 | 2 | 0 | 4 | −0.265 | −0.025 | 0.240 | 1.08 |
| 1,4,6,9 | 3.93 × 10−16 | 3.70 × 10−12 | 3.20 × 10−12 | 4 | 0 | 2 | 0 | 0 | 0 | 2 | 4 | −0.269 | −0.024 | 0.244 | 0.00 |
| 1,4,7,8 | 1.24 × 10−13 | 1.35 × 10−12 | 4.06 × 10−12 | 2 | 2 | 1 | 1 | 1 | 0 | 1 | 4 | −0.264 | −0.024 | 0.241 | 2.21 |
| 2,3,7,8 | 1.72 × 10−13 | 1.35 × 10−15 | 5.20 × 10−12 | 0 | 4 | 0 | 2 | 2 | 0 | 0 | 4 | −0.261 | −0.022 | 0.239 | 0.00 |
| Compound | kα | kβ | kγ | Nα | Nβ | N1,9 | N2,8 | No | Nm | Np | N | EHOMO | ELUMO | ΔEHOMO-LUMO | D |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Penta-CDDs | |||||||||||||||
| 1,2,3,4,6 | 4.48 × 10−14 | 9.83 × 10−13 | 5.07 × 10−12 | 3 | 2 | 1 | 1 | 3 | 2 | 1 | 5 | −0.267 | −0.028 | 0.240 | 3.14 |
| 1,2,3,4,7 | 4.09 × 10−14 | 6.84 × 10−13 | 7.14 × 10−12 | 2 | 3 | 1 | 1 | 3 | 2 | 1 | 5 | −0.265 | −0.028 | 0.237 | 1.70 |
| 1,2,3,6,7 | 1.34 × 10−13 | 2.34 × 10−13 | 6.10 × 10−12 | 2 | 3 | 1 | 1 | 3 | 1 | 0 | 5 | −0.266 | −0.028 | 0.238 | 1.51 |
| 1,2,3,6,8 | 2.05 × 10−13 | 1.09 × 10−12 | 7.71 × 10−12 | 2 | 3 | 1 | 2 | 2 | 2 | 0 | 5 | −0.267 | −0.029 | 0.238 | 1.12 |
| 1,2,3,6,9 | 1.10 × 10−13 | 6.93 × 10−13 | 4.24 × 10−12 | 3 | 2 | 2 | 1 | 2 | 1 | 1 | 5 | −0.269 | −0.028 | 0.240 | 2.49 |
| 1,2,3,7,8 | 1.49 × 10−13 | 1.24 × 10−15 | 5.21 × 10−12 | 1 | 4 | 1 | 2 | 3 | 1 | 0 | 5 | −0.265 | −0.028 | 0.237 | 1.06 |
| 1,2,3,7,9 | 1.66 × 10−13 | 7.56 × 10−13 | 6.46 × 10−12 | 2 | 3 | 2 | 1 | 2 | 2 | 0 | 5 | −0.267 | −0.029 | 0.238 | 1.84 |
| 1,2,3,8,9 | 1.01 × 10−13 | 5.26 × 10−13 | 6.51 × 10−12 | 2 | 3 | 2 | 2 | 3 | 1 | 0 | 5 | −0.266 | −0.028 | 0.238 | 3.18 |
| 1,2,4,6,7 | 3.66 × 10−14 | 1.98 × 10−12 | 5.72 × 10−12 | 3 | 2 | 1 | 1 | 2 | 1 | 1 | 5 | −0.268 | −0.030 | 0.239 | 1.39 |
| 1,2,4,6,8 | 2.38 × 10−13 | 2.50 × 10−12 | 1.17 × 10−11 | 3 | 2 | 1 | 2 | 1 | 2 | 1 | 5 | −0.267 | −0.029 | 0.238 | 1.12 |
| 1,2,4,6,9 | 7.33 × 10−16 | 1.56 × 10−12 | 4.79 × 10−12 | 4 | 1 | 2 | 1 | 1 | 1 | 2 | 5 | −0.271 | −0.030 | 0.241 | 1.51 |
| 1,2,4,7,8 | 7.27 × 10−14 | 5.78 × 10−13 | 4.89 × 10−12 | 2 | 3 | 1 | 2 | 2 | 1 | 1 | 5 | −0.267 | −0.030 | 0.238 | 0.93 |
| 1,2,4,7,9 | 8.16 × 10−14 | 2.05 × 10−12 | 7.50 × 10−12 | 3 | 2 | 2 | 1 | 1 | 2 | 1 | 5 | −0.270 | −0.031 | 0.239 | 1.02 |
| 1,2,4,8,9 | 3.71 × 10−14 | 1.12 × 10−12 | 6.03 × 10−12 | 3 | 2 | 2 | 2 | 2 | 1 | 1 | 5 | −0.268 | −0.030 | 0.238 | 2.41 |
| Hexa-CDDs | |||||||||||||||
| 1,2,3,4,6,7 | 1.60 × 10−14 | 2.54 × 10−13 | 6.16 × 10−12 | 3 | 3 | 1 | 1 | 4 | 2 | 1 | 6 | −0.270 | −0.033 | 0.237 | 2.26 |
| 1,2,3,4,6,8 | 1.10 × 10−13 | 8.46 × 10−13 | 7.87 × 10−12 | 3 | 3 | 1 | 2 | 3 | 3 | 1 | 6 | −0.271 | −0.034 | 0.237 | 1.19 |
| 1,2,3,4,6,9 | 7.74 × 10−16 | 7.82 × 10−13 | 4.45 × 10−12 | 4 | 2 | 2 | 1 | 3 | 2 | 2 | 6 | −0.273 | −0.034 | 0.239 | 2.36 |
| 1,2,3,4,7,8 | 6.63 × 10−14 | 9.51 × 10−16 | 5.04 × 10−12 | 2 | 4 | 1 | 2 | 4 | 2 | 1 | 6 | −0.269 | −0.033 | 0.236 | 0.19 |
| 1,2,3,6,7,8 | 2.56 × 10−13 | 1.15 × 10−15 | 6.27 × 10−12 | 2 | 4 | 1 | 2 | 4 | 2 | 0 | 6 | −0.270 | −0.033 | 0.237 | 0.00 |
| 1,2,3,6,7,9 | 1.07 × 10−13 | 6.23 × 10−13 | 6.05 × 10−12 | 3 | 3 | 2 | 1 | 3 | 2 | 1 | 6 | −0.272 | −0.034 | 0.237 | 0.97 |
| 1,2,3,6,8,9 | 1.20 × 10−13 | 5.56 × 10−13 | 6.32 × 10−12 | 3 | 3 | 2 | 2 | 3 | 2 | 1 | 6 | −0.272 | −0.034 | 0.237 | 1.71 |
| 1,2,3,7,8,9 | 1.46 × 10−13 | 1.13 × 10−15 | 6.19 × 10−12 | 2 | 4 | 2 | 2 | 4 | 2 | 0 | 6 | −0.269 | −0.033 | 0.237 | 2.01 |
| 1,2,4,6,7,9 | 6.30 × 10−16 | 6.19 × 10−13 | 6.07 × 10−12 | 4 | 2 | 2 | 1 | 2 | 2 | 2 | 6 | −0.274 | −0.036 | 0.238 | 0.00 |
| 1,2,4,6,8,9 | 6.55 × 10−16 | 1.22 × 10−12 | 5.86 × 10−12 | 4 | 2 | 2 | 2 | 2 | 2 | 2 | 6 | −0.274 | −0.036 | 0.238 | 1.02 |
| Hepta-CDDs | |||||||||||||||
| 1,2,3,4,6,7,8 | 7.21 × 10−14 | 9.28 × 10−16 | 5.71 × 10−12 | 3 | 4 | 1 | 2 | 5 | 3 | 1 | 7 | −0.273 | −0.038 | 0.236 | 0.95 |
| 1,2,3,4,6,7,9 | 6.81 × 10−16 | 4.15 × 10−13 | 5.41 × 10−12 | 4 | 3 | 2 | 1 | 4 | 3 | 2 | 7 | −0.276 | −0.039 | 0.237 | 1.06 |
| OCDD | |||||||||||||||
| 1,2,3,4,6,7,8,9 | 7.95 × 10−16 | 7.51 × 10−16 | 5.98 × 10−12 | 4 | 4 | 2 | 2 | 6 | 4 | 2 | 8 | −0.277 | −0.043 | 0.235 | 0.00 |



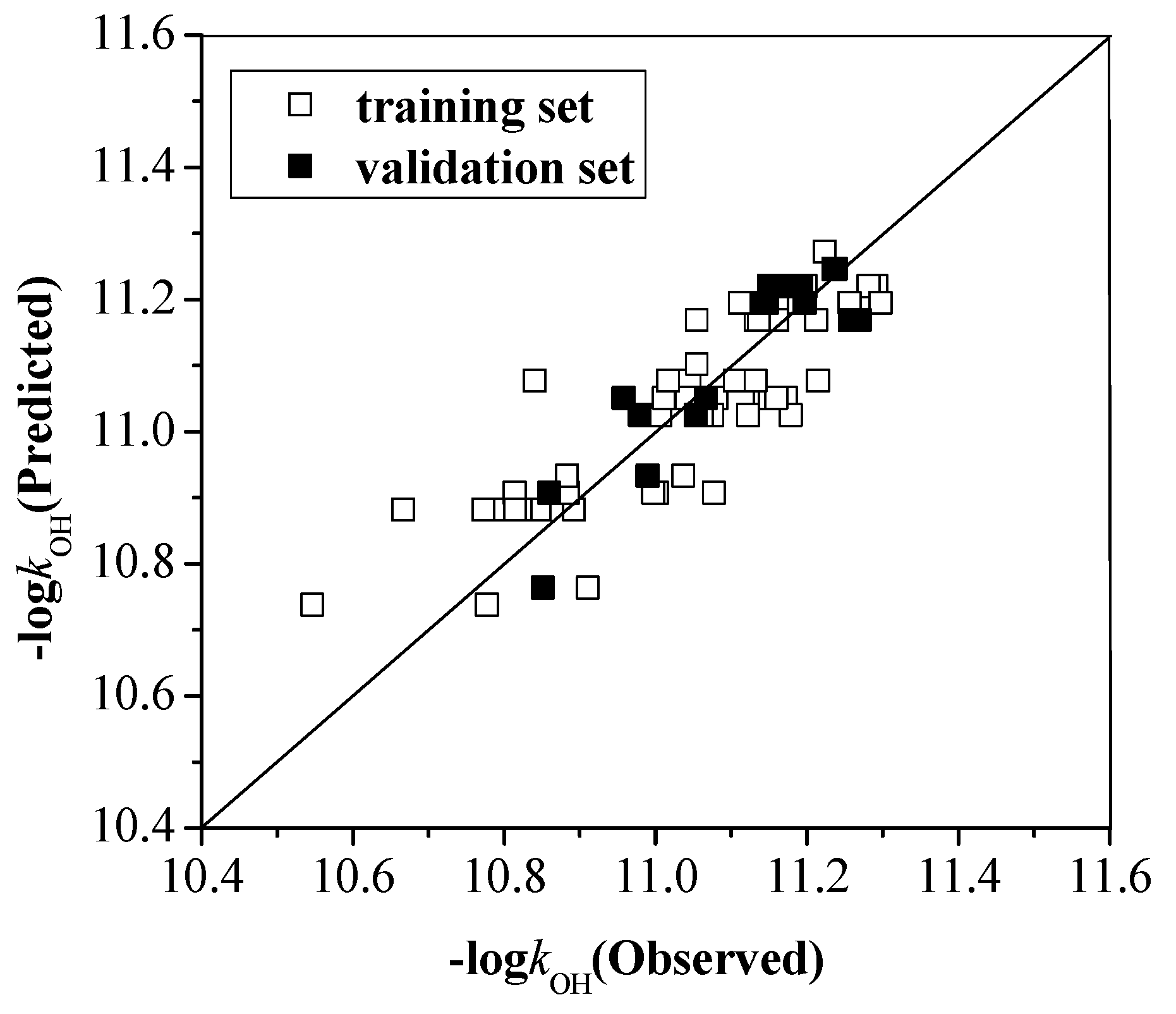
2.2.2. The Total Rate Constant kOH with the Configuration Parameters

3. Computational Methods
3.1. Geometry Optimization
3.2. Kinetic Calculation
3.3. Quantitative Structure–Activity Relationship
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interests
References
- Chu, S.; Zheng, M.; Xu, X. Characterization of the combustion products of polyethylene. Chemosphere 1999, 39, 1497–1512. [Google Scholar]
- Yasuhara, A.; Katami, T.; Okuda, T.; Ohno, N.; Adriaens, P. Formation of dioxins during the combustion of newspapers in the presence of sodium chloride and poly(vinyl chloride). Environ. Sci. Technol. 2001, 35, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K.; Lin, C.; Lin, Y.C.; Wang, L.C.; Chang-Chien, G.P. Polychlorinated dibenzo-p-dioxins/dibenzofuran mass distribution in both start-up and normal condition in the whole municipal solid waste incinerator. J. Hazard. Mater. 2008, 160, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Hoyos, A.; Cobo, M.; Aristizábal, B.; Córdoba, F.; de Correa, C.M. Total suspended particulate (TSP), polychlorinated dibenzodioxin (PCDD) and polychlorinated dibenzofuran (PCDF) emissions from medical waste incinerators in Antioquia, Colombia. Chemosphere 2008, 73, S137–S142. [Google Scholar] [CrossRef] [PubMed]
- Stalling, D.L.; Norstrom, R.J.; Smith, L.M.; Simon, M. Patterns of PCDD, PCDF, and PCB components analysis. Chemosphere 1985, 14, 627–643. [Google Scholar] [CrossRef]
- Hays, S.M.; Aylward, L.L. Dioxin risks in perspective: Past, present, and future. Regul. Toxicol. Pharmacol. 2003, 37, 202–217. [Google Scholar] [CrossRef]
- Kwok, E.S.C.; Atkinson, R. Estimation of hydroxyl radical reaction-rate constants for gas-phase organic-compounds using a structure-reactivity relationship: An update. Atmos. Environ. 1995, 29, 1685–1695. [Google Scholar] [CrossRef]
- Atkinson, R. Atmospheric chemistry of PCBs, PCDDs and PCDFs. Environ. Sci. Technol. 1996, 6, 53–72. [Google Scholar]
- Brubaker, W.W.; Hites, R.A. Polychlorinated dibenzo-p-dioxins and dibenzo-furans: Gas-phase hydroxyl radical reactions and related atmospheric removal. Environ. Sci. Technol. 1997, 31, 1805–1810. [Google Scholar] [CrossRef]
- Taylor, P.H.; Yamada, T.; Neuforth, A. Kinetics of OH radical reactions with dibenzo-p-dioxin and selected chlorinated dibenzo-p-dioxins. Chemosphere 2005, 58, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Altarawneh, M.; Kennedy, E.M.; Dlugogorski, B.Z.; Mackie, J.C. Computational study of the oxidation and decomposition of dibenzofuran under atmospheric conditions. J. Phys. Chem. A 2008, 112, 6960–6967. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, A. The oxidation mechanisms of polychlorinated dibenzo-p-dioxins under atmospheric conditions: A theoretical study. Chemosphere 2012, 89, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Zeng, X.L.; Wang, Z.Y.; Liu, H.X. QSPR modeling of n-octanol/water partition coefficients and water solubility of PCDEs by the method of Cl substitution position. Sci. Total. Environ. 2007, 382, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Zeng, X.L.; Zhai, Z.C. Prediction of supercooled liquid vapor pressures and n-octanol/air partition coefficients for polybrominated diphenyl ethers by means of molecular descriptors from DFT method. Sci. Total Environ. 2008, 389, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Q.; Zhang, X.S.; Qu, R.J.; Xu, Y.; Wang, Z.Y. Synthesis and QSPR study on environment-related properties of polychlorinated diphenyl sulfides (PCDPSs). Chemosphere 2012, 88, 844–854. [Google Scholar]
- Oberg, T. A QSAR for the hydroxyl radical reaction rate constant: Validation, domain of application, and prediction. Atmos. Environ. 2005, 39, 2189–2200. [Google Scholar]
- Wang, Y.N.; Chen, J.W.; Li, X.H.; Wang, B.; Cai, X.Y.; Huang, L.P. Predicting rate constants of hydroxyl radical reactions with organic pollutants: Algorithm, validation, applicability domain, and mechanistic interpretation. Atmos. Environ. 2009, 43, 1131–1135. [Google Scholar] [CrossRef]
- Lee, J.E.; Choi, W.; Mhin, B.J.; Balasubramanian, K. Theoretical study on the reaction of OH radicals with polychlorinated dibenzo-p-dioxin. J. Phys. Chem. A 2004, 108, 607–614. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Pople, J.A. Gaussian 03, Revision B. 02; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: The MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and Van der waals interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Zheng, J.J.; Zhao, Y.; Truhlar, D.G. The DBH24/08 database and its use to assess electronic structure model chemistries for chemical reaction barrier heights. J. Chem. Theory Comput. 2009, 5, 808–821. [Google Scholar] [CrossRef]
- Zhang, C.X.; Sun, T.L.; Sun, X.M. Mechanism for OH-initiated degradation of 2,3,7,8-tetrachlorinated dibenzo-p-dioxins in the presence of O2 and NO/H2O. Environ. Sci. Technol. 2011, 45, 4756–4762. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.M.; Zhang, C.X.; Zhao, Y.Y.; Bai, J.; Zhang, Q.Z.; Wang, W.X. Atmospheric chemical reactions of 2,3,7,8-tetrachlorinated dibenzofuran initiated by an OH radical: Mechanism and kinetics study. Environ. Sci. Technol. 2012, 46, 8148–8155. [Google Scholar] [CrossRef] [PubMed]
- Merrick, J.P.; Moran, D.; Radom, L. An evaluation of harmonic vibrational frequency scale factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Hou, H.; Wang, B.S. Ab initio study of the reaction of propionyl (C2H5CO) radical with oxygen (O2). J. Chem. Phys. 2007, 127, 054306. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hopfinger, A.J. Application of genetic function approximation to quantitative structure–activity relationships and quantitative structure–property relationships. J. Chem. Inf. Comput. Sci. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Materials Studio, version 5.0; Software for simulating and modeling materials; Accelrys Software Inc.: San Diego, CA, USA, 2006.
- Roy, K.; Mitra, I.; Kar, S.; Ojha, P.K.; Das, R.N.; Kabir, H. Comparative studies on some metrics for external validation of QSPR models. J. Chem. Inf. Model. 2012, 52, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Papa, E.; Villa, F.; Gramatica, P. Statistically validated QSARs, based on theoretical descriptors, for modeling aquatic toxicity of organic chemicals in Pimephales promelas (fathead minnow). J. Chem. Inf. Model. 2005, 45, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, C.; Zhang, C.; Sun, X. Quantitative Structure-Activity Relationships Study on the Rate Constants of Polychlorinated Dibenzo-p-Dioxins with OH Radical. Int. J. Mol. Sci. 2015, 16, 18812-18824. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160818812
Qi C, Zhang C, Sun X. Quantitative Structure-Activity Relationships Study on the Rate Constants of Polychlorinated Dibenzo-p-Dioxins with OH Radical. International Journal of Molecular Sciences. 2015; 16(8):18812-18824. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160818812
Chicago/Turabian StyleQi, Chuansong, Chenxi Zhang, and Xiaomin Sun. 2015. "Quantitative Structure-Activity Relationships Study on the Rate Constants of Polychlorinated Dibenzo-p-Dioxins with OH Radical" International Journal of Molecular Sciences 16, no. 8: 18812-18824. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160818812