
Biomarkers of Chondrocyte Apoptosis and Autophagy in Osteoarthritis

 ,
,  , ,
, ,
Abstract
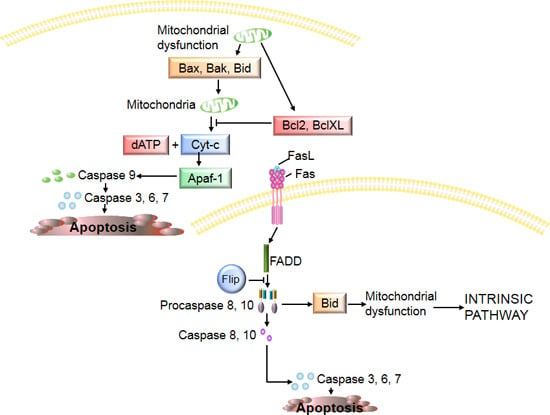
:
1. Introduction
2. Methods
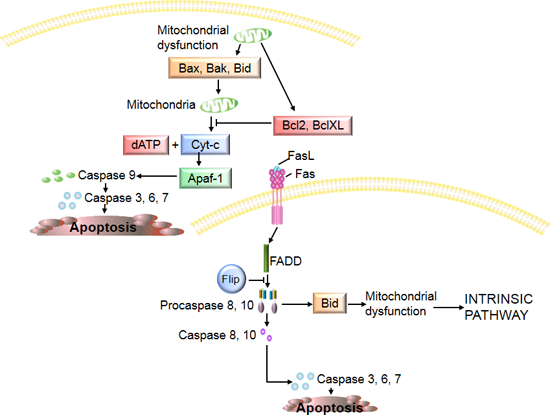
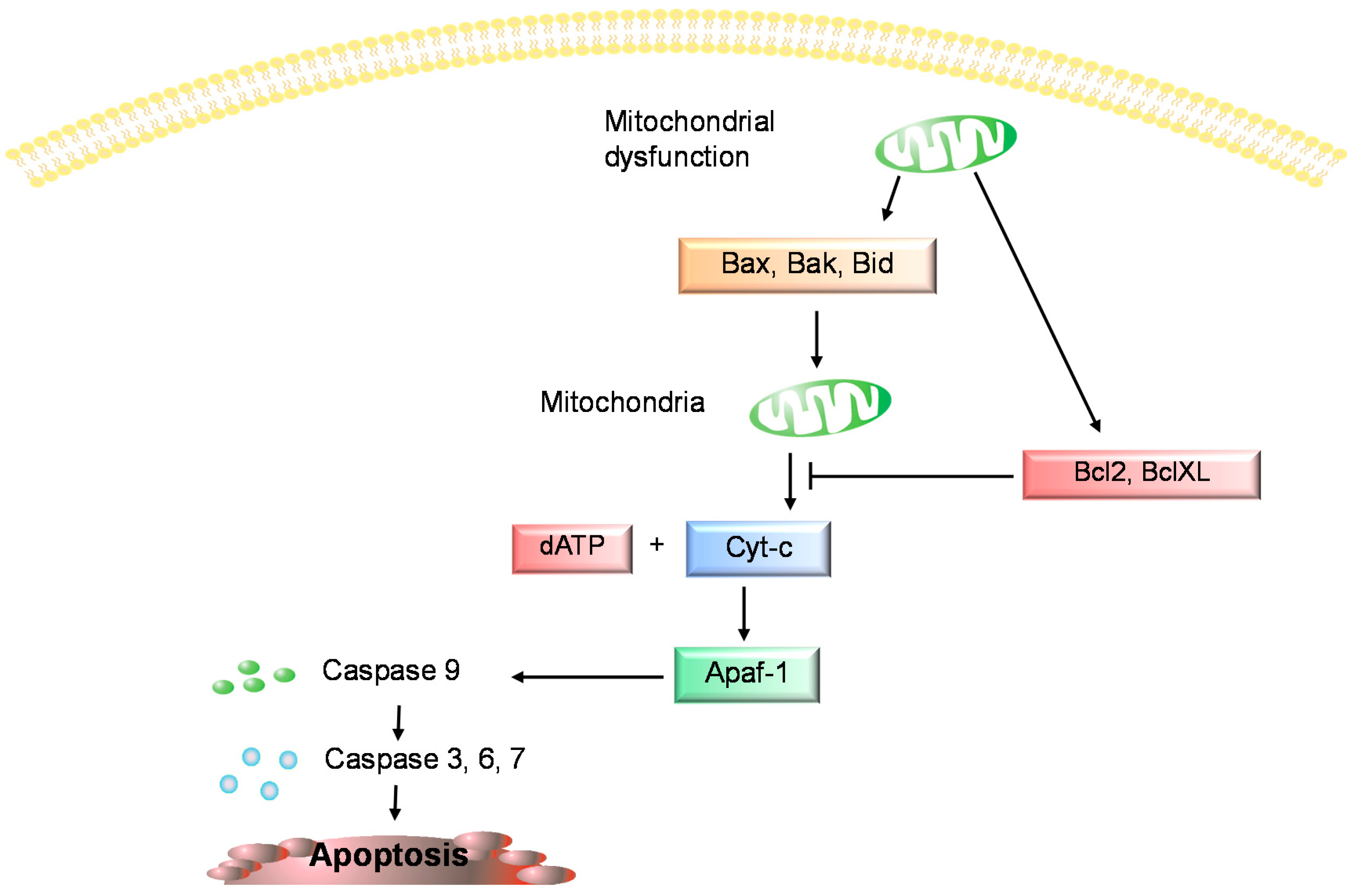
3. Chondrocyte Apoptosis in Osteoarthritis

4. Biomarkers of Chondrocyte Degeneration

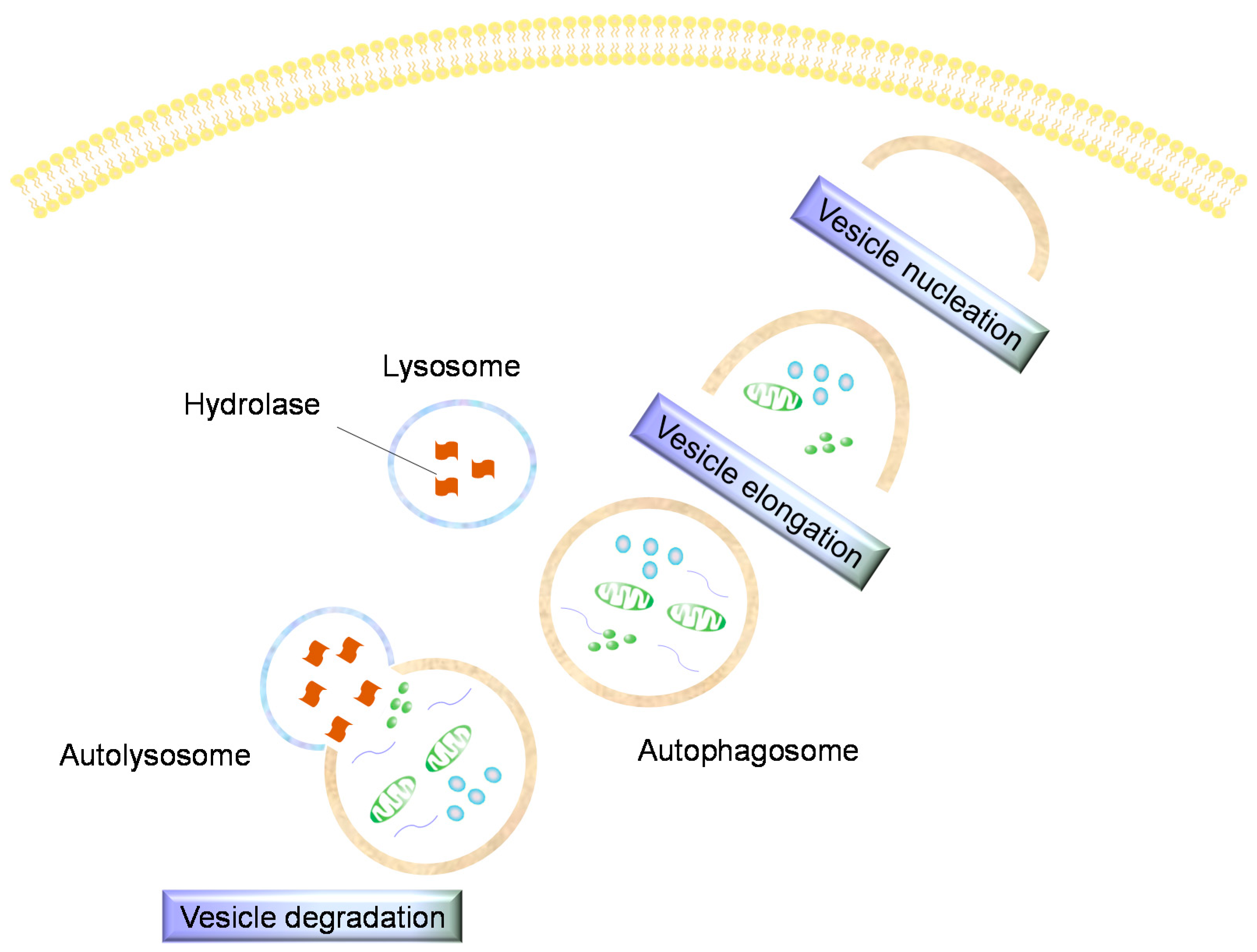
5. Autophagy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apoptosis | Necrosis | Autophagy |
|---|---|---|
| Rounding of cells; Plasma membrane blebbing; Nuclear fragmentation; Chromatin condensation; Reduction in cellular and nuclear volume; Apoptosis body formation; Mitochondrial swelling (rare) | Plasma membrane rupture; Mitochondrial and cytoplasmic swelling; No vesicle formation; Moderate chromatin condensation | Accumulation of autophagic vacuoles; Lack of chromatin condensation; Late-stage mitochondrial swelling |
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Musumeci, G.; Aiello, F.C.; Szychlinska, M.A.; di Rosa, M.; Castrogiovanni, P.; Mobasheri, A. Osteoarthritis in the XXIst century: Risk factors and behaviours that influence disease onset and progression. Int. J. Mol. Sci. 2015, 16, 6093–6112. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.; Szychlinska, M.A.; Tibullo, D.; Malaguarnera, L.; Musumeci, G. Expression of CHI3L1 and CHIT1 in osteoarthritic rat cartilage model. A morphological study. Eur. J. Histochem. 2014, 16, 2423. [Google Scholar] [CrossRef] [PubMed]
- Galanti, C.; Musumeci, G.; Valentino, J.; Giunta, S.; Castorina, S. A role for apoptosis in temporomandibular joint disc degeneration. A contemporary review. Ital. J. Anat. Embryol. 2013, 118, 151–158. [Google Scholar] [PubMed]
- Musumeci, G.; Loreto, C.; Carnazza, M.L.; Martinez, G. Characterization of apoptosis in articular cartilage derived from the knee joints of patients with osteoarthritis. Knee Surg. Sports Traumatol. Arthrosc. 2011, 19, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, B.; Frank, H.G.; Kaufmann, P. The apoptosis cascade—Morphological and immunohistochemical methods for its visualization. Anat. Embryol. 1999, 200, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zamli, Z.; Sharif, M. Chondrocyte apoptosis: A cause or consequence of osteoarthritis? Int. J. Rheum. Dis. 2011, 14, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Loreto, C.; Carnazza, M.L.; Strehin, I.; Elisseeff, J. OA cartilage derived chondrocytes encapsulated in poly (ethylene glycol) diacrylate (PEGDA) for the evaluation of cartilage restoration and apoptosis in an in vitro model. Histol. Histopathol. 2011, 26, 1265–1278. [Google Scholar] [PubMed]
- Thomas, C.M.; Murray, R.; Sharif, M. Chondrocyte apoptosis determined by caspase-3 expression varies with fibronectin distribution in equine articular cartilage. Int. J. Rheum. Dis. 2011, 14, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Castrogiovanni, P.; Mazzone, V.; Szychlinska, M.A.; Castorina, S.; Loreto, C. Histochemistry as a unique approach for investigating normal and osteoarthritic cartilage. Eur. J. Histochem. 2014, 58, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Castrogiovanni, P.; Loreto, C.; Castorina, S.; Pichler, K.; Weinberg, A.M. Post-traumatic caspase-3 expression in the adjacent areas of growth plate injury site. A morphological study. Int. J. Mol. Sci. 2013, 14, 15767–15784. [Google Scholar] [CrossRef] [PubMed]
- Shakibaei, M.; John, T.; Seifarth, C.; Mobasheri, A. Resveratrol inhibits IL-1β-induced stimulation of caspase-3 and cleavage of PARP in human articular chondrocytes in vitro. Ann. N. Y. Acad. Sci. 2007, 1095, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Giunta, S.; Castorina, A.; Marzagalli, R.; Szychlinska, M.A.; Pichler, K.; Mobasheri, A.; Musumeci, G. Ameliorative effects of PACAP against cartilage degeneration. Morphological, immunohistochemical and biochemical evidence from in vivo and in vitro models of rat osteoarthritis. Int. J. Mol. Sci. 2015, 16, 5922–5944. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.M. Apoptosis signaling in tumor therapy. Ann. N. Y. Acad. Sci. 2004, 1028, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, R.; Pierce, G.N. The toxicity of dietary trans fats. Food Chem. Toxicol. 2015, 78, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Sallem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Akl, H.; Vervloessem, T.; Kiviluoto, S.; Bittremieux, M.; Parys, J.B.; De Smedt, H.; Bultynck, G. A dual role for the anti-apoptotic Bcl-2 protein in cancer: Mitochondria versus endoplasmic reticulum. Biochim. Biophys. Acta 2014, 1843, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Loreto, C.; Rapisarda, V.; Carnazza, M.L.; Musumeci, G.; D’Agata, V.; Valentino, M.; Martinez, G. Bitumen products alter Bax, Bcl-2 and cytokeratin expression: An in vivo study of chronically exposed road pavers. J. Cutan. Pathol. 2007, 34, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Witzig, T.E.; Adjei, A.A. Targeting apoptosis pathways in cancer therapy. CA Cancer J. Clin. 2005, 55, 178–194. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Hashimoto, S.; Kubo, T.; Hirasawa, Y.; Lotz, M.; Amiel, D. Effect of hyaluronan on chondrocyte apoptosis and nitric oxide production in experimentally induced osteoarthritis. J. Rheumatol. 2000, 27, 1713–1720. [Google Scholar] [PubMed]
- Mobasheri, A.; Matta, C.; Zákány, R.; Musumeci, G. Chondrosenescence: Definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas 2015, 80, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Grogan, S.P.; D’Lima, D.D. Joint aging and chondrocyte cell death. Int. J. Clin. Rheumtol. 2010, 5, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.J.; Chen, T.G.; Chang, H.C.; Chiu, W.T.; Chang, C.C.; Chen, R.M. Nitric oxide from both exogenous and endogenous sources activates mitochondria-dependent events and induces insults to human chondrocytes. J. Cell. Biochem. 2007, 101, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Maneiro, E.; Lopez-Armada, M.J.; de Andres, M.C.; Caramés, B.; Martín, M.A.; Bonilla, A.; del Hoyo, P.; Galdo, F.; Arenas, J.; Blanco, F.J. Effect of nitric oxide on mitochondrial respiratory activity of human articular chondrocytes. Ann. Rheum. Dis. 2005, 64, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Cherng, Y.G.; Chang, H.C.; Lin, Y.L.; Kuo, M.L.; Chiu, W.T.; Chen, R.M. Apoptotic insults to human chondrocytes induced by sodium nitroprusside are involved in sequential events, including cytoskeletal remodeling, phosphorylation of mitogen-activated protein kinase kinase kinase-1/c-Jun N-terminal kinase, and Bax-mitochondria-mediated caspase activation. J. Orthop. Res. 2008, 26, 1018–1026. [Google Scholar] [PubMed]
- Kuhn, K.; Hashimoto, S.; Lotz, M. IL-1β protects human chondrocytes from CD95-induced apoptosis. J. Immunol. 2000, 164, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Del Carlo, M.; Loeser, R.F. Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum. 2002, 46, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Tanaka, M.; Suda, T.; Tomita, T.; Hayashida, K.; Takeuchi, E.; Kaneko, M.; Takano, H.; Nagata, S.; Ochi, T. Soluble Fas ligand in the joints of patients with rheumatoid arthritis and osteoarthritis. Arthritis Rheum. 1998, 41, 657–662. [Google Scholar] [CrossRef]
- Hashimoto, S.; Ochs, R.L.; Rosen, F.; Quach, J.; McCabe, G.; Solan, J.; Seegmiller, J.E.; Terkeltaub, R.; Lotz, M. Chondrocyte-derived apoptotic bodies and calcification of articular cartilage. Proc. Natl. Acad. Sci. USA 1998, 95, 3094–3099. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Hwang, S.G.; Shin, D.Y.; Kang, S.S.; Chun, J.S. P38 kinase regulates nitric oxide-induced apoptosis of articular chondrocytes by accumulating p53 via NFkappa B-dependent transcription and stabilization by serine 15 phosphorylation. J. Biol. Chem. 2002, 277, 33501–33508. [Google Scholar] [CrossRef] [PubMed]
- Yatsugi, N.; Tsukazaki, T.; Osaki, M.; Koji, T.; Yamashita, S.; Shindo, H. Apoptosis of articular chondrocytes in rheumatoid arthritis and osteoarthritis: Correlation of apoptosis with degree of cartilage destruction and expression of apoptosis-related proteins of p53 and c-myc. J. Orthop. Sci. 2000, 5, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.P.; Faure, M.P.; DiBattista, J.A.; Wilhelm, S.; Visco, D.; Martel-Pelletier, J. Coordinate synthesis of stromelysin, interleukin-1, and oncogene proteins in experimental osteoarthritis. An immunohistochemical study. Am. J. Pathol. 1993, 142, 95–105. [Google Scholar] [PubMed]
- Loreto, C.; Barbagli, G.; Djinovic, R.; Vespasiani, G.; Carnazza, M.L.; Miano, R.; Musumeci, G.; Sansalone, S. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and its death receptor (DR5) in Peyroniet’s disease. A Biomolecular Study of Apoptosis Activation. J. Sex. Med. 2011, 8, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Shakibaei, M.; Csaki, C.; Nebrich, S.; Mobasheri, A. Resveratrol suppresses interleukin-1β-induced inflammatory signaling and apoptosis in human articular chondrocytes: Potential for use as a novel nutraceutical for the treatment of osteoarthritis. Biochem. Pharmacol. 2008, 76, 1426–1439. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Kalamegam, G.; Musumeci, G.; Batt, M.E. Chondrocyte and mesenchymal stem cell-based therapies for cartilage repair in osteoarthritis and related orthopaedic conditions. Maturitas 2014, 78, 188–98. [Google Scholar] [CrossRef] [PubMed]
- Sillat, T.; Barreto, G.; Clarijs, P.; Soininen, A.; Ainola, M.; Pajarinen, J.; Korhonen, M.; Konttinen, Y.T.; Sakalyte, R.; Hukkanen, M.; et al. Toll-like receptors in human chondrocytes and osteoarthritic cartilage. Acta Orthop. 2013, 84, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Bobacz, K.; Sunk, I.G.; Hofstaetter, J.G.; Amoyo, L.; Toma, C.D.; Akira, S.; Weichhart, T.; Saemann, M.; Smolen, J.S. Toll-like receptors and chondrocytes: The lipopolysaccharide-induced decrease in cartilage matrix synthesis is dependent on the presence of toll-like receptor 4 and antagonized by bone morphogenetic protein 7. Arthritis Rheum. 2007, 56, 1880–1893. [Google Scholar] [CrossRef] [PubMed]
- Liu-Bryan, R. Synovium and the innate inflammatory network in osteoarthritis progression. Curr. Rheumatol. Rep. 2013, 15, 323. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.A.; Mundle, S.; Cole, A.A. Tumor necrosis factor-α induced DNA cleavage in human articular chondrocytes may involve multiple endonucleolytic activities during apoptosis. Microsc. Res. Tech. 2000, 50, 236–242. [Google Scholar] [CrossRef]
- Kühn, K.; Lotz, M. Regulation of CD95 (Fas/APO-1)-induced apoptosis in human chondrocytes. Arthritis Rheum. 2001, 44, 1644–1653. [Google Scholar] [CrossRef]
- López-Armada, M.J.; Caramés, B.; Lires-Deán, M.; Cillero-Pastor, B.; Ruiz-Romero, C.; Galdo, F.; Blanco, F.J. Cytokines, tumor necrosis factor-α and interleukin-1β, differentially regulate apoptosis in osteoarthritis cultured human chondrocytes. Osteoarthr. Cartil. 2006, 14, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Caramés, B.; López-Armada, M.J.; Cillero-Pastor, B.; Lires-Dean, M.; Vaamonde, C.; Galdo, F.; Blanco, F.J. Differential effects of tumor necrosis factor-α and interleukin-1β on cell death in human articular chondrocytes. Osteoarthr. Cartil. 2008, 16, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Madej, W.; van Caam, A.; Blaney Davidson, E.N.; van der Kraan, P.M.; Buma, P. Physiological and excessive mechanical compression of articular cartilage activates Smad2/3P signaling. Osteoarthr. Cartil. 2014, 22, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Itayem, R.; Mengarelli-Widholm, S.; Reinholt, F.P. The long-term effect of a short course of transforming growth factor-β1 on rat articular cartilage. APMIS 1999, 107, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Van Beuningen, H.M.; Glansbeek, H.L.; van der Kraan, P.M.; van den Berg, W.B. Osteoarthritis-like changes in the murine knee joint resulting from intra-articular transforming growth factor-β injections. Osteoarthr. Cartil. 2000, 8, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, W.B. Osteoarthritis year 2010 in review: Pathomechanisms. Osteoarthr. Cartil. 2011, 19, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Yammani, R.R.; Long, D.; Loeser, R.F. Interleukin-7 stimulates secretion of S100A4 by activating the JAK/STAT signaling pathway in human articular chondrocytes. Arthritis Rheum. 2009, 60, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Senolt, L.; Grigorian, M.; Lukanidin, E.; Simmen, B.; Michel, B.A.; Pavelka, K.; Gay, R.E.; Gay, S.; Neidhart, M. S100A4 is expressed at site of invasion in rheumatoid arthritis synovium and modulates production of matrix metalloproteinases. Ann. Rheum. Dis. 2006, 65, 1645–1648. [Google Scholar] [CrossRef] [PubMed]
- Grigorian, M.; Andresen, S.; Tulchinsky, E.; Kriajevska, M.; Carlberg, C.; Kruse, C.; Cohn, M.; Ambartsumian, N.; Christensen, A.; Selivanova, G.; et al. Tumor suppressor p53 protein is a new target for the metastasis-associated Mts1/S100A4 protein: Functional consequences of their interaction. J. Biol. Chem. 2001, 276, 22699–22708. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Loreto, C.; Leonardi, R.; Castorina, S.; Giunta, S.; Carnazza, M.L.; Trovato, F.M.; Pichler, K.; Weinberg, A.M. The effects of physical activity on apoptosis and lubricin expression in articular cartilage in rats with glucocorticoid-induced osteoporosis. J. Bone Miner. Metab. 2013, 31, 274–84. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Loreto, C.; Carnazza, M.L.; Coppolino, F.; Cardile, V.; Leonardi, R. Lubricin is expressed in chondrocytes derived from osteoarthritic cartilage encapsulated in poly (ethylene glycol) diacrylate scaffold. Eur. J. Histochem. 2011, 55, e31. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Mobasheri, A.; Trovato, F.M.; Szychlinska, M.A.; Graziano, A.C.E.; Lo Furno, D.; Avola, R.; Mangano, S.; Giuffrida, R.; Cardile, R. Biosynthesis of collagen I, II, RUNX2 and lubricin at different time points of chondrogenic differentiation in a 3D in vitro model of human mesenchymal stem cells derived from adipose tissue. Acta Histochem. 2014, 116, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Lo Furno, D.; Loreto, C.; Giuffrida, R.; Caggia, S.; Leonardi, R.; Cardile, V. Mesenchymal stem cells from adipose tissue which have been differentiated into chondrocytes in three-dimensional culture express lubricin. Exp. Biol. Med. 2011, 236, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.; Rusu, M.C.; Loreto, F.; Loreto, C.; Musumeci, G. Immunolocalization and expression of lubricin in the bilaminar zone of the human temporomandibular joint disc. Acta Histochem. 2011, 114, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.; Musumeci, G.; Sicurezza, E.; Loreto, C. Lubricin in human temporomandibular joint disc: An immunohistochemical study. Arch. Oral Biol. 2012, 57, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.; Loreto, C.; Talic, N.; Caltabiano, R.; Musumeci, G. Immunolocalization of lubricin in the rat periodontal ligament during experimental tooth movement. Acta Histochem. 2012, 114, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Trovato, F.M.; Loreto, C.; Leonardi, R.; Szychlinska, M.A.; Castorina, S.; Mobasheri, A. Lubricin expression in human osteoarthritic knee meniscus and synovial fluid: A morphological, immunohistochemical and biochemical study. Acta Histochem. 2014, 116, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Castrogiovanni, P.; Trovato, F.M.; Imbesi, R.; Giunta, S.; Szychlinska, M.A.; Loreto, C.; Castorina, S.; Mobasheri, A. Moderate physical activity ameliorates cartilage degeneration in a rat model of aging: A study on lubricin expression. Scand. J. Med. Sci. Sports 2015, 25, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Trovato, F.M.; Pichler, K.; Weinberg, A.M.; Loreto, C.; Castrogiovanni, P. Extra-virgin olive oil diet and mild physical activity prevent cartilage degeneration in an osteoarthritis model: An in vivo and in vitro study on lubricin expression. J. Nutr. Biochem. 2013, 24, 2064–2075. [Google Scholar] [CrossRef] [PubMed]
- Pichler, K.; Loreto, C.; Leonardi, R.; Reuber, T.; Weinberg, A.M.; Musumeci, G. In rat with glucocorticoid-induced osteoporosis, RANKL is downregulated in bone cells by physical activity (treadmill and vibration stimulation training). Histol. Histopathol. 2013, 28, 1185–1196. [Google Scholar] [PubMed]
- Kurz, B.; Lemke, A.; Kehn, M.; Domm, C.; Patwari, P.; Frank, E.H.; Grodzinsky, A.J.; Schünke, M. Influence of tissue maturation and antioxidants on the apoptotic response of articular cartilage after injurious compression. Arthritis Rheum. 2004, 50, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Lee, J.H.; DiMicco, M.A.; Vanderploeg, E.J.; Blake, S.M.; Hung, H.H.; Plaas, A.H.; James, I.E.; Song, X.Y.; Lark, M.W.; et al. Mechanical injury potentiates proteoglycan catabolism induced by interleukin-6 with soluble interleukin-6 receptor and tumor necrosis factor α in immature bovine and adult human articular cartilage. Arthritis Rheum. 2009, 60, 2985–2996. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.L.; Wishnok, J.S.; Chai, D.H.; Grodzinsky, A.J.; Tannenbaum, S.R. A sodium dodecyl sulfate-polyacrylamide gel electrophoresis-liquid chromatography tandem mass spectrometry analysis of bovine cartilage tissue response to mechanical compression injury and the inflammatory cytokines tumor necrosis factor α and interleukin-1β. Arthritis Rheum. 2008, 58, 489–500. [Google Scholar] [PubMed]
- Dang, A.C.; Kim, H.T. Chondrocyte apoptosis after simulated intraarticular fracture: A comparison of histologic detection methods. Clin. Orthop. Relat. Res. 2009, 467, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Zheng, T.; Zhang, M.; Wang, D.; Du, S.; Li, X.; Fang, J.; Cao, X. Static mechanical stress induces apoptosis in rat endplate chondrocytes through MAPK and mitochondria-dependent caspase activation signaling pathways. PLoS ONE 2013, 8, 69403. [Google Scholar] [CrossRef] [PubMed]
- Zaman, F.; Chrysis, D.; Huntjens, K.; Chagin, A.; Takigawa, M.; Fadeel, B.; Sävendahl, L. Dexamethasone differentially regulates Bcl-2 family proteins in human proliferative chondrocytes: Role of pro-apoptotic Bid. Toxicol. Lett. 2014, 224, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Echtermeyer, F.; Bertrand, J.; Dreier, R.; Meinecke, I.; Neugebauer, K.; Fuerst, M.; Lee, Y.J.; Song, Y.W.; Herzog, C.; Theilmeier, G.; Pap, T. Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nat. Med. 2009, 15, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Peluso, D.; Kanki, K.; Yang, Z.; Majumdar, M.K.; et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005, 434, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Kon, S.; Ikesue, M.; Kimura, C.; Aoki, M.; Nakayama, Y.; Saito, Y.; Kurotaki, D.; Diao, H.; Matsui, Y.; Segawa, T.; et al. Syndecan-4 protects against osteopontin-mediated acute hepatic injury by masking functional domains of osteopontin. J. Exp. Med. 2008, 205, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Iwasaki, N.; Kon, S.; Takahashi, D.; Morimoto, J.; Matsui, Y.; Denhardt, D.T.; Rittling, S.; Minami, A.; Uede, T. Accelerated development of aging-associated and instability-induced osteoarthritis in osteopontin-deficient mice. Arthritis Rheum. 2009, 60, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Galluzzi, L.; Tajeddine, N.; Ortiz, C.; Criollo, A.; Tasdemir, E.; Morselli, E.; Ben Younes, A.; Maiuri, M.C.; Lavandero, S.; et al. Senescence, apoptosis or autophagy? When a damaged cell must decide its path—A mini-review. Gerontology 2008, 54, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Almonte-Becerril, M.; Navarro-Garcia, F.; Gonzalez-Robles, A.; Vega-Lopez, M.A.; Lavalle, C.; Kouri, J.B. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis 2010, 15, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Szychlinska, M.A.; Mobasheri, A. Age-related degeneration of articular cartilage in the pathogenesis of osteoarthritis: Molecular markers of senescent chondrocytes. Histol. Histopathol. 2015, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Caramés, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [PubMed]
- Lotz, M.K.; Caramés, B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat. Rev. Rheumatol. 2011, 7, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, F.; Liang, W.; Ye, H.; Li, H.; Yu, F.; Chen, J.; Chen, W.; Lin, R.; Zheng, C.; et al. Tougu Xiaotong capsule promotes chondrocyte autophagy by regulating the Atg12/LC3 conjugation systems. Int. J. Mol. Med. 2014, 34, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.J.; Luo, W.; Lei, G.H. Role of HIF-1α and HIF-2α in osteoarthritis. Jt. Bone Spine 2015, 82, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Kawakami, Y.; Kobayashi, M.; Greco, N.; Cummins, J.H.; Matsushita, T.; Kuroda, R.; Kurosaka, M.; Fu, F.H.; Huard, J. Local intra-articular injection of rapamycin delays articular cartilage degeneration in a murine model of osteoarthritis. Arthritis Res. Ther. 2014, 16, 482. [Google Scholar] [CrossRef] [PubMed]
- Caramés, B.; Kiosses, W.B.; Akasaki, Y.; Brinson, D.C.; Eap, W.; Koziol, J.; Lotz, M.K. Glucosamine activates autophagy in vitro and in vivo. Arthritis Rheum. 2013, 65, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musumeci, G.; Castrogiovanni, P.; Trovato, F.M.; Weinberg, A.M.; Al-Wasiyah, M.K.; Alqahtani, M.H.; Mobasheri, A. Biomarkers of Chondrocyte Apoptosis and Autophagy in Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 20560-20575. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160920560
Musumeci G, Castrogiovanni P, Trovato FM, Weinberg AM, Al-Wasiyah MK, Alqahtani MH, Mobasheri A. Biomarkers of Chondrocyte Apoptosis and Autophagy in Osteoarthritis. International Journal of Molecular Sciences. 2015; 16(9):20560-20575. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160920560
Chicago/Turabian StyleMusumeci, Giuseppe, Paola Castrogiovanni, Francesca Maria Trovato, Annelie Martina Weinberg, Mohammad K. Al-Wasiyah, Mohammed H. Alqahtani, and Ali Mobasheri. 2015. "Biomarkers of Chondrocyte Apoptosis and Autophagy in Osteoarthritis" International Journal of Molecular Sciences 16, no. 9: 20560-20575. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160920560