
Gene Expression Variations of Red—White Skin Coloration in Common Carp (Cyprinus carpio)

Abstract
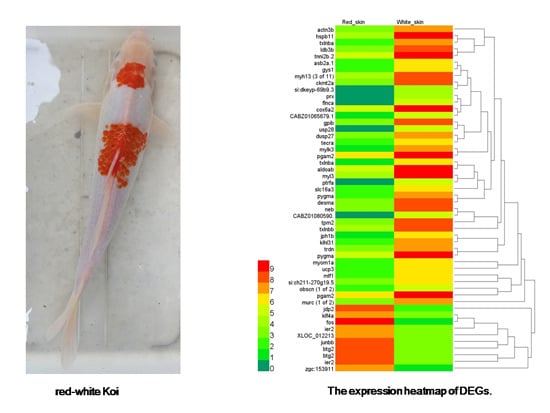
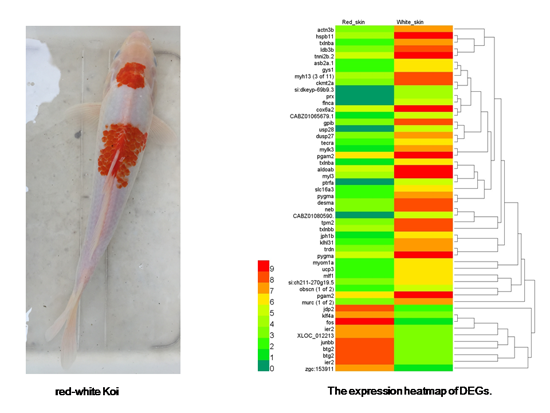
:
1. Introduction
2. Results and Discussion
2.1. Transcriptome Assembly of Red—White Skin in Koi

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
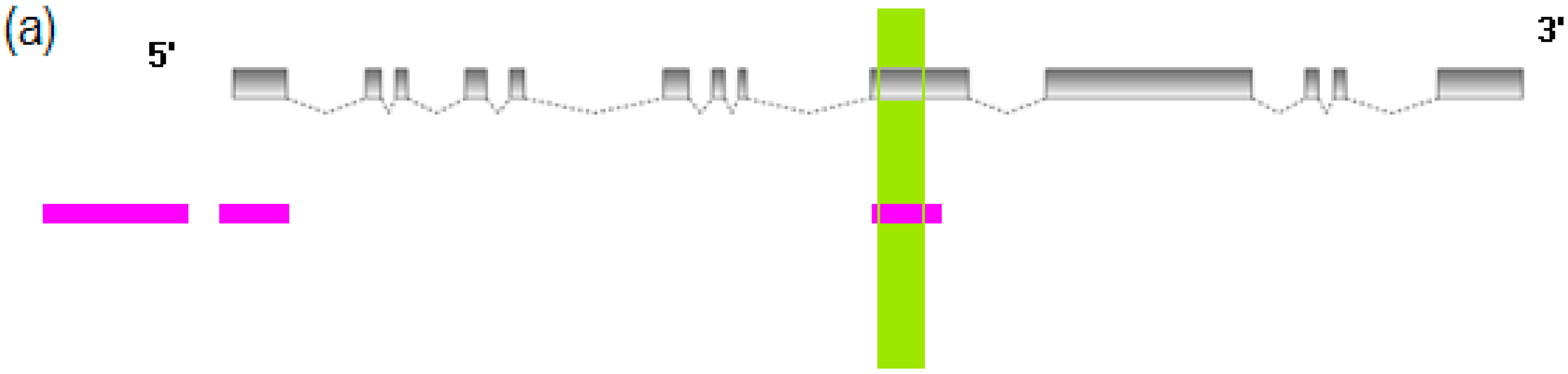{kind=link}
{kind=link}
| Class Code | Transcript Number | Percentage | Description |
|---|---|---|---|
| = | 54,270 | 63.2% | Complete match of intron chain |
| j | 27,689 | 32.3% | At least one splice junction was shared with a reference transcript |
| u | 3331 | 3.9% | Unknown, intergenic transcript |
| x | 533 | 0.6% | Antisense transcripts to the reference annotation |
| Total | 85,823 | 100% |
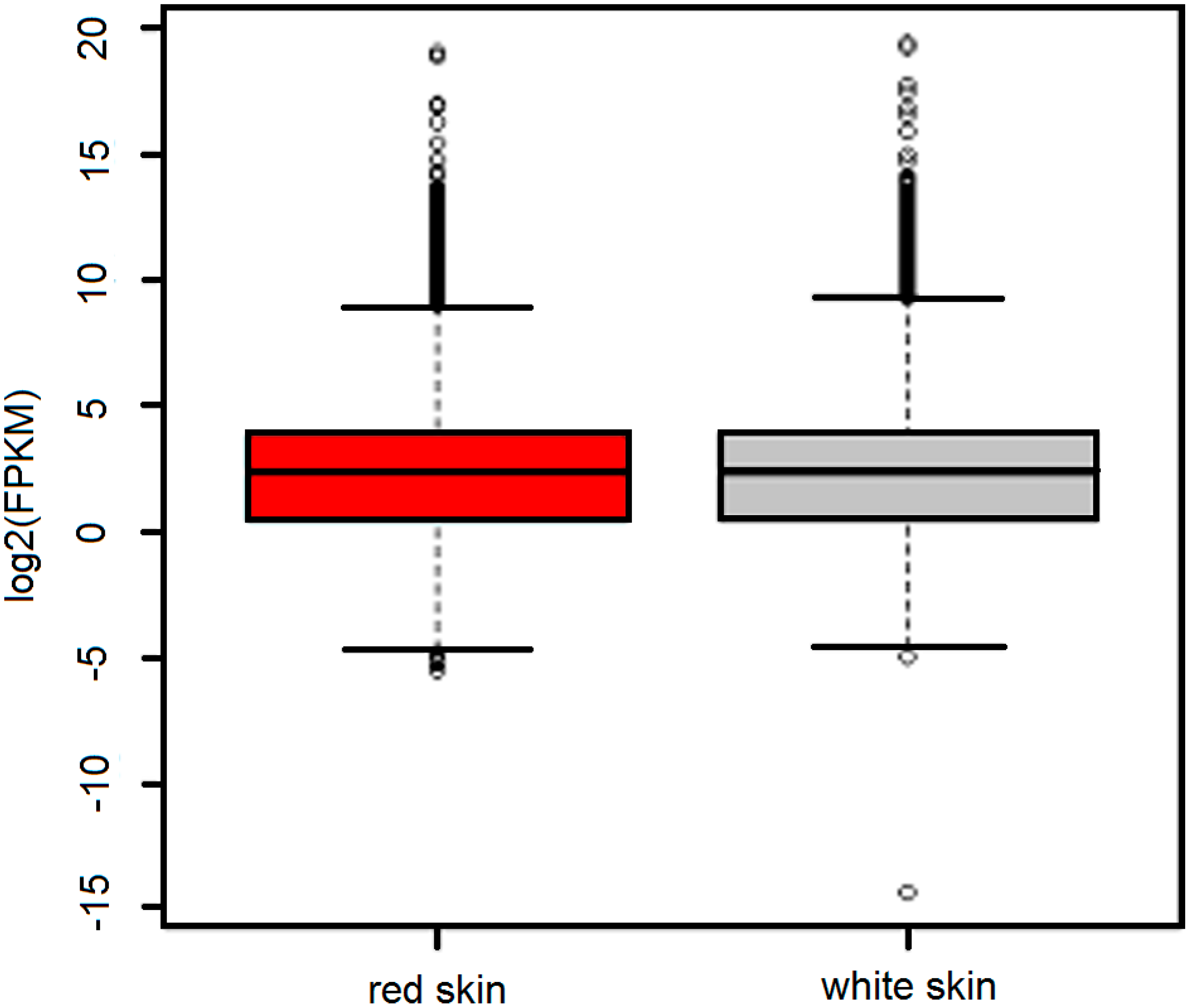
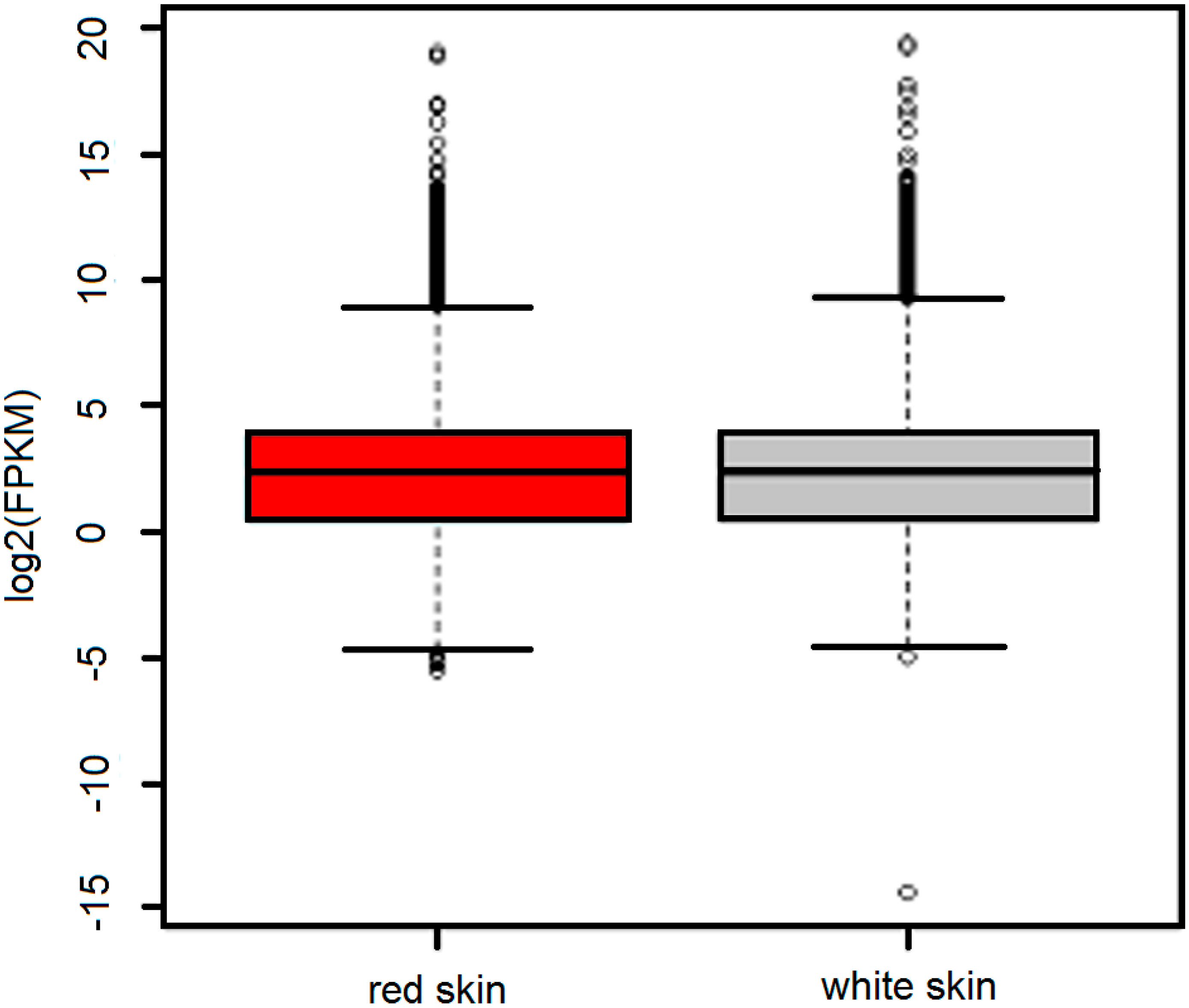
2.2. Overview of the Transcriptome in Red Skin and White Skin
| Type | Red Skin | White Skin | |
|---|---|---|---|
| Published protein-coding genes | 26,914 | 26,812 | |
| Novel protein-coding genes | 2258 | 2277 | |
| Putative ncRNAs | Homologous ncRNAs | 117 | 118 |
| ncRNAs without homolog | 733 | 734 | |
| Total | 850 | 852 | |
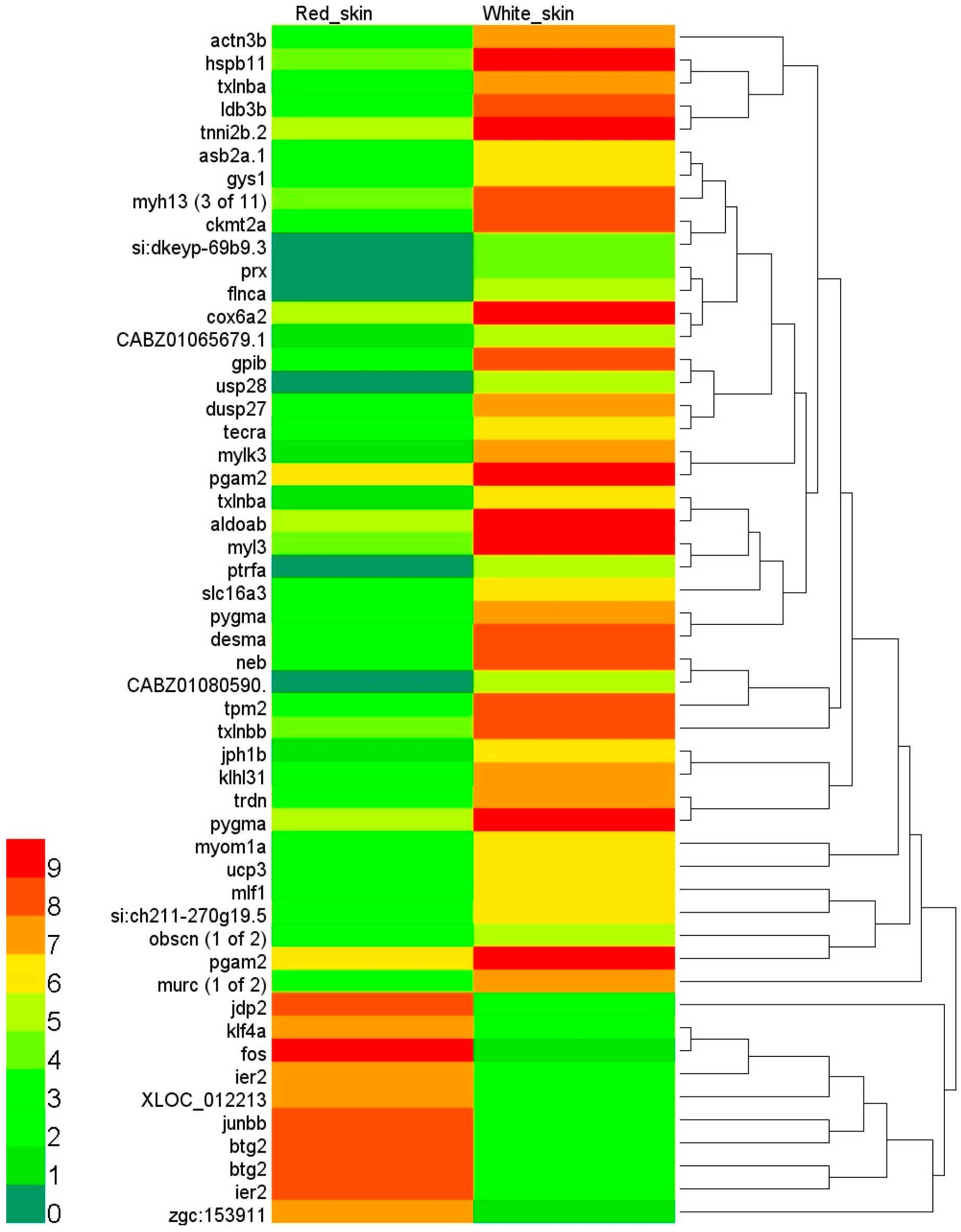
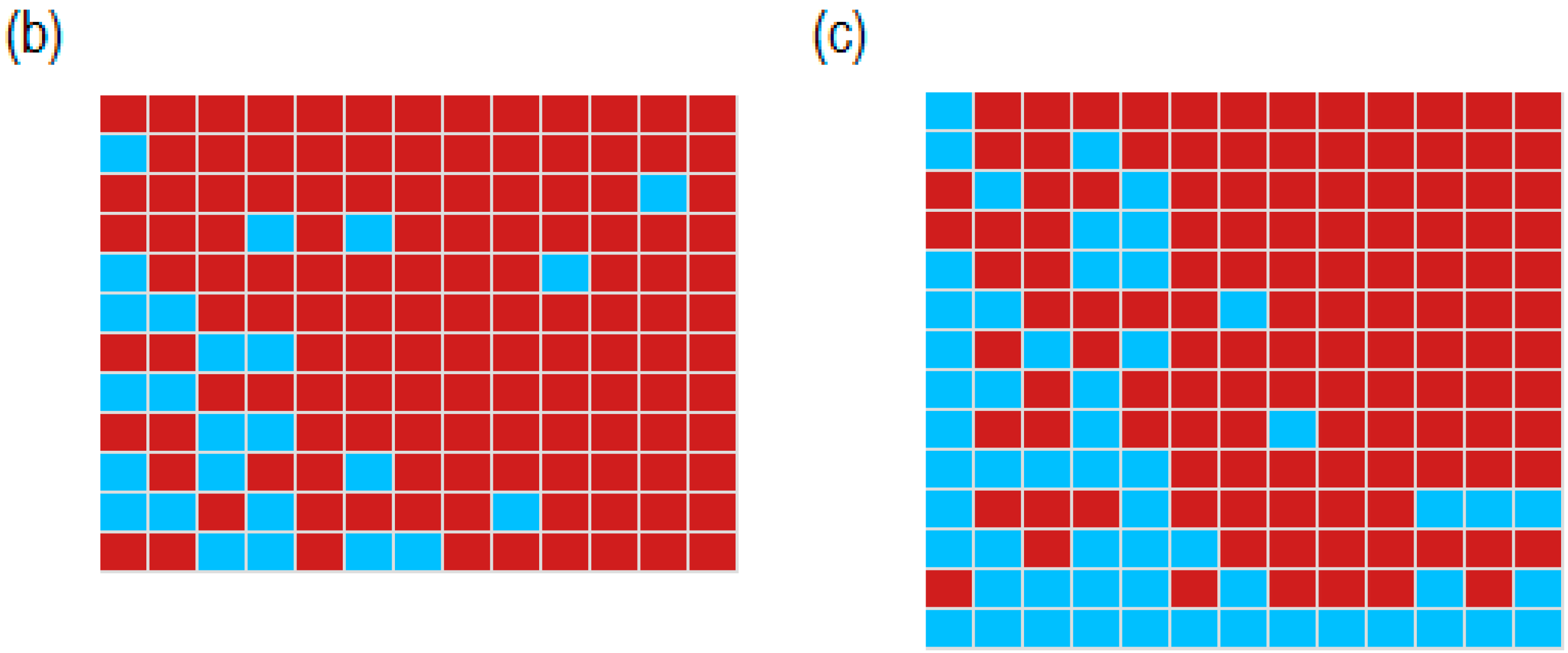
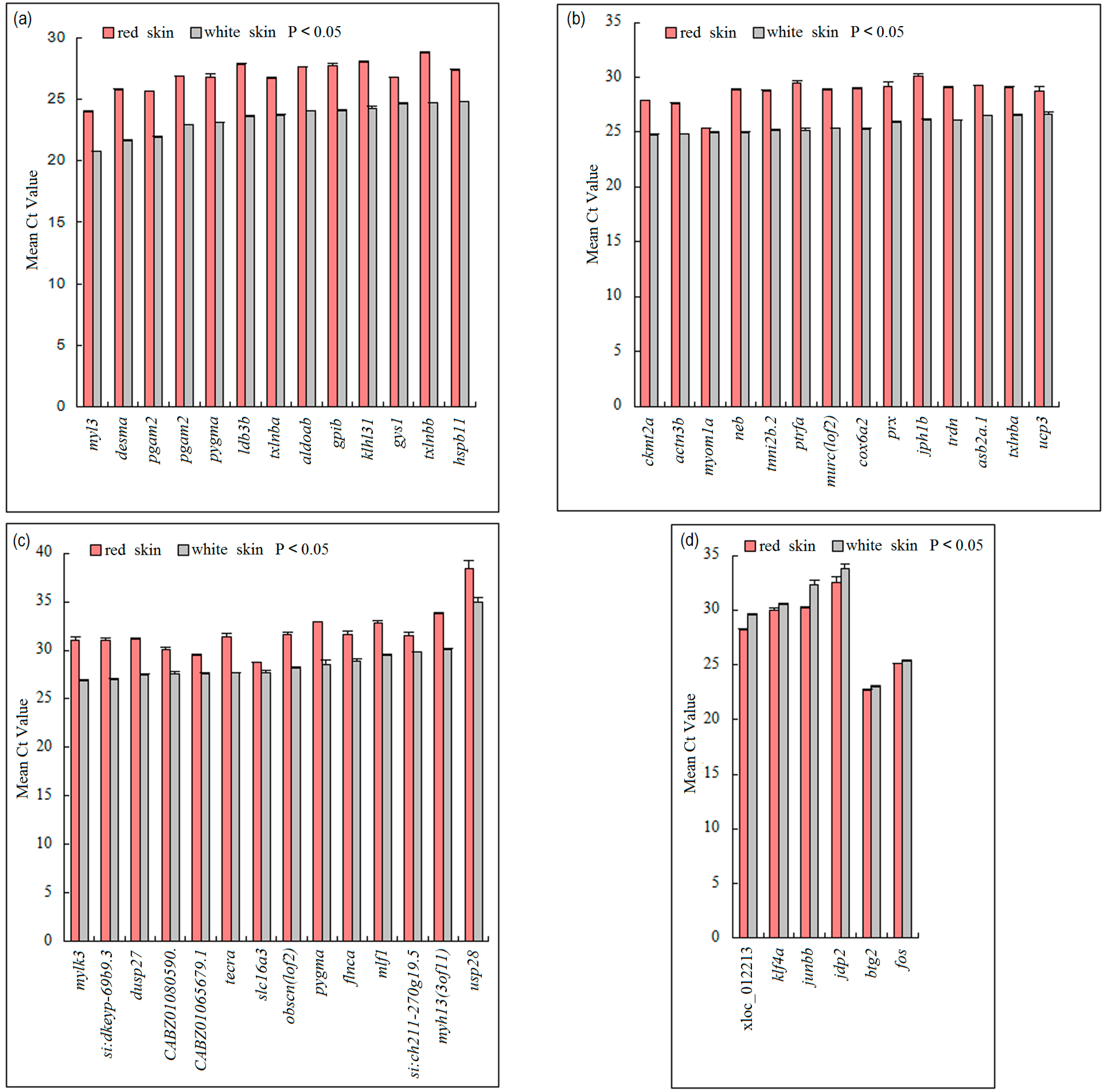
2.3. Identification of Differentially Expressed Genes
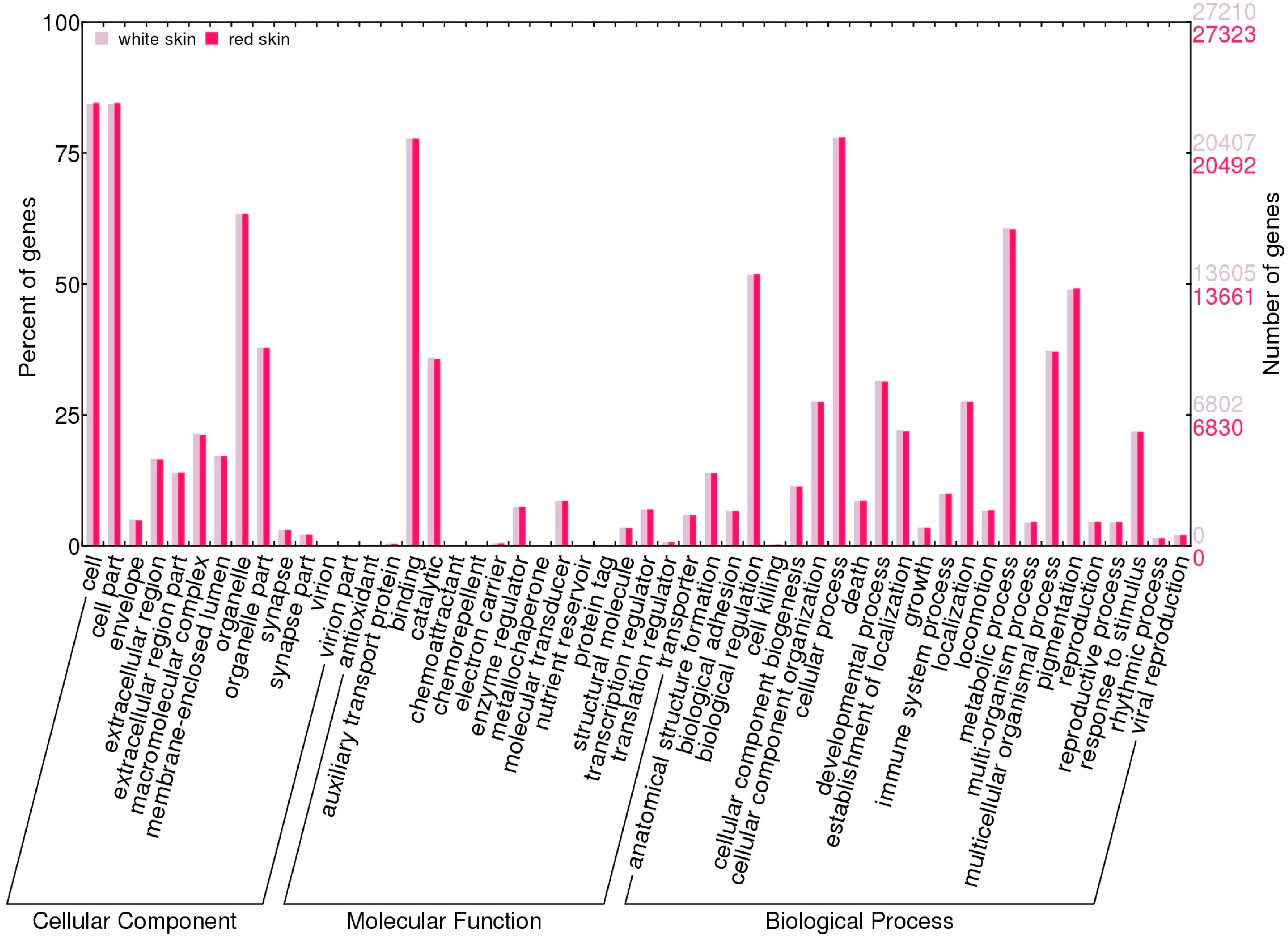
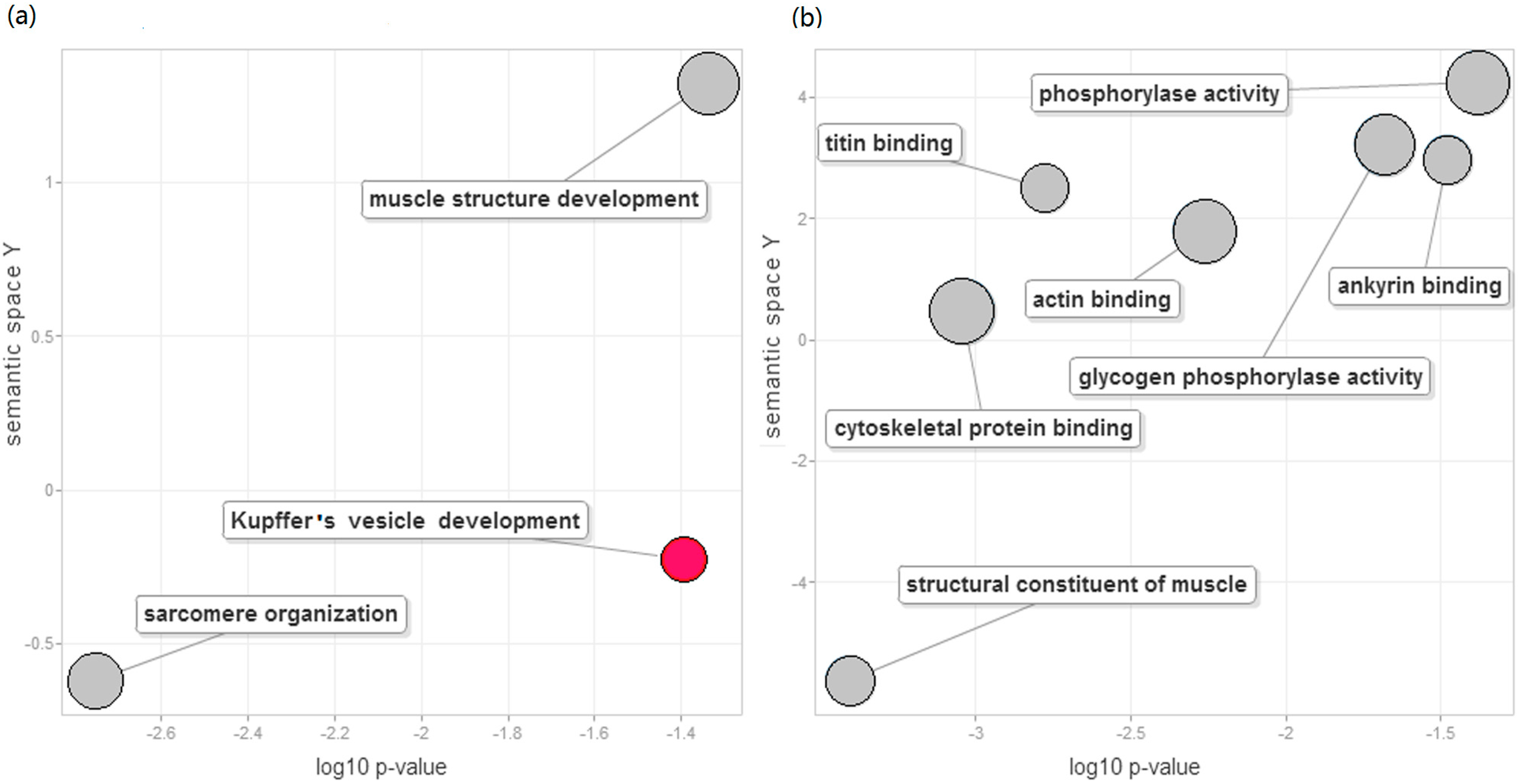
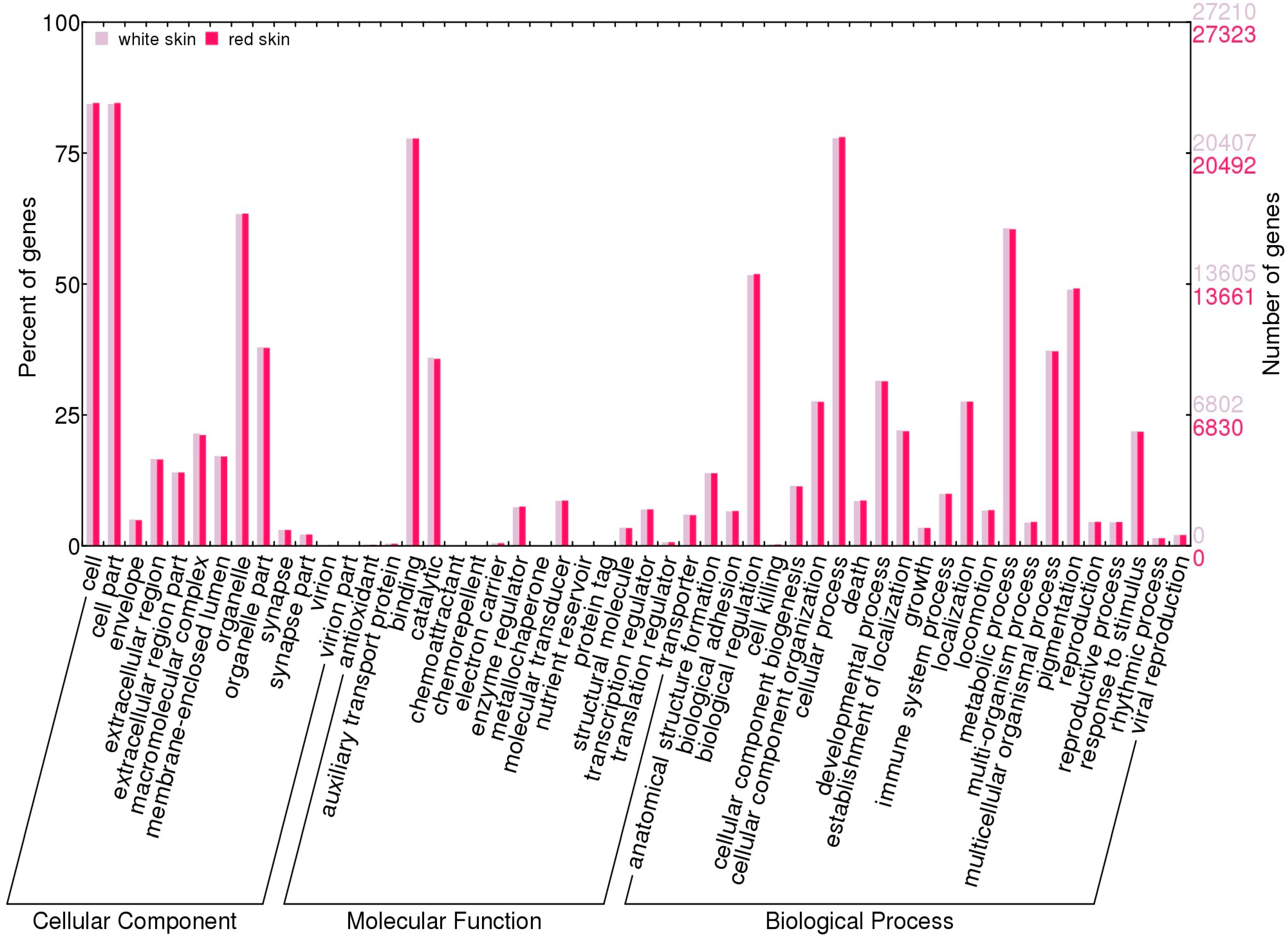
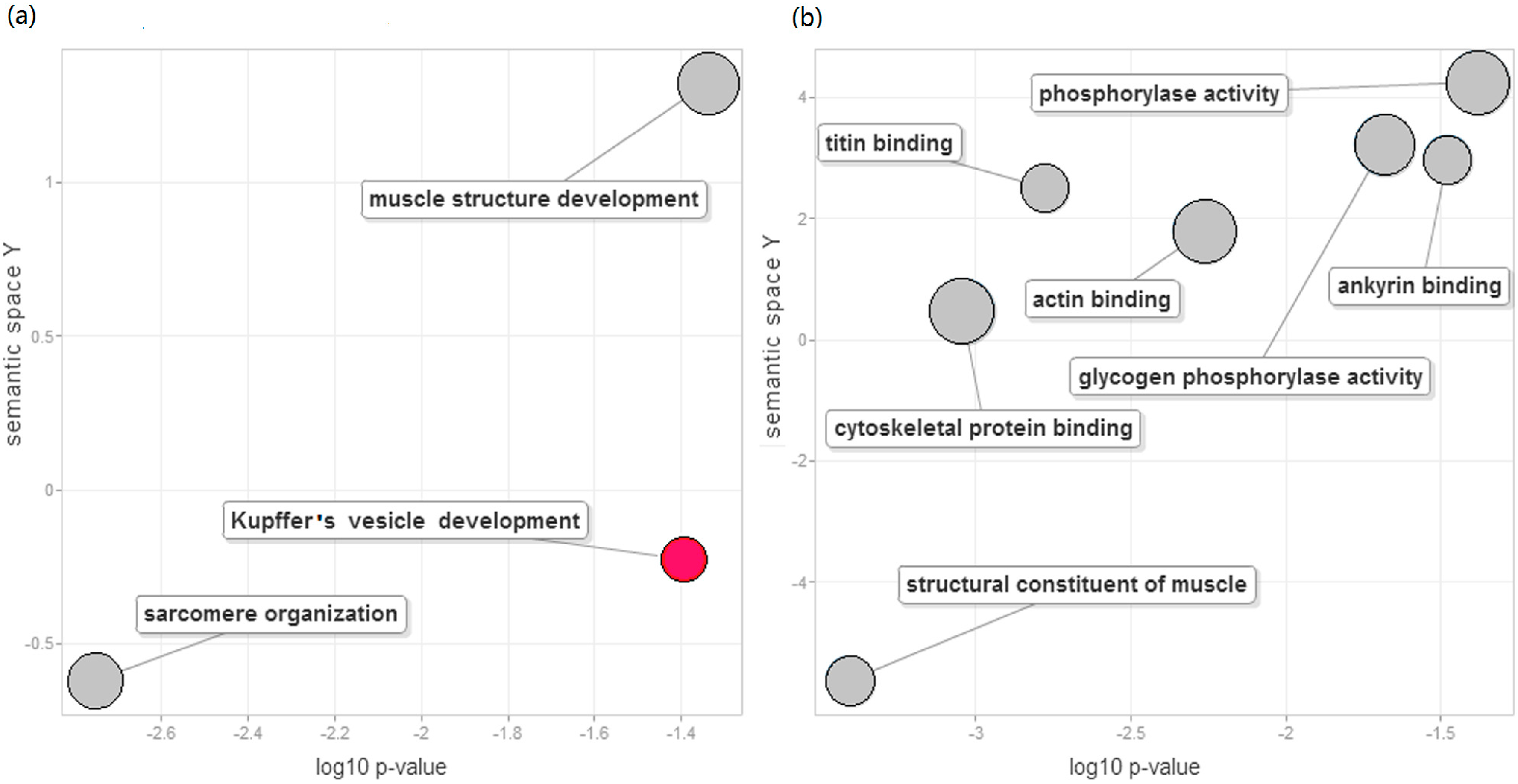
2.4. Function Enrichment Analysis

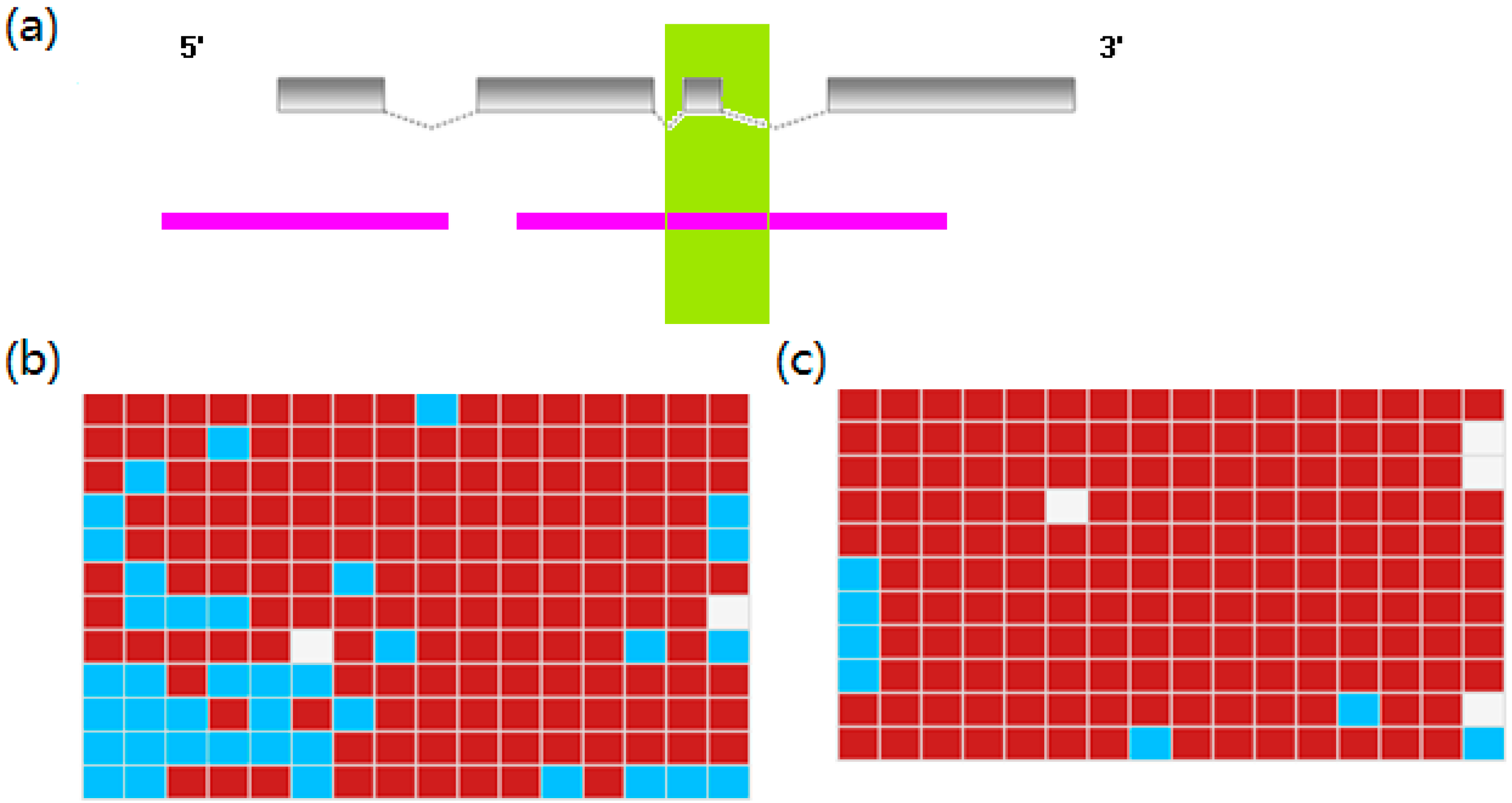
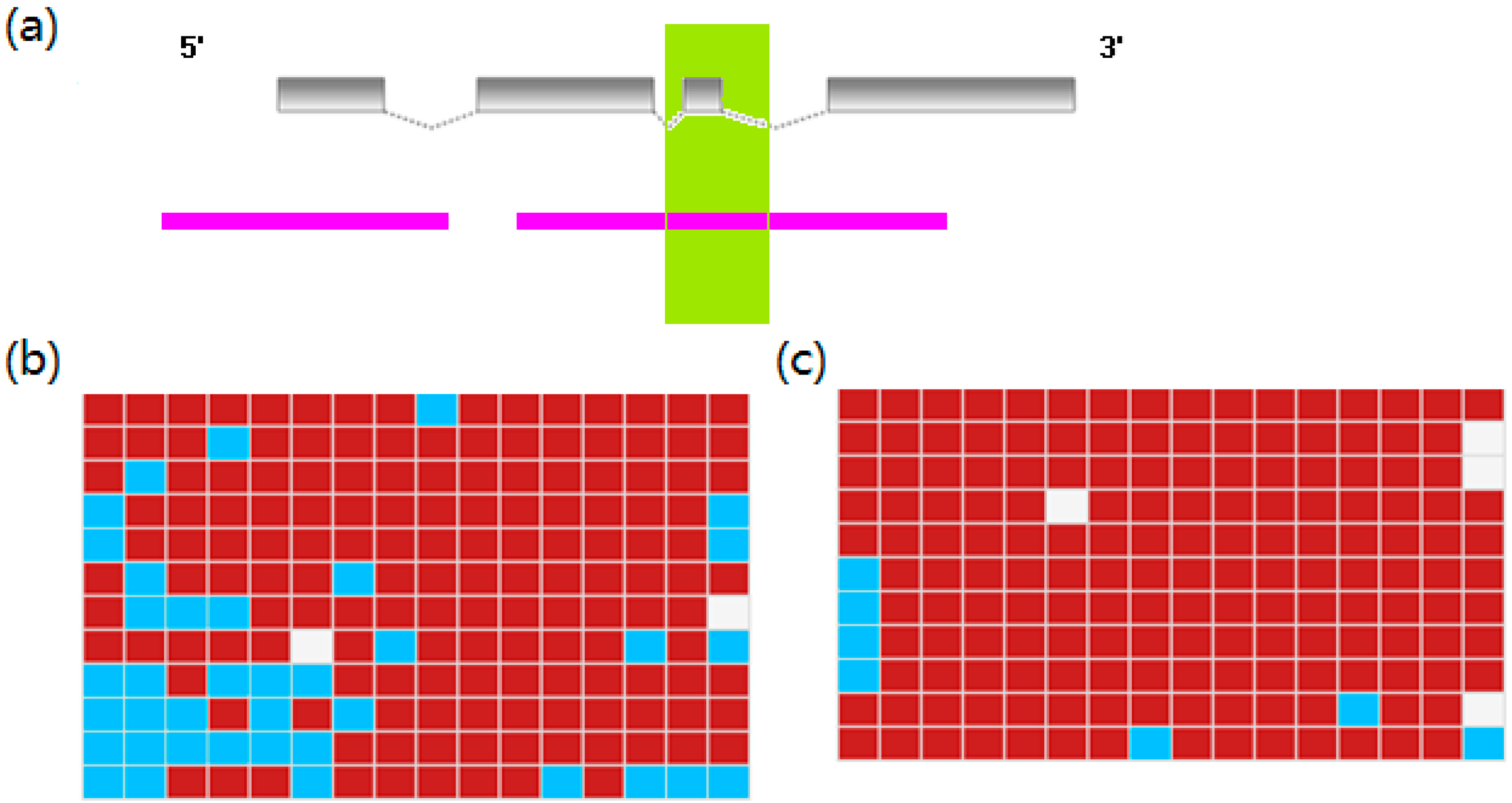
2.5. Differential DNA Methylation Status of DEGs


3. Experimental Section
3.1. Sample Collection and Transcriptome Sequencing
3.2. Identification of Transcriptional Loci in the Skin
3.3. Identifying Differentially Expressed Genes
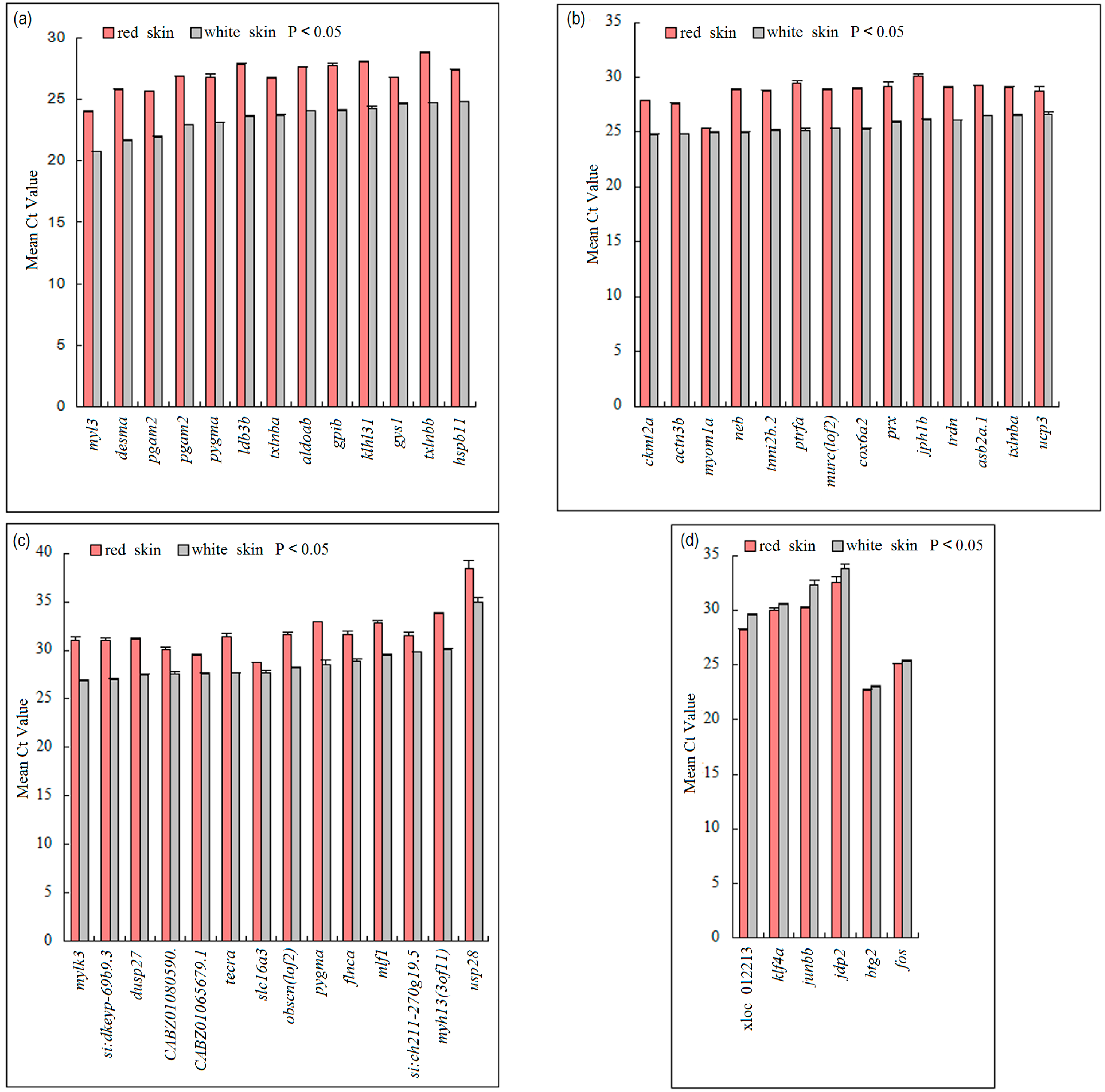
3.4. Validation of DEGs
3.5. Gene Ontology Enrichment Analysis
3.6. BS-PCR Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Steiner, C.C.; Weber, J.N.; Hoekstra, H.E. Adaptive variation in beach mice produced by two interacting pigmentation genes. PLoS Biol. 2007, 5, e219. [Google Scholar] [CrossRef] [PubMed]
- Protas, M.E.; Patel, N.H. Evolution of coloration patterns. Annu. Rev. Cell Dev. Biol. 2008, 24, 425–446. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S. Skin colour: No hiding in the dark. Nature 2014, 515, S121–S123. [Google Scholar] [CrossRef] [PubMed]
- Oetting, W.S.; King, R.A. Molecular basis of albinism: Mutations and polymorphisms of pigmentation genes associated with albinism. Hum. Mutat. 1999, 13, 99–115. [Google Scholar] [CrossRef]
- Parichy, D.M.; Mellgren, E.M.; Rawls, J.F.; Lopes, S.S.; Kelsh, R.N.; Johnson, S.L. Mutational analysis of endothelin receptor b1 (rose) during neural crest and pigment pattern development in the zebrafish danio rerio. Dev. Biol. 2000, 227, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Gariepy, C.E.; Cass, D.T.; Yanagisawa, M. Null mutation of endothelin receptor type b gene in spotting lethal rats causes aganglionic megacolon and white coat color. Proc. Natl. Acad. Sci. USA 1996, 93, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Jackson, I.J. Homologous pigmentation mutations in human, mouse and other model organisms. Hum. Mol. Genet. 1997, 6, 1613–1624. [Google Scholar] [CrossRef] [PubMed]
- Le Guellec, D.; Morvan-Dubois, G.; Sire, J.Y. Skin development in bony fish with particular emphasis on collagen deposition in the dermis of the zebrafish (danio rerio). Int. J. Dev. Biol. 2004, 48, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Braasch, I.; Schartl, M.; Volff, J.N. Evolution of pigment synthesis pathways by gene and genome duplication in fish. BMC Evol. Biol. 2007, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Quigley, I.K.; Turner, J.M.; Nuckels, R.J.; Manuel, J.L.; Budi, E.H.; MacDonald, E.L.; Parichy, D.M. Pigment pattern evolution by differential deployment of neural crest and post-embryonic melanophore lineages in danio fishes. Development 2004, 131, 6053–6069. [Google Scholar] [CrossRef] [PubMed]
- Mellgren, E.M.; Johnson, S.L. The evolution of morphological complexity in zebrafish stripes. Trends Genet. 2002, 18, 128–134. [Google Scholar] [CrossRef]
- Odenthal, J.; Rossnagel, K.; Haffter, P.; Kelsh, R.N.; Vogelsang, E.; Brand, M.; van Eeden, F.J.; Furutani-Seiki, M.; Granato, M.; Hammerschmidt, M.; et al. Mutations affecting xanthophore pigmentation in the zebrafish, danio rerio. Development 1996, 123, 391–398. [Google Scholar] [PubMed]
- Haffter, P.; Odenthal, J.; Mullins, M.C.; Lin, S.; Farrell, M.J.; Vogelsang, E.; Haas, F.; Brand, M.; van Eeden, F.J.; Furutani-Seiki, M.; et al. Mutations affecting pigmentation and shape of the adult zebrafish. Dev. Genes Evol. 1996, 206, 260–276. [Google Scholar] [CrossRef] [PubMed]
- Balon, E.K. Origin and domestication of the wild carp, cyprinus carpio: From roman gourmets to the swimming flowers. Aquaculture 1995, 129, 3–48. [Google Scholar] [CrossRef]
- Bar, I.; Kaddar, E.; Velan, A.; David, L. Melanocortin receptor 1 and black pigmentation in the japanese ornamental carp (cyprinus carpio var. Koi). Front. Genet. 2013, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Balon, E.K. About the oldest domesticates among fishes. J. Fish Biol. 2004, 65, 1–27. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. Tophat: Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhang, X.; Wang, X.; Li, J.; Liu, G.; Kuang, Y.; Xu, J.; Zheng, X.; Ren, L.; Wang, G.; et al. Genome sequence and genetic diversity of the common carp, cyprinus carpio. Nat. Genet. 2014, 46, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with tophat and cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, F.; Khanin, R.; Liang, Y.; Pirun, M.; Krek, A.; Zumbo, P.; Mason, C.E.; Socci, N.D.; Betel, D. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013, 14, R95. [Google Scholar] [CrossRef] [PubMed]
- Henning, F.; Jones, J.C.; Franchini, P.; Meyer, A. Transcriptomics of morphological color change in polychromatic midas cichlids. BMC Genom. 2013, 14, 171. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, I. The pteridine pathway in zebrafish: Regulation and specification during the determination of neural crest cell-fate. Pigment Cell Res. 2003, 16, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Chavan, B.; Gillbro, J.M.; Rokos, H.; Schallreuter, K.U. GTP cyclohydrolase feedback regulatory protein controls cofactor 6-tetrahydrobiopterin synthesis in the cytosol and in the nucleus of epidermal keratinocytes and melanocytes. J. Investig. Dermatol. 2006, 126, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Muffley, L.A.; Zhu, K.Q.; Engrav, L.H.; Gibran, N.S.; Hocking, A.M. Spatial and temporal localization of the melanocortin 1 receptor and its ligand alpha-melanocyte-stimulating hormone during cutaneous wound repair. J. Histochem. Cytochem. 2011, 59, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Obika, M. Morphological and biochemical characterization of goldfish erythrophores and their pterinosomes. J. Cell Biol. 1968, 39, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Bagnara, J.T.; Kreutzfeld, K.L.; Fernandez, P.J.; Cohen, A.C. Presence of pteridine pigments in isolated iridophores. Pigment Cell Res. 1988, 1, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Essner, J.J.; Amack, J.D.; Nyholm, M.K.; Harris, E.B.; Yost, H.J. Kupffer’s vesicle is a ciliated organ of asymmetry in the zebrafish embryo that initiates left-right development of the brain, heart and gut. Development 2005, 132, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. Quickgo: A web-based tool for gene ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.K.; Dawid, I.B. Fgf-dependent left-right asymmetry patterning in zebrafish is mediated by ier2 and fibp1. Proc. Natl. Acad. Sci. USA 2009, 106, 2230–2235. [Google Scholar] [CrossRef] [PubMed]
- Zarnescu, O. Ultrastructure of the skin melanophores and iridophores in paddlefish, polyodon spathula. Micron 2007, 38, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Fujii, R. Cytophysiology of fish chromatophores. Int. Rev. Cytol. 1993, 143, 191–255. [Google Scholar]
- Obika, M.; Meyer-Rochow, V.B. Dermal and epidermal chromatophores of the antarctic teleost trematomus bernacchii. Pigment Cell Res. 1990, 3, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Rohrlich, S.T.; Porter, K.R. Fine structural observations relating to the production of color by the iridophores of a lizard. Anolis carolinensis. J. Cell Biol. 1972, 53, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Rochow, V.B.; Royuela, M.; Fraile, B.; Paniagua, R. Smooth muscle proteins as intracellular components of the chromatophores of the antarctic fishes pagothenia borchgrevinki and trematomus bernacchii (nototheniidae). Protoplasma 2001, 218, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Uribe, R.A.; Yieh, L.; Nuckels, R.; Gross, J.M. Zebrafish mutations in gart and paics identify crucial roles for de novo purine synthesis in vertebrate pigmentation and ocular development. Development 2009, 136, 2601–2611. [Google Scholar] [CrossRef] [PubMed]
- Higdon, C.W.; Mitra, R.D.; Johnson, S.L. Gene expression analysis of zebrafish melanocytes, iridophores, and retinal pigmented epithelium reveals indicators of biological function and developmental origin. PLoS ONE 2013, 8, e67801. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.N. Glycogen phosphorylase: Control by phosphorylation and allosteric effectors. FASEB J. 1992, 6, 2274–2282. [Google Scholar] [PubMed]
- Robertson, K.D.; Jones, P.A. DNA methylation: Past, present and future directions. Carcinogenesis 2000, 21, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Segre, J.A.; Bauer, C.; Fuchs, E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 1999, 22, 356–360. [Google Scholar] [PubMed]
- Chen, X.; Whitney, E.M.; Gao, S.Y.; Yang, V.W. Transcriptional profiling of kruppel-like factor 4 reveals a function in cell cycle regulation and epithelial differentiation. J. Mol. Biol. 2003, 326, 665–677. [Google Scholar] [CrossRef]
- Tarkowski, A.K.; Jagiello, K.; Czolowska, R.; Ozdzenski, W. Mouse chimaeras developed from electrofused blastocysts: New evidence for developmental plasticity of the inner cell mass. Int. J. Dev. Biol. 2005, 49, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Lee, H.M.; Qin, H.; Li, J.W.; Gao, Z.; Liu, X.; Chan, L.L.; Kl Lam, V.; So, W.Y.; Wang, Y.; et al. Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation. Genome Biol. 2014, 15, 408. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The miqe guidelines: Minimum information for publication of quantitative real-time pcr experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J.; et al. The human pancreatic islet transcriptome: Expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012, 8, e1002552. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Shannon, C.P.; Fishbane, N.; Ruan, J.; Zhou, M.; Balshaw, R.; Wilson-McManus, J.E.; Ng, R.T.; McManus, B.M.; Tebbutt, S.J. Variation in RNA-seq transcriptome profiles of peripheral whole blood from healthy individuals with and without globin depletion. PLoS ONE 2014, 9, e91041. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Li, J.T.; Kuang, Y.Y.; Xu, R.; Zhao, Z.X.; Hou, G.Y.; Liang, H.W.; Sun, X.W. The transcriptomes of the crucian carp complex (Carassius auratus) provide insights into the distinction between unisexual triploids and sexual diploids. Int. J. Mol. Sci. 2014, 15, 9386–9406. [Google Scholar] [CrossRef] [PubMed]
- Li, J.T.; Hou, G.Y.; Kong, X.F.; Li, C.Y.; Zeng, J.M.; Li, H.D.; Xiao, G.B.; Li, X.M.; Sun, X.W. The fate of recent duplicated genes following a fourth-round whole genome duplication in a tetraploid fish, common carp (Cyprinus carpio). Sci. Rep. 2015, 5, 8199. [Google Scholar] [CrossRef] [PubMed]
- Trans Decoder. Available online: http://transdecoder.github.io (accessed on 14th January 2015).
- Cunningham, F.; Amode, M.R.; Barrell, D.; Beal, K.; Billis, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fitzgerald, S.; et al. Ensembl 2015. Nucleic Acids Res. 2015, 43, D662–D669. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Yuan, J.; Li, H.; Li, M.; Zhao, G.; Bu, D.; Zhu, W.; Wu, W.; Chen, R.; Zhao, Y. Noncodev4: Exploring the world of long non-coding RNA genes. Nucleic Acids Res. 2014, 42, D98–D103. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Gerhardt, D.J.; Dinger, M.E.; Crawford, J.; Trapnell, C.; Jeddeloh, J.A.; Mattick, J.S.; Rinn, J.L. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol. 2012, 30, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Komori, H.K.; LaMere, S.; Podshivalova, K.; Salomon, D.R. Finding the active genes in deep RNA-seq gene expression studies. BMC Genom. 2013, 14, 778. [Google Scholar] [CrossRef] [PubMed]
- Yoav Benjamini, D.Y. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- The R Project. Available online: http://www.r-project.org/ (accessed on 12 February 2015).
- Kong, H.; Zhu, M.; Cui, F.; Wang, S.; Gao, X.; Lu, S.; Wu, Y.; Zhu, H. Quantitative assessment of short amplicons in FFPE-derived long-chain RNA. Sci. Rep. 2014, 4, 7246. [Google Scholar] [CrossRef] [PubMed]
- Savan, R.; Igawa, D.; Sakai, M. Cloning, characterization and expression analysis of interleukin-10 from the common carp, cyprinus carpio l. Eur. J. Biochem. 2003, 270, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Hu, W.; Hao, J.; Zhu, Z. Developmental expression of steroidogenic factor-1, cyp19a1a and cyp19a1b from common carp (Cyprinus carpio). Gen. Comp. Endocrinol. 2010, 167, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Li, J.T.; Zhang, X.F.; Sun, X.W. Transcriptome analysis reveals the time of the fourth round of genome duplication in common carp (Cyprinus carpio). BMC Genom. 2012, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.P.; Xue, W.; Wang, J.T.; Wan, Y.M.; Wang, S.L.; Xu, P.; Zhang, Y.; Li, J.T.; Sun, X.W. Identification of common carp (Cyprinus carpio) micrornas and microRNA-related SNPs. BMC Genom. 2012, 13, 413. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- GOatools. Available online: https://github.com/tanghaibao/goatools (accessed on 23 May 2015).
- Hocheberg, Y. A sharper bonferroni procedure for multiple tests of significance. Biometrika 1988, 75, 800–802. [Google Scholar] [CrossRef]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Guo, W.; Romano, J. A generalized Sidak-Holm procedure and control of generalized error rates under independence. Stat. Appl. Genet. Mol. Biol. 2007, 6, Article3. [Google Scholar] [CrossRef] [PubMed]
- Rohde, C.; Zhang, Y.; Reinhardt, R.; Jeltsch, A. BISMA—fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinform. 2010, 11, 230. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.-M.; Song, Y.-N.; Xiao, G.-B.; Zhu, B.-H.; Xu, G.-C.; Sun, M.-Y.; Xiao, J.; Mahboob, S.; Al-Ghanim, K.A.; Sun, X.-W.; et al. Gene Expression Variations of Red—White Skin Coloration in Common Carp (Cyprinus carpio). Int. J. Mol. Sci. 2015, 16, 21310-21329. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160921310
Li X-M, Song Y-N, Xiao G-B, Zhu B-H, Xu G-C, Sun M-Y, Xiao J, Mahboob S, Al-Ghanim KA, Sun X-W, et al. Gene Expression Variations of Red—White Skin Coloration in Common Carp (Cyprinus carpio). International Journal of Molecular Sciences. 2015; 16(9):21310-21329. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160921310
Chicago/Turabian StyleLi, Xiao-Min, Ying-Nan Song, Gui-Bao Xiao, Bai-Han Zhu, Gui-Cai Xu, Ming-Yuan Sun, Jun Xiao, Shahid Mahboob, Khalid A. Al-Ghanim, Xiao-Wen Sun, and et al. 2015. "Gene Expression Variations of Red—White Skin Coloration in Common Carp (Cyprinus carpio)" International Journal of Molecular Sciences 16, no. 9: 21310-21329. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160921310