
Physiological, Ultrastructural and Proteomic Responses in the Leaf of Maize Seedlings to Polyethylene Glycol-Stimulated Severe Water Deficiency

Abstract
:
1. Introduction
2. Results and Discussion
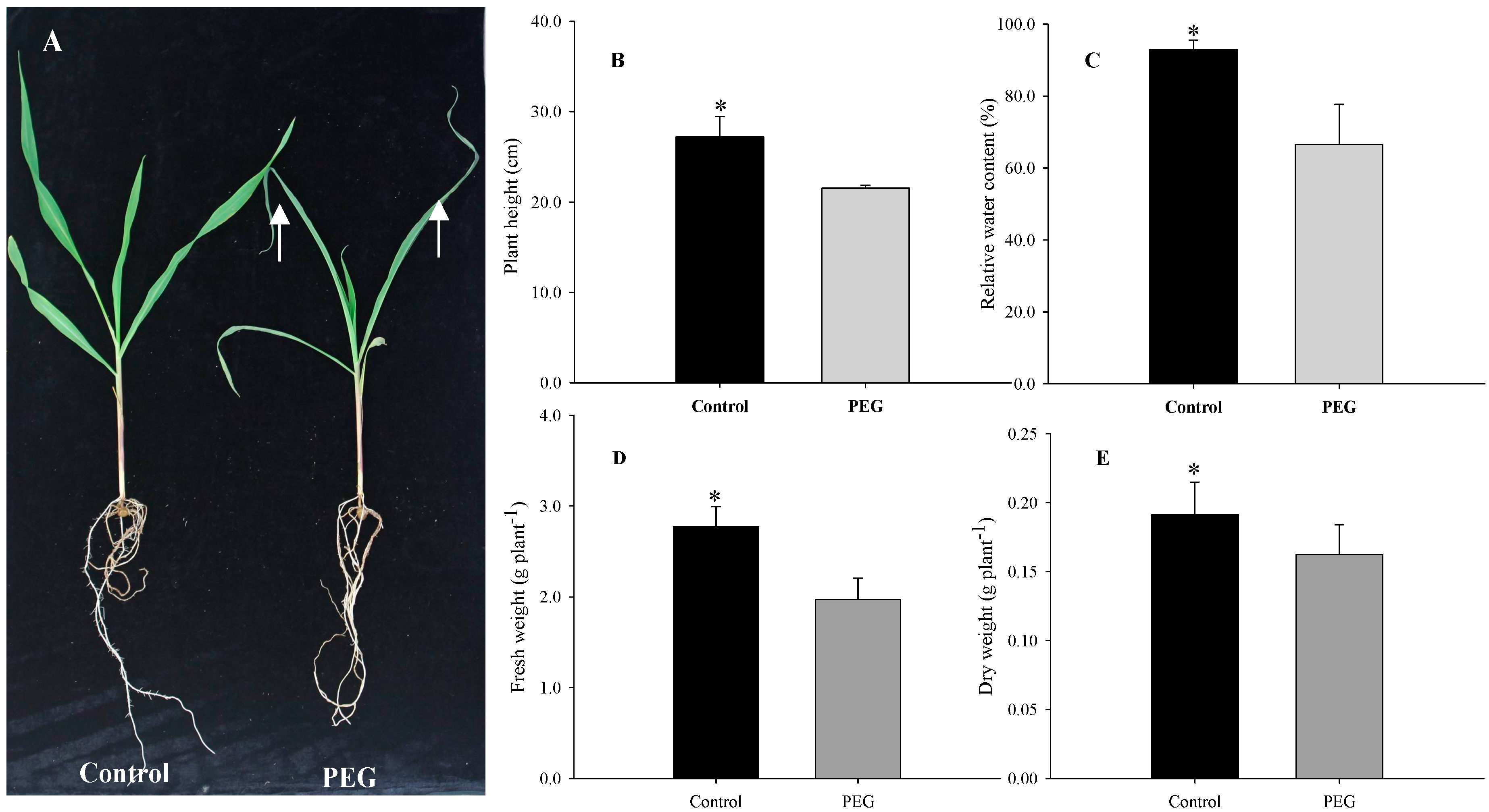
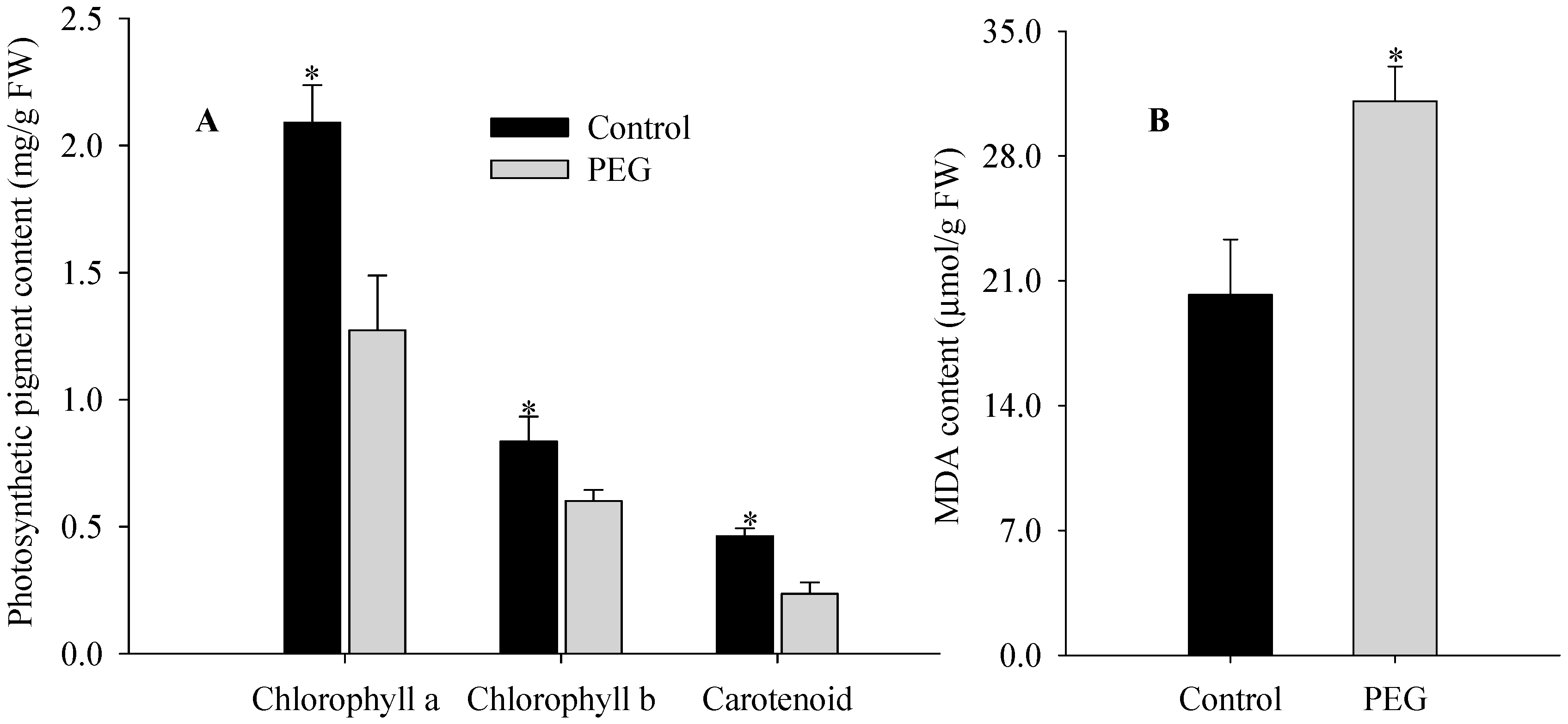
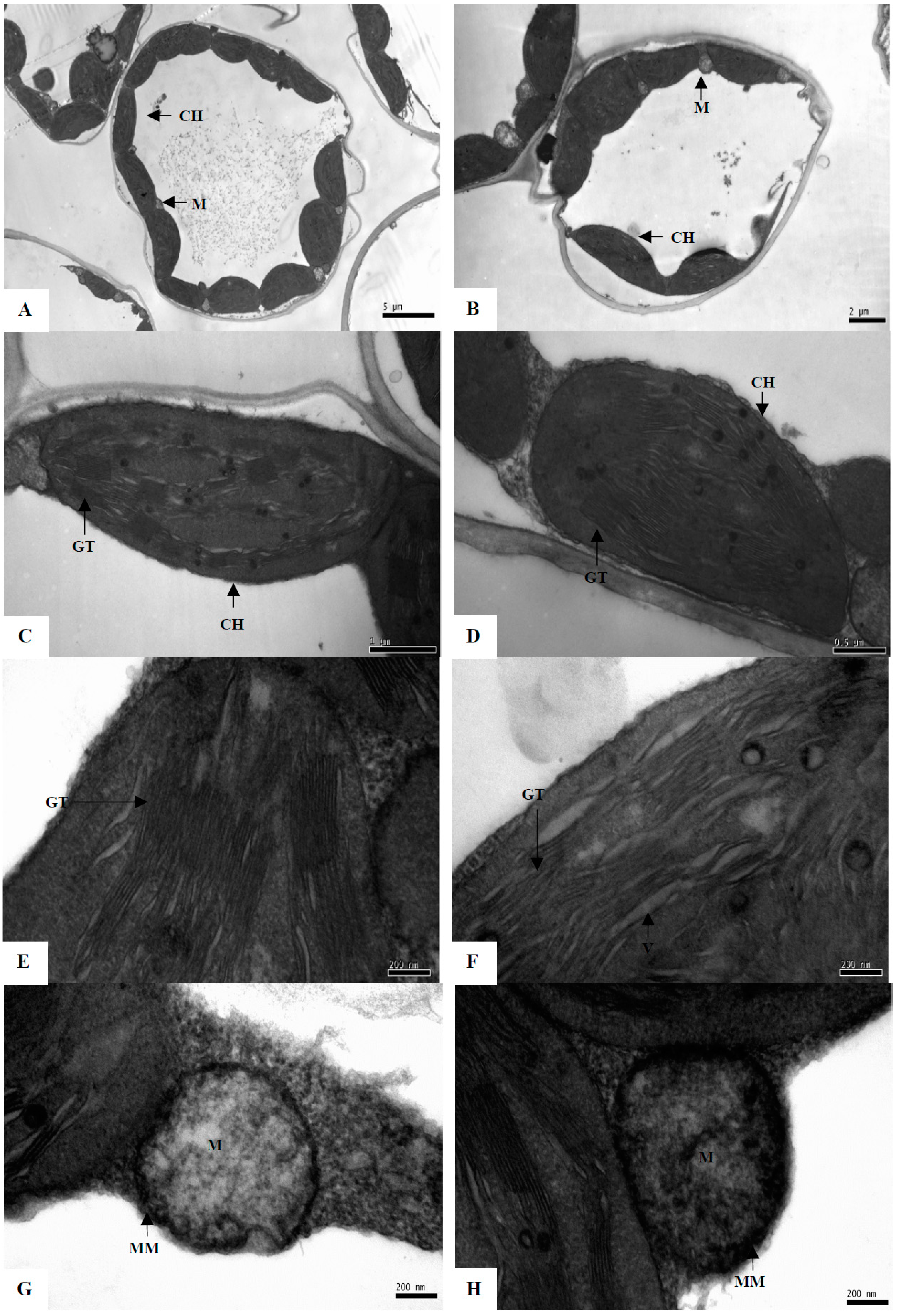
2.1. Morphological, Physiological, and Ultrastructural Changes of Maize Seedlings under PEG-Stimulated Water Deficiency



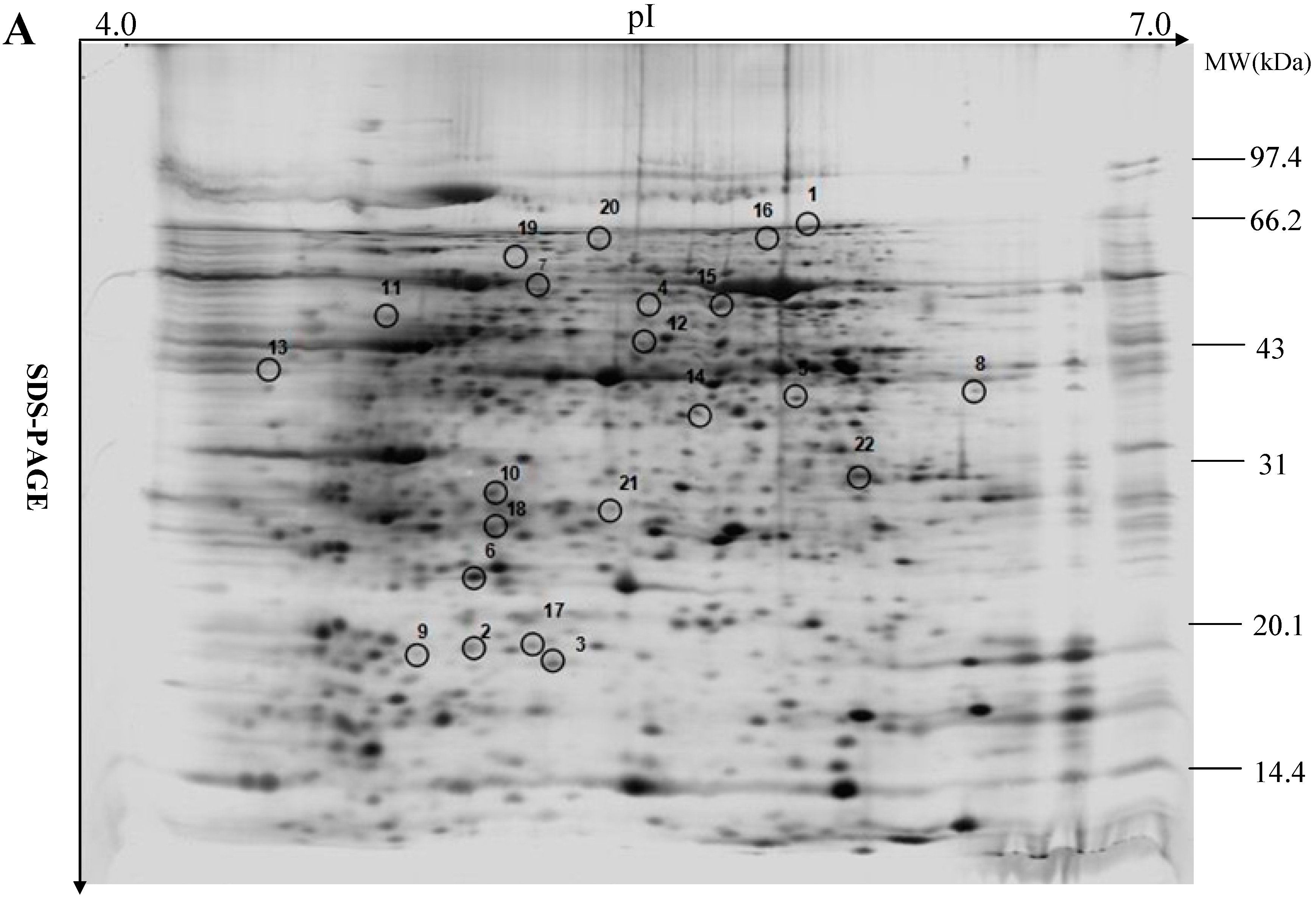
2.2. Leaf Proteome Analysis of Maize Seedlings Exposed to PEG-stimulated Water Deficiency



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot No. a | Protein Names (Species) | Accession No. b | Protein PI/Mw | Coverage (%) | Prot. Score /Ion Score c | Pep. Count | p-Value | Ratio d | Function e | Subcellular Location f |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | β-d-glucan exohydrolase (Triticum aestivum) | gi12227502 | 6.38/64.12 | 33 | 336/285 | 10 | 0.0062 | −6.06 | Carbohydrate metabolism | Other |
| 2 | 17.4 kDa class I heat shock (Zea mays) | gi296512087 | 5.8/17.93 | 24 | 289/221 | 8 | 0.0021 | +5.46 | Stress defense | C |
| 3 | 16.9 kDa class I heat shock protein 1 (Zea mays) | gi296512071 | 5.54/17.15 | 27 | 477/419 | 7 | 0.0017 | +5.16 | Stress defense | C |
| 4 | WD40-like β propeller repeat family protein (Zea mays) | gi413932479 | 5.76/74.24 | 24 | 454/367 | 15 | 0.0017 | −4.98 | Signal transduction | SP |
| 5 | Ribulose-1,5-bisphosphate carboxylase/oxygenase L-subunit (Zea mays) | gi11467200 | 6.33/53.29 | 26 | 489/405 | 14 | 0.0082 | −4.90 | Photosynthesis | C |
| 6 | Ribulose-1,5-bisphosphate carboxylase/oxygenase L-subunit (Zea mays) | gi11467200 | 9.77/21.62 | 35 | 365/259 | 6 | 0.0059 | −3.96 | Photosynthesis | C |
| 7 | Natterin-4 (Triticum urartu) | gi414587984 | 5.72/41.35 | 47 | 246/210 | 7 | 0.0042 | +3.82 | Unknown function | C |
| 9 | Electron carrier/electron transporter iron ion binding protein (Zea mays) | gi414886715 | 7.57/21.20 | 16 | 74/62 | 2 | 0.0014 | +3.44 | Signal transduction | C |
| 10 | Fumarylacetoacetate hydrolase domain-containing protein 1 (Zea mays) | gi226495395 | 5.75/24.74 | 11 | 308/275 | 5 | 0.0037 | +3.32 | Protein metabolism | SP |
| 11 | Ribulose bisphosphate carboxylase/oxygenase activase (Zea mays) | gi413920135 | 5.89/51.23 | 42 | 196/149 | 11 | 0.0023 | −3.48 | Photosynthesis | C |
| 12 | Translation elongation/initiation factor family protein (Zea mays) | gi414873962 | 6.11/48.68 | 45 | 498/383 | 15 | 0.0066 | +3.28 | Protein metabolism | M |
| 13 | Fructose-bisphosphate aldolase (Zea mays) | gi223975775 | 6.37/38.41 | 16 | 76/44 | 6 | 0.0087 | +3.28 | Carbohydrate metabolism | M |
| 14 | RNA-binding protein FUS-like isoform X2 (Setaria italica) | gi414876731 | 5.96/33.59 | 32 | 752/663 | 11 | 0.0017 | +3.16 | Signal transduction | Other |
| 15 | Pore-forming toxin-like protein Hfr-2 (Triticum aestivum) | gi414587991 | 6.53/42.29 | 49 | 959/789 | 19 | 0.0029 | +3.08 | Unknown function | C |
| 16 | Ribulose-1,5-bisphosphate carboxylase/oxygenase L-subunit (Zea mays) | gi11467200 | 6.33/53.29 | 31 | 100/259 | 16 | 0.0032 | +3.04 | Photosynthesis | C |
| 17 | Eukaryotic translation initiation factor 5A (Zea mays) | gi162464130 | 5.61/17.71 | 29 | 77/40 | 5 | 0.0024 | −3.02 | Protein metabolism | Other |
2.3. Subcellular Location of the Identified Protein Species
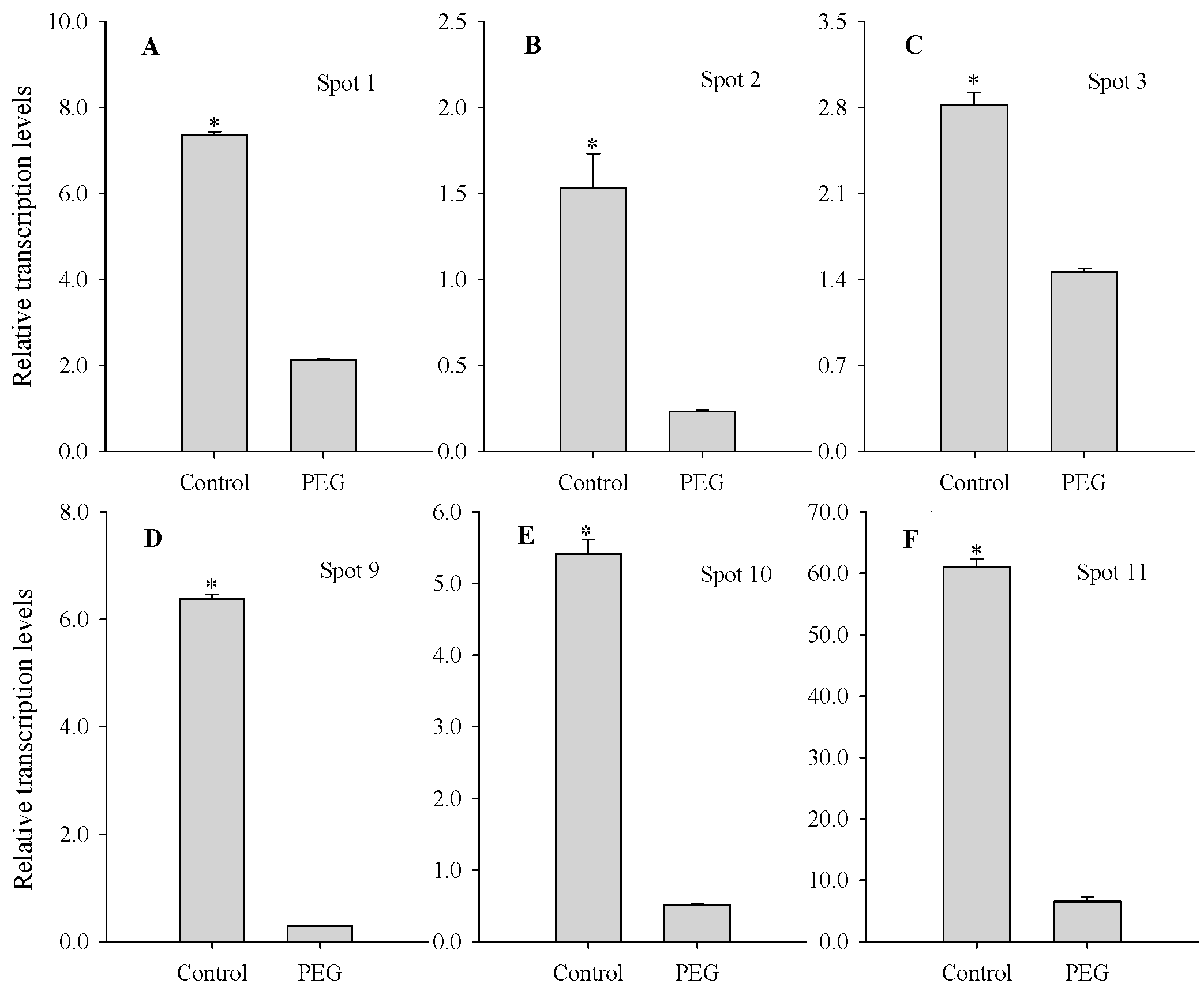
2.4. Comparison between mRNA and Protein Levels of PEG Stress-Responsive Protein Species

2.5. Comparisons of Proteome Expression Patterns between this Study and Others
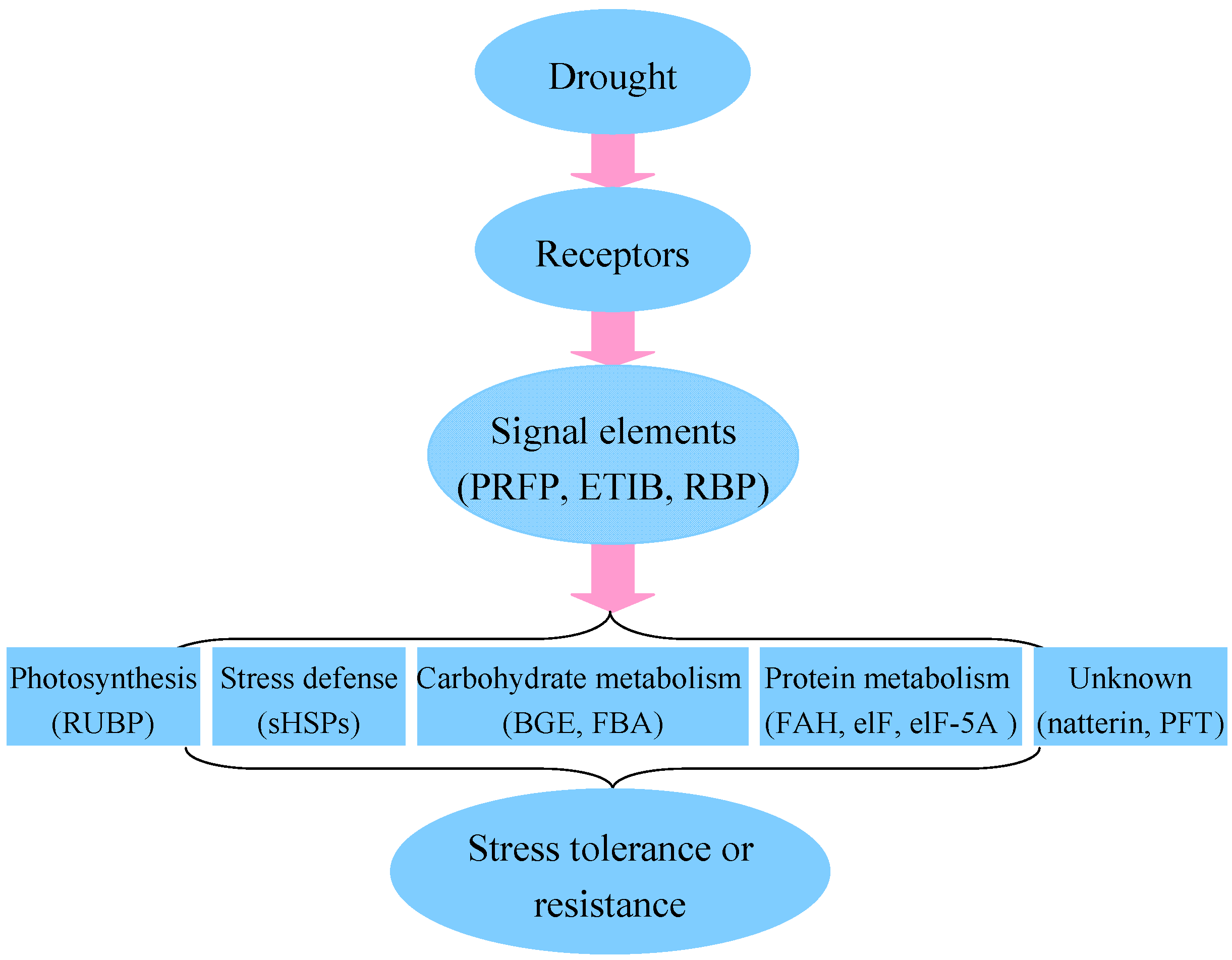
2.6. Putative Mechanism of Water Deficiency in Higher Plants

3. Experimental Section
3.1. Plant Materials
3.2. PEG-Stimulated Water Deficiency
3.3. Growth Parameters
3.4. Transmission Electron Microscopy
3.5. Measurement of Photosynthetic Pigments and MDA Content
3.6. Protein Extraction
3.7. 2-DE Separation and Image Analysis
3.8. In-Gel Digestion and MALDI-TOF-TOF MS/MS Analysis
3.9. qPCR
| Spot No. | Accession No. of Protein Species | Accession No. of the Related Genes | Forward Primer | Reverse Primer | Product Lengths (bp) |
|---|---|---|---|---|---|
| 1 | gi12227501 | AX053136 | GTGGAACGCATAACGGAATC | GTGGAGACACCTCGGATGAT | 109 |
| 2 | gi296512087 | CBM39185 | GGAGGAGAAGAGGGACACCT | ATGGACGCACTGATCTGCT | 108 |
| 3 | gi296512071 | CBM39177 | ATCCCTTCGACACCATGTTC | CCTTGACCTCCTCCTTCTTG | 159 |
| 9 | gi414886715 | NM_001147896 | GAAGGCAAGGAAATGCTGTC | TATCTTCCACTCCGGCAACT | 153 |
| 10 | gi226495395 | NP_001168488 | TCGTCACCAGCATCATCATC | TGCTCCGTCTCCTTGTTCAC | 109 |
| 11 | gi413920135 | NC_001666 | CTACGCGGTGGACTTGATTT | CAGTTTCGGCTTGTGCTTTA | 121 |
| β-actin | gi168403 | J01238 | GTTTCCTGGGATTGCCGAT | CTGCTGAAAAGTGCTGAG | 130 |
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fulda, S.; Mikkats, S.; Stegmanns, H.; Horn, R. Physiology and proteomics of drought stress acclimation in sunflower (Helianthus annuus L.). Plant Biol. 2011, 13, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra Reddy, A.; Chaitanya, K.V.; Vivekanandan, M. Drought-induced responses of photosynthesis and antioxidant metabolism in higher plants. J. Plant Physiol. 2004, 161, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Achievements and challenges in understanding plant abiotic stress responses and tolerance. Plant Cell Physiol. 2011, 52, 1569–1582. [Google Scholar] [CrossRef] [PubMed]
- De Faria Müller, B.S.; Sakamoto, T.; Silveira, R.D.D.; Zambussi-Carvalho, P.F.; Pereira, M.; Pappas, G.J., Jr.; do Carmo Costa, M.M.; Guimarães, C.M.; Pereira, W.J.; Brondani, C.; et al. Differentially expressed genes during flowering and grain filling in common bean (Phaseolus vulgaris) grown under drought stress conditions. Plant Mol. Biol. Rep. 2014, 32, 438–451. [Google Scholar] [CrossRef]
- Hao, J.H.; Dong, C.J.; Zhang, Z.G.; Wang, X.L.; Shang, Q.M. Insights into salicylic acid responses in cucumber (Cucumis sativus L.) cotyledons based on a comparative proteomic analysis. Plant Sci. 2012, 187, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Kosová, K.; Vítámvása, P.; Prášila, I.T.; Renaut, J. Plant proteome changes under abiotic stress-contribution of proteomics studies to understanding plant stress response. J. Proteom. 2011, 74, 1301–1322. [Google Scholar] [CrossRef] [PubMed]
- Bazargani, M.M.; Sarhadi, E.; Bushehri, A.A.; Matros, A.; Mock, H.P.; Naghavi, M.R.; Hajihoseini, V.; Mardi, M.; Hajirezaei, M.R.; Moradi, F.; et al. A proteomics view on the role of drought-induced senescence and oxidative stress defense in enhanced stem reserves remobilization in wheat. J. Proteom. 2011, 74, 1959–1973. [Google Scholar] [CrossRef] [PubMed]
- Valdés, A.E.; Irar, S.; Majada, J.P.; Rodríguez, A.; Fernández, B.; Pagès, M. Drought tolerance acquisition in Eucalyptus globulus (Labill.): A research on plant morphology, physiology and proteomics. J. Proteom. 2013, 21, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Kausar, R.; Arshad, M.; Shahzad, A.; Komatsu, S. Proteomics analysis of sensitive and tolerant barley genotypes under drought stress. Amino Acids 2013, 44, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Shou, H.; Bordallo, P.; Wang, K. Expression of the Nicotiana protein kinase (NPK1) enhanced drought tolerance in transgenic maize. J. Exp. Bot. 2004, 55, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Lu, M.; Li, C.; Liu, T.; Wang, W.; Wu, J.; Tai, F.; Li, X.; Zhang, J. Differential expression of proteins in maize roots in response to abscisic acid and drought. Acta Physiol. Plant. 2011, 33, 2437–2446. [Google Scholar] [CrossRef]
- Tai, F.J.; Yuan, Z.L.; Wu, X.L.; Zhao, P.F.; Hu, X.L.; Wang, W. Identification of membrane proteins in maize leaves, altered in expression under drought stress through polyethylene glycol treatment. Plant Omics J. 2011, 4, 250–256. [Google Scholar]
- Mexal, J.; Fisher, J.T.; Osteryoung, J.; Reid, C.P.P. Oxygen availability in polyethylene glycol solutions and its implication in plant-water relations. Plant Physiol. 1975, 55, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Z.; Zhou, G.S.; Shimizu, H. Effects of soil drought with nocturnal warming on leaf stomatal traits and mesophyll cell ultrastructure of a perennial grass. Crop Sci. 2009, 49, 1843–1851. [Google Scholar] [CrossRef]
- Giles, K.L.; Cohen, D.; Beardsell, M.F. Effects of water stress on the ultrastructure of leaf cells of Sorghum Bicolor. Plant Physiol. 1976, 57, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, H.; Apweiler, R.; Holzhütter, H.G.; Jungblut, P.R. Finding one’s way in proteomics: A protein species nomenclature. Chem. Cent. J. 2009, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhou, Y.; Zhu, W.; Li, X.; Fan, L.; Zhang, G. Proteomic analysis of cold stress-responsive proteins in Thellungiella rosette leaves. Planta 2009, 230, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ma, Z.; Hu, Y.; Zhang, H.; Wang, C.; Wang, Q.; Ji, W. Expression of special genes inhibited by powdery mildew (Blumeria graminis f. sp. tritici) in wheat germplasm N9436. Front. Agric. China 2010, 4, 10–17. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, W.Y.; Kwak, K.J.; Oh, S.H.; Han, Y.S.; Kang, H. Zinc finger-containing glycine-rich RNA-binding protein in Oryza sativa has an RNA chaperone activity under cold stress conditions. Plant Cell Environ. 2010, 33, 759–768. [Google Scholar] [PubMed]
- Ananieva, E.A.; Gillaspy, G.E.; Ely, A.; Bumette, R.N.; Les Erickson, F. Interaction of the WD40 domain of a myoinositol polyphosphate 5-phosphatase with SnRK1 links inositol, sugar, and stress signaling. Plant Physiol. 2008, 148, 1868–1882. [Google Scholar] [CrossRef] [PubMed]
- Degenkolbe, T.; Do, P.T.; Zuther, E.; Repsilber, D.; Walther, D.; Hincha, D.K.; Köhl, K.I. Expression profiling of rice cultivars differing in their tolerance to long-term drought stress. Plant Mol. Biol. 2009, 69, 133–153. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, B.; Wang, Y.; Liu, G.; Yang, C. Overexpression of a heat shock protein (ThHSP18.3) from Tamarix hispida confers stress tolerance to yeast. Mol. Biol. Rep. 2012, 39, 4889–4897. [Google Scholar] [CrossRef] [PubMed]
- Grigorova, B.; Vaseva, I.I.; Demirevska, K.; Feller, U. Expression of selected heat shock proteins after individually applied and combined drought and heat stress. Acta Physiol. Plant. 2011, 33, 2041–2049. [Google Scholar] [CrossRef]
- Fatehi, F.; Hosseinzadeh, A.; Alizadeh, H.; Brimavandi, T.; Sturuik, P.C. The proteome response of salt-resistant and salt-sensitive barley genotypes to long-term salinity stress. Mol. Biol. Rep. 2012, 39, 6387–6397. [Google Scholar] [CrossRef] [PubMed]
- Pelloux, J.; Jolivet, Y.; Fontaine, V.; Banvoy, J.; Dizengremel, P. Changes in Rubisco and Rubisco activase gene expression and polypeptide content in Pinus halepensis M. subjected to ozone and drought. Plant Cell Environ. 2001, 24, 123–131. [Google Scholar] [CrossRef]
- Xu, L.; Yu, J.; Han, L.; Huang, B. Photosynthetic enzyme activities and gene expression associated with drought tolerance and post-drought recovery in Kentucky bluegrass. Environ. Exp. Bot. 2013, 89, 28–35. [Google Scholar] [CrossRef]
- Huang, G.T.; Ma, S.L.; Bai, L.P.; Zhang, L.; Ma, H.; Jia, P.; Liu, J.; Zhong, M.; Guo, Z.F. Signal transduction during cold, salt, and drought stresses in plants. Mol. Biol. Rep. 2012, 39, 969–987. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Ren, C.; Zhi, T.; Zhou, Z.; Liu, Y.; Chen, F.; Peng, W.; Xie, D. Disruption of fumarylacetoacetate hydrolase causes spontaneous cell death under short-day conditions in Arabidopsis. Plant Physiol. 2013, 162, 1956–1964. [Google Scholar] [CrossRef] [PubMed]
- Syntichaki, P.; Troulinaki, K.; Tavernarakis, N. eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature 2007, 445, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Mahe, A.; Brangeon, J.; Prioul, J.L. A maize vacuolur invertase, IVR2, is induced by water stress. Organ/tissue specificity and diurnal modulation of expression. Plant Physiol. 2000, 124, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Hrmova, M.; Harvey, A.J.; Wang, J.; Shirley, N.J.; Jones, G.P.; Stone, B.A.; Høj, P.B.; Fincher, G.B. Barley beta-d-glucan exohydrolases with beta-d-glucosidase activity. Purification, characterization, and determination of primary structure from a cDNA clone. J. Biol. Chem. 1996, 271, 5277–5286. [Google Scholar] [PubMed]
- Prasad, C.S.; Gupta, S.; Kumar, H.; Tiwari, M. Evolutionary and functional analysis of fructose bisphosphate aldolase of plant parasitic nematodes. Bioinformation 2013, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Liu, X.; Pang, Y.; Yu, T.; Xiao, R.; Jin, M.; Han, Y.; Su, P.; Wang, J.; Lv, L.; et al. Characterization, phylogenetic analysis and cDNA cloning of natterin-like gene from the blood of lamprey, Lampetra japonica. Immunol. Lett. 2012, 148, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, X.; Wang, C.; Chen, X.; Qu, Z.; Yu, X.; Han, Q.; Zhao, J.; Guo, J.; Huang, L.; et al. Wheat defense genes in fungal (Puccinia striiformis) infection. Funct. Integr. Genom. 2010, 10, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, Signal P and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef] [PubMed]
- Abogadallah, G.M. Differential regulation of photorespiratory gene expression by moderate and severe salt and drought stress in relation to oxidative stress. Plant Sci. 2011, 180, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Elberse, I.A.M.; van Damme, J.M.M.; van Tienderen, P.H. Plasticity of growth characteristics in wild barley (Hordeum spontaneum) in response to nutrient limitation. J. Ecol. 2003, 91, 371–382. [Google Scholar] [CrossRef]
- Murata, N.; Iwanaga, F.; Maimaiti, A.; Imada, S.; Mori, N.; Tanaka, K.; Yamanaka, N. Significant improvement of salt tolerance with 2-day acclimatization treatment in Elaeagnus oxycarpa seedlings. Environ. Exp. Bot. 2012, 77, 170–174. [Google Scholar] [CrossRef]
- Barrs, H.D.; Weatherley, P.E. A reexamination of relative turgidity for estimating the water deficits in leaves. Aust. J. Biol. Sci. 1962, 15, 413–428. [Google Scholar]
- Lichtenthaler, H.K. Chlorophylls and carotenoids: Pigments of photosynthetic biomembranes. Method Enzymol. 1987, 148, 360–370. [Google Scholar]
- Zheng, Y.H.; Jia, A.J.; Ning, T.Y.; Xu, J.L.; Li, Z.J.; Jiang, G.M. Potassium nitrate application alleviates sodium chloride stress in winter wheat cultivars differing in salt tolerance. J. Plant Physiol. 2008, 165, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Jagadish, S.V.K.; Muthurajan, R.; Oane, R.; Wheeler, T.R.; Heuer, S.; Bennett, J.; Craufurd, P.Q. Physiological and proteomic approaches to address heat tolerance during anthesis in rice (Oryza sativa L.). J. Exp. Bot. 2010, 61, 143–156. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, R.; Xin, L.; Mao, J.; Li, L.; Kang, G.; Yang, Q. Physiological, Ultrastructural and Proteomic Responses in the Leaf of Maize Seedlings to Polyethylene Glycol-Stimulated Severe Water Deficiency. Int. J. Mol. Sci. 2015, 16, 21606-21625. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160921606
Shao R, Xin L, Mao J, Li L, Kang G, Yang Q. Physiological, Ultrastructural and Proteomic Responses in the Leaf of Maize Seedlings to Polyethylene Glycol-Stimulated Severe Water Deficiency. International Journal of Molecular Sciences. 2015; 16(9):21606-21625. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160921606
Chicago/Turabian StyleShao, Ruixin, Longfei Xin, Jun Mao, Leilei Li, Guozhang Kang, and Qinghua Yang. 2015. "Physiological, Ultrastructural and Proteomic Responses in the Leaf of Maize Seedlings to Polyethylene Glycol-Stimulated Severe Water Deficiency" International Journal of Molecular Sciences 16, no. 9: 21606-21625. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160921606