
Dual Inhibition of MEK and PI3K Pathway in KRAS and BRAF Mutated Colorectal Cancers



Abstract
:
1. Introduction
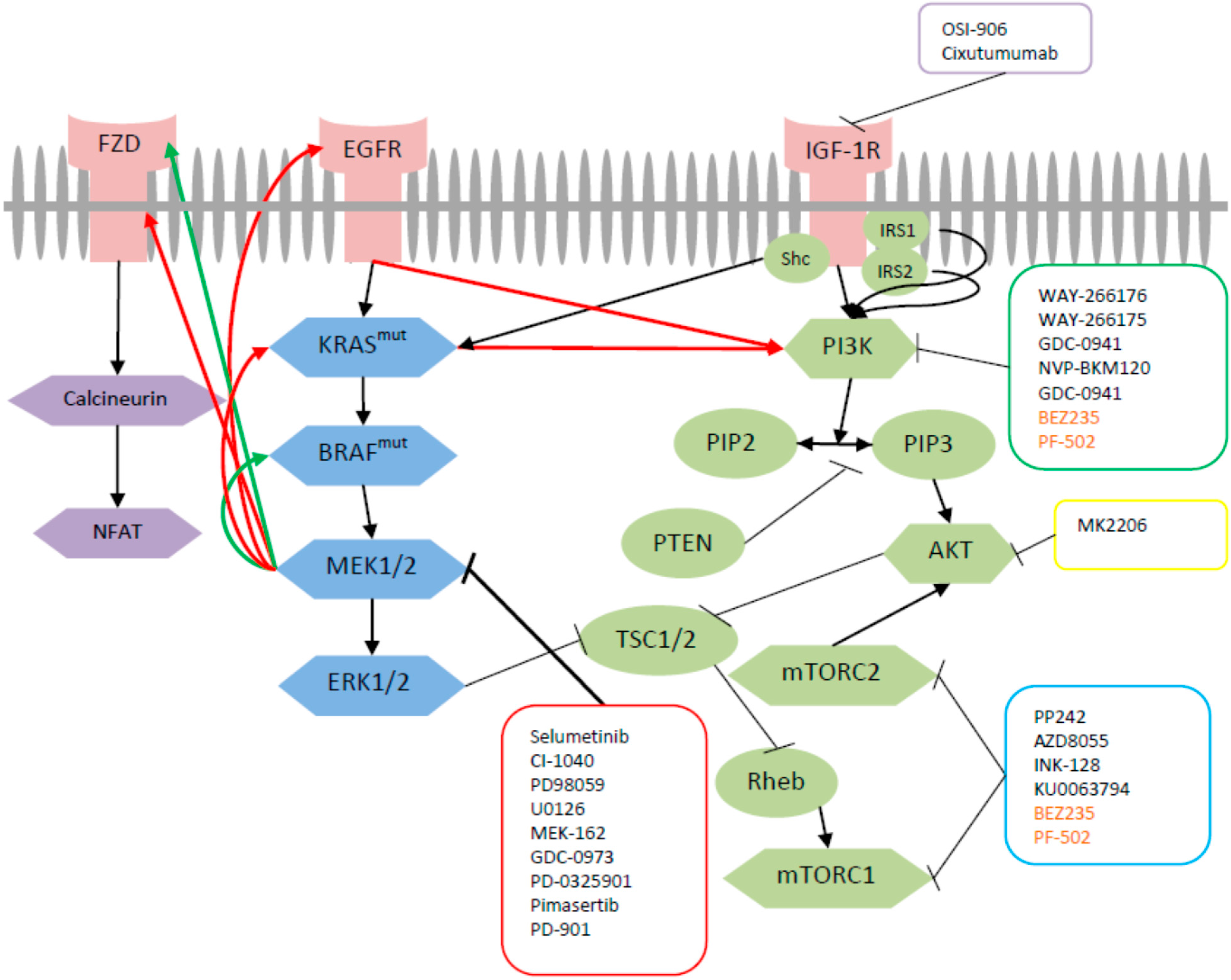
2. Cross-Talk between MEK and PI3K Pathway

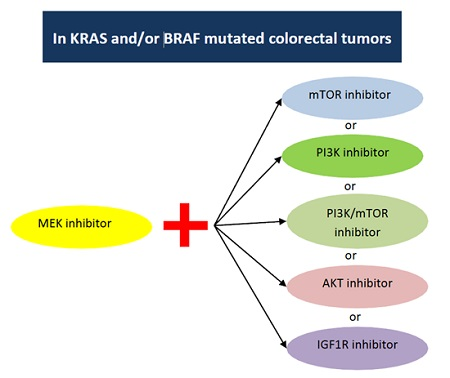
3. Pre-Clinical Data on Dual Targeted Inhibition with MEK
3.1. Combined Inhibition of MEK and mTOR
3.2. Combined MEK and PI3K Inhibition
3.3. Combined MEK and P13K/mTOR Inhibition
3.4. Combined MEK and AKT Inhibition
3.5. Combined Inhibition of MEK and IGFR
4. Clinical Trials on Dual Targeted Inhibition with MEK

{kind=link}
{kind=link}
| Trial | MEK Inhibitor | PI3K Inhibitor | Phase | Population |
|---|---|---|---|---|
| NCT01363232 | MEK162 | BKM120 | Phase I | Patients with advanced solid cancers (colorectal cancer, triple-negative breast cancer, pancreatic cancer, and other cancers harboring KRAS, BRAF and NRAS mutations) |
| NCT01392521 | BAY86-9766 | BAY80-6946 | Phase Ib | Patients with advanced solid cancers |
| NCT01449058 | MEK162 | BYL719 | Phase II | Patients with advanced solid cancers (colorectal cancer, esophageal cancer, pancreatic cancer, non-small cell lung cancer, and other advanced solid tumors harboring RAS or BRAF mutations) |
| Trial | MEK Inhibitor | PI3K/mTOR Inhibitor | Phase | Population |
| NCT00996892 | GDC-0973 | Pictilisb (GDC-0941) | Phase I | Patients with advanced solid cancers |
| NCT01337765 | MEK162 | BEZ235 | Phase 1 | Patients with advanced solid cancers (colorectal cancer, triple-negative breast cancer, pancreatic cancer, malignant melanoma, non-small cell lung cancer, and other cancers harboring KRAS, BRAF and NRAS mutation) |
| NCT01390818 | Pimasertib (MSC1936369B) | SAR245409 | Phase 1 | Patients with advanced solid cancers (colorectal cancer, pancreatic cancer, thyroid cancer, non-small cell lung cancer, renal cancer, breast cancer, melanoma, ovarian cancer) and any cancer diagnosed with aberrations in one or more the following genes: PTEN, BRAF, KRAS, NRAS, PI3KCA, ErbB1, ErbB2) |
| NCT01347866 | PD0325901 | PF-05212384 | Phase 1 | Patients with advanced solid cancers harboring KRAS or BRAF mutation and patients with KRAS mutation with no more than one prior systemic therapy regimen |
| Trial | MEK Inhibitor | AKT Inhibitor | Phase | Population |
| NCT01562275 | GDC-0973 | GDC-0068 | Phase 1 | Patients with locally advanced or metastatic solid tumors |
5. Conclusions
Author Contributions
Conflicts of Interest
References
- LeRoith, D.; Roberts, C.T., Jr. The insulin-like growth factor system and cancer. Cancer Lett. 2003, 195, 127–137. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; de Vriendt, V.; Normanno, N.; Ciardiello, F.; Tejpar, S. KRAS, BRAF, PIK3CA, and PTEN mutations: Implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011, 12, 594–603. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian map kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Khokhlatchev, A.V.; Canagarajah, B.; Wilsbacher, J.; Robinson, M.; Atkinson, M.; Goldsmith, E.; Cobb, M.H. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 1998, 93, 605–615. [Google Scholar] [CrossRef]
- Fremin, C.; Meloche, S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J. Hematol. Oncol. 2010, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Banerji, U.; Camidge, D.R.; Verheul, H.M.; Agarwal, R.; Sarker, D.; Kaye, S.B.; Desar, I.M.; Timmer-Bonte, J.N.; Eckhardt, S.G.; Lewis, K.D.; et al. The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): A phase I open-label multicenter trial in patients with advanced cancer. Clin. Cancer Res. 2010, 16, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, P.M.; Adjei, A.A.; Varterasian, M.; Gadgeel, S.; Reid, J.; Mitchell, D.Y.; Hanson, L.; DeLuca, P.; Bruzek, L.; Piens, J.; et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 5281–5293. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.; Adjei, A.A.; Lorusso, P.M.; Waterhouse, D.; Hecht, J.R.; Natale, R.B.; Hamid, O.; Varterasian, M.; Asbury, P.; Kaldjian, E.P.; et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 2004, 22, 4456–4462. [Google Scholar] [CrossRef] [PubMed]
- Bennouna, J.; Lang, I.; Valladares-Ayerbes, M.; Boer, K.; Adenis, A.; Escudero, P.; Kim, T.Y.; Pover, G.M.; Morris, C.D.; Douillard, J.Y. A phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Investig. New Drug. 2011, 29, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Solit, D.B. Resistance to MEK inhibitors: Should we co-target upstream? Sci. Signal. 2011, 4, pe16. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Song, Y.; Costa, C.; Cook, R.; Arteaga, C.L.; Asara, J.M.; Engelman, J.A. MEK inhibition leads to PI3K/Akt activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012, 72, 3228–3237. [Google Scholar] [CrossRef] [PubMed]
- Britten, C.D. PI3K and MEK inhibitor combinations: Examining the evidence in selected tumor types. Cancer Chemother. Pharm. 2013, 71, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Temraz, S.; Mukherji, D.; Shamseddine, A. Sequencing of treatment in metastatic colorectal cancer: Where to fit the target. World J. Gastroenterol. 2014, 20, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. Braf mutation predicts sensitivity to MEK inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.R.; Logie, A.; McKay, J.S.; Martin, P.; Steele, S.; Jenkins, R.; Cockerill, M.; Cartlidge, S.; Smith, P.D. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: Mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol. Cancer Ther. 2007, 6, 2209–2219. [Google Scholar] [PubMed]
- Cui, D.; Cao, D.; Yang, Y.; Qiu, M.; Huang, Y.; Yi, C. Effect of BRAF V600E mutation on tumor response of anti-EGFR monoclonal antibodies for first-line metastatic colorectal cancer treatment: A meta-analysis of randomized studies. Mol. Biol. Rep. 2014, 41, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Sorich, M.J.; Wiese, M.D.; Rowland, A.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized, controlled trials. Ann. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, Q.; Wang, W.; Liu, J.; Ren, J.; Li, Q.; Hou, F. KRAS status and resistance to epidermal growth factor receptor tyrosine-kinase inhibitor treatment in patients with metastatic colorectal cancer: A meta-analysis. Colorectal Dis. 2014, 16, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Little, A.S.; Balmanno, K.; Sale, M.J.; Newman, S.; Dry, J.R.; Hampson, M.; Edwards, P.A.; Smith, P.D.; Cook, S.J. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci. Signal. 2011, 4, ra17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; van Becelaere, K.; Jiang, P.; Przybranowski, S.; Omer, C.; Sebolt-Leopold, J. A role for K-ras in conferring resistance to the MEK inhibitor, CI-1040. Neoplasia 2005, 7, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Nallapareddy, S.; Tan, A.C.; Spreafico, A.; Pitts, T.M.; Morelli, M.P.; Selby, H.M.; Kachaeva, M.I.; Flanigan, S.A.; Kulikowski, G.N.; et al. Identification of predictive markers of response to the MEK1/2 inhibitor selumetinib (AZD6244) in K-ras-mutated colorectal cancer. Mol. Cancer Ther. 2010, 9, 3351–3362. [Google Scholar] [CrossRef] [PubMed]
- Nosho, K.; Kawasaki, T.; Ohnishi, M.; Suemoto, Y.; Kirkner, G.J.; Zepf, D.; Yan, L.; Longtine, J.A.; Fuchs, C.S.; Ogino, S. PIK3CA mutation in colorectal cancer: Relationship with genetic and epigenetic alterations. Neoplasia 2008, 10, 534–541. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Claes, B.; Bernasconi, D.; de Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Halilovic, E.; She, Q.B.; Ye, Q.; Pagliarini, R.; Sellers, W.R.; Solit, D.B.; Rosen, N. PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res. 2010, 70, 6804–6814. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; Greshock, J.; Holbrook, J.D.; Gilmartin, A.; Zhang, X.; McNeil, E.; Conway, T.; Moy, C.; Laquerre, S.; Bachman, K.; et al. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol. Cancer Ther. 2012, 11, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Jagani, Z.; Xiang, K.X.; Loo, A.; Dorsch, M.; Yao, Y.M.; Sellers, W.R.; Lengauer, C.; Stegmeier, F. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009, 69, 4286–4293. [Google Scholar] [CrossRef] [PubMed]
- Balmanno, K.; Chell, S.D.; Gillings, A.S.; Hayat, S.; Cook, S.J. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int. J. Cancer 2009, 125, 2332–2341. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Tsimberidou, A.M.; Garrido-Laguna, I.; Wang, X.; Luthra, R.; Hong, D.S.; Naing, A.; Falchook, G.S.; Moroney, J.W.; Piha-Paul, S.A.; et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol. Cancer Ther. 2011, 10, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Spreafico, A.; Tentler, J.J.; Pitts, T.M.; Tan, A.C.; Gregory, M.A.; Arcaroli, J.J.; Klauck, P.J.; McManus, M.C.; Hansen, R.J.; Kim, J.; et al. Rational combination of a MEK inhibitor, selumetinib, and the Wnt/calcium pathway modulator, cyclosporin A, in preclinical models of colorectal cancer. Clin. Cancer Res. 2013, 19, 4149–4162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Tian, X.Q.; Sun, D.F.; Zhao, S.L.; Xiong, H.; Fang, J.Y. Combined inhibition of MEK and mTOR signaling inhibits initiation and progression of colorectal cancer. Cancer Investig. 2009, 27, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef] [PubMed]
- Blaser, B.; Waselle, L.; Dormond-Meuwly, A.; Dufour, M.; Roulin, D.; Demartines, N.; Dormond, O. Antitumor activities of ATP-competitive inhibitors of mTOR in colon cancer cells. BMC Cancer 2012, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.V.; Logie, A.; Davies, B.R.; Alferez, D.; Runswick, S.; Fenton, S.; Chresta, C.M.; Gu, Y.; Zhang, J.; Wu, Y.L.; et al. Enhanced apoptosis and tumor growth suppression elicited by combination of MEK (selumetinib) and mTOR kinase inhibitors (AZD8055). Cancer Res. 2012, 72, 1804–1813. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Cui, J.F.; Chen, M.B.; Liu, C.Y.; Liu, F.; Zhang, Q.D.; Zou, J.; Lu, P.H. The preclinical evaluation of the dual mTORC1/2 inhibitor INK-128 as a potential anti-colorectal cancer agent. Cancer Biol. Ther. 2015, 16, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Toral-Barza, L.; Shi, C.; Zhang, W.G.; Zask, A. Response and determinants of cancer cell susceptibility to PI3K inhibitors: Combined targeting of PI3K and MEK1 as an effective anticancer strategy. Cancer Biol. Ther. 2008, 7, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Merchant, M.; Orr, C.; Chan, J.; Den Otter, D.; Berry, L.; Kasman, I.; Koeppen, H.; Rice, K.; Yang, N.Y.; et al. Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res. 2012, 72, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Roper, J.; Sinnamon, M.J.; Coffee, E.M.; Belmont, P.; Keung, L.; Georgeon-Richard, L.; Wang, W.V.; Faber, A.C.; Yun, J.; Yilmaz, O.H.; et al. Combination PI3K/MEK inhibition promotes tumor apoptosis and regression in PIK3CA wild-type, KRAS mutant colorectal cancer. Cancer Lett. 2014, 347, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; Troiani, T.; D’Aiuto, E.; Morgillo, F.; Vitagliano, D.; Capasso, A.; Costantino, S.; Ciuffreda, L.P.; Merolla, F.; Vecchione, L.; et al. Antitumor activity of pimasertib, a selective MEK 1/2 inhibitor, in combination with PI3K/mTOR inhibitors or with multi-targeted kinase inhibitors in pimasertib-resistant human lung and colorectal cancer cells. Int. J. Cancer. 2013, 133, 2089–2101. [Google Scholar] [CrossRef] [PubMed]
- Migliardi, G.; Sassi, F.; Torti, D.; Galimi, F.; Zanella, E.R.; Buscarino, M.; Ribero, D.; Muratore, A.; Massucco, P.; Pisacane, A.; et al. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin. Cancer Res. 2012, 18, 2515–2525. [Google Scholar] [CrossRef] [PubMed]
- Pitts, T.M.; Newton, T.P.; Bradshaw-Pierce, E.L.; Addison, R.; Arcaroli, J.J.; Klauck, P.J.; Bagby, S.M.; Hyatt, S.L.; Purkey, A.; Tentler, J.J.; et al. Dual pharmacological targeting of the map kinase and PI3K/mTOR pathway in preclinical models of colorectal cancer. PLoS ONE 2014, 9, e113037. [Google Scholar] [CrossRef] [PubMed]
- E, J.; Xing, J.; Gong, H.; He, J.; Zhang, W. Combine MEK inhibition with PI3K/mTOR inhibition exert inhibitory tumor growth effect on KRAS and PIK3CA mutation CRC xenografts due to reduced expression of vegf and matrix metallopeptidase-9. Tumor Biol. 2015, 36, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Haagensen, E.J.; Kyle, S.; Beale, G.S.; Maxwell, R.J.; Newell, D.R. The synergistic interaction of MEK and PI3K inhibitors is modulated by mTOR inhibition. Br. J. Cancer 2012, 106, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Roy, H.K.; Olusola, B.F.; Clemens, D.L.; Karolski, W.J.; Ratashak, A.; Lynch, H.T.; Smyrk, T.C. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis 2002, 23, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Petronella, B.A.; Cooke, A.; Kadalbajoo, M.; Siu, K.W.; Kleinberg, A.; May, E.W.; Gokhale, P.C.; Schulz, R.; Kahler, J.; et al. Discovery of novel insulin-like growth factor-1 receptor inhibitors with unique time-dependent binding kinetics. ACS Med. Chem. Lett. 2013, 4, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Pitts, T.M.; Tan, A.C.; Kulikowski, G.N.; Tentler, J.J.; Brown, A.M.; Flanigan, S.A.; Leong, S.; Coldren, C.D.; Hirsch, F.R.; Varella-Garcia, M.; et al. Development of an integrated genomic classifier for a novel agent in colorectal cancer: Approach to individualized therapy in early development. Clin. Cancer Res. 2010, 16, 3193–3204. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, S.A.; Pitts, T.M.; Newton, T.P.; Kulikowski, G.N.; Tan, A.C.; McManus, M.C.; Spreafico, A.; Kachaeva, M.I.; Selby, H.M.; Tentler, J.J.; et al. Overcoming IGF1R/IR resistance through inhibition of MEK signaling in colorectal cancer models. Clin. Cancer Res. 2013, 19, 6219–6229. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B.; et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Wilky, B.A.; Rudek, M.A.; Ahmed, S.; Laheru, D.A.; Cosgrove, D.; Donehower, R.C.; Nelkin, B.; Ball, D.; Doyle, L.A.; Chen, H.; et al. A phase I trial of vertical inhibition of IGF signalling using cixutumumab, an anti-IGF-1R antibody, and selumetinib, an MEK 1/2 inhibitor, in advanced solid tumours. Br. J. Cancer 2015, 112, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Do, K.; Speranza, G.; Bishop, R.; Khin, S.; Rubinstein, L.; Kinders, R.J.; Datiles, M.; Eugeni, M.; Lam, M.H.; Doyle, L.A.; et al. Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer. Investig. New Drug 2015, 33, 720–728. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Temraz, S.; Mukherji, D.; Shamseddine, A. Dual Inhibition of MEK and PI3K Pathway in KRAS and BRAF Mutated Colorectal Cancers. Int. J. Mol. Sci. 2015, 16, 22976-22988. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160922976
Temraz S, Mukherji D, Shamseddine A. Dual Inhibition of MEK and PI3K Pathway in KRAS and BRAF Mutated Colorectal Cancers. International Journal of Molecular Sciences. 2015; 16(9):22976-22988. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160922976
Chicago/Turabian StyleTemraz, Sally, Deborah Mukherji, and Ali Shamseddine. 2015. "Dual Inhibition of MEK and PI3K Pathway in KRAS and BRAF Mutated Colorectal Cancers" International Journal of Molecular Sciences 16, no. 9: 22976-22988. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160922976