
Resistance to Recombinant Human Erythropoietin Therapy in a Rat Model of Chronic Kidney Disease Associated Anemia

 , , ,
, , ,
Abstract
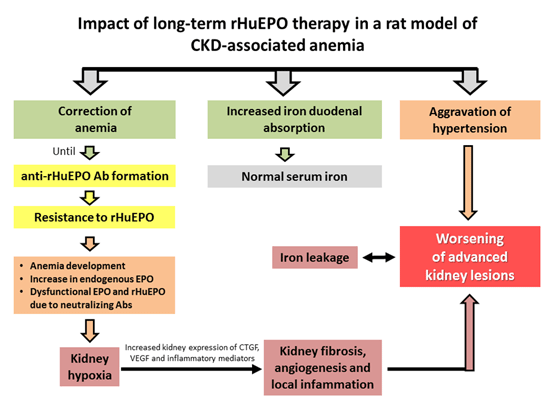
:
1. Introduction
2. Results
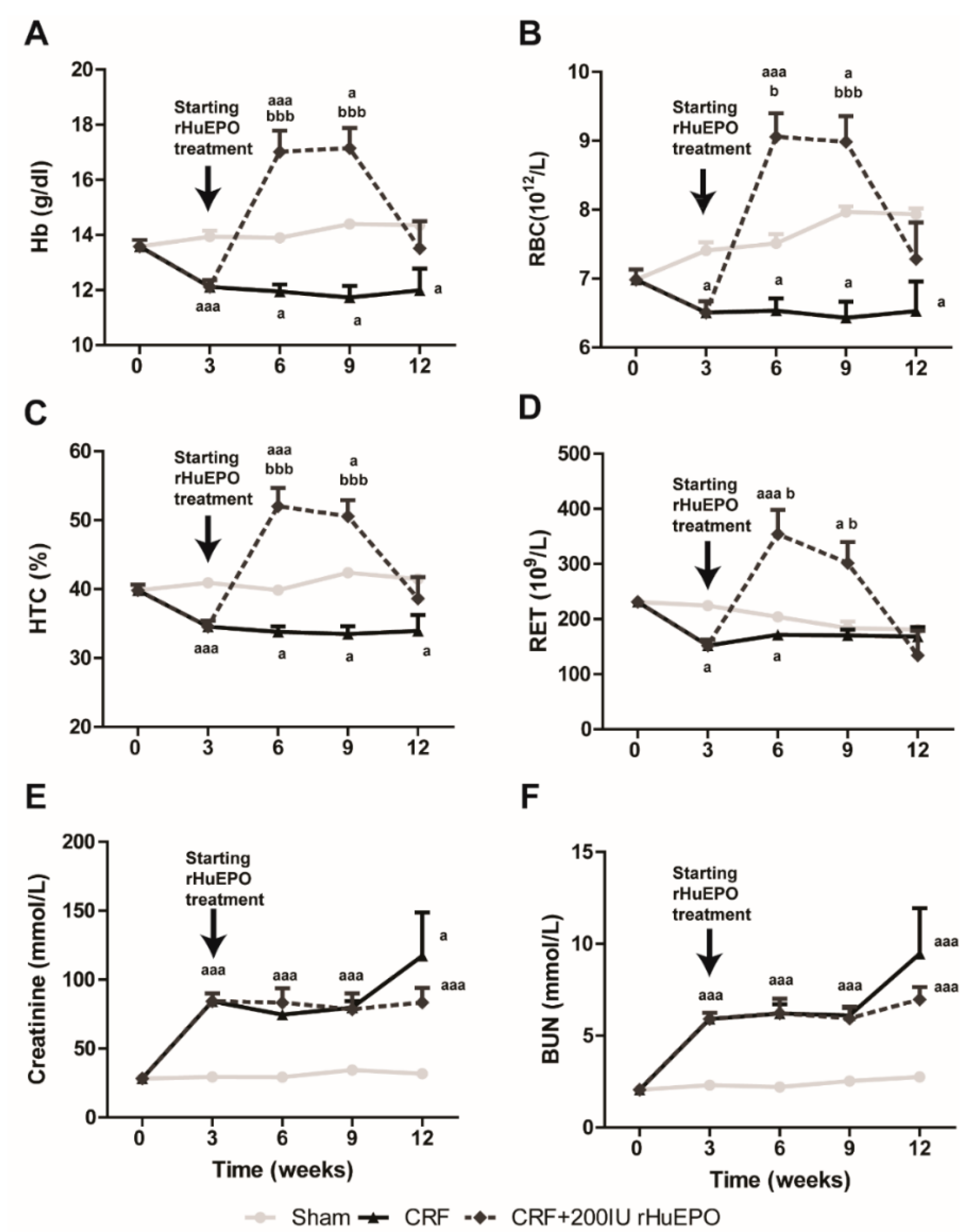
2.1. Body and Tissue Weights and Blood Pressure

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Sham | CRF | CRF + 200 IU rHuEPO |
|---|---|---|---|
| Body and tissues weights | |||
| BW (kg) | 0.45 ± 0.02 | 0.36 ± 0.01 a,1 | 0.39 ± 0.01 |
| KW (g) | 1.22 ± 0.03 | 1.65 ± 0.04 | 1.69 ± 0.08 a |
| KW/BW (g/kg) | 2.72 ± 0.05 | 4.61 ± 0.22 aa | 4.34 ± 0.20 aa,b |
| HW (g) | 1.16 ± 0.03 | 1.24 ± 0.07 | 1.25 ± 0.04 |
| HW/BW (g/kg) | 2.58 ± 0.08 | 3.48 ± 0.25 aa | 3.21 ± 0.14 a |
| LW (g/kg) | 13.33 ± 0.48 | 11.32 ± 0.34 | 13.44 ± 0.42 |
| LW/BW (g/kg) | 29.61 ± 0.65 | 31.43 ± 0.71 | 34.41 ± 1.14 a |
| Blood pressure and heart rate | |||
| SBP (mmHg) | 117.7 ± 1.15 | 134.1 ± 4.6 aa | 169.1 ± 1.5 aaa,bbb |
| DBP (mmHg) | 110.1 ± 0.59 | 113.2 ± 2.08 | 133.3 ± 7.2 aaa,bb |
| MBP (mmHg) | 115.0 ± 0.97 | 117.5 ± 1.21 | 143.9 ± 4.8 aaa,bbb |
| HR (beats/min) | 357.7 ± 2.74 | 367.6 ± 5.19 | 376.4 ± 3.2 |
| Hematological data | |||
| WBC (×103/µL) | 1.78 ± 0.30 | 5.01 ± 1.76 | 3.16 ± 0.70 |
| MCV (fL) | 52.52 ± 0.53 | 51.93 ± 0.69 | 52.83 ± 0.81 |
| MCH (pg) | 18.08 ± 0.18 | 18.36 ± 0.24 | 18.60 ± 0.19 |
| MCHC (g/dL) | 34.60 ± 0.08 | 35.37 ± 0.19 aa | 35.26 ± 0.30 |
| RDW (%) | 11.48 ± 2.53 | 18.34 ± 3.23 | 17.88 ± 0.67 a,b |
| PLT (×103/µL) | 713.75 ± 15.19 | 769.00 ± 73.17 | 786.00 ± 50.83 |
| PDW (%) | 16.34 ± 0.18 | 16.44 ± 0.20 | 16.44 ± 0.24 |
| Biochemical parameters | |||
| TGs (mmol/L) | 1.05 ± 0.14 | 1.58 ± 0.32 | 1.58 ± 0.29 |
| Total-c (mmol/L) | 1.25 ± 0.06 | 2.44 ± 0.54 a | 2.74 ± 0.25 |
| CK (U/L) | 540.57 ± 58.94 | 473.00 ± 85.57 | 294.86 ± 35.86 a,b |
| ALT (U/L) | 35.17 ± 2.21 | 42.00 ± 18.53 a | 26.14 ± 1.18 a |
| AST (U/L) | 80.57 ± 7.84 | 139.43 ± 70.70 | 54.57 ± 3.02 a,b |
| Bilirubin (µmol/L) | 8.04 × 10−5 ± 1.03 × 10−5 | 1.03 × 10−5 ± 1.71 × 10−5 | 1.20 × 10−5 ± 1.71 × 10−5 |
| IL-6 (pg/mL) | 132.29 ± 4.28 | 138.33 ± 4.22 | 138.73 ± 1.70 |
| hsCRP (µg/mL) | 262.25 ± 12.43 | 225.31 ± 7.95 a | 244.23 ± 7.99 |
| INF-γ (pg/mL) | 23.30 ± 3.10 | 25.51 ± 2.26 | 25.51 ± 1.22 |
| TGF-β1 (ng/mL) | 75.74 ± 5.62 | 84.13 ± 3.85 | 72.37 ± 4.55 |
| VEGF (pg/mL) | 4.23 ± 0.94 | 14.16 ± 2.24 a | 10.03 ± 1.00 a |
2.2. Hematological and Biochemical Data

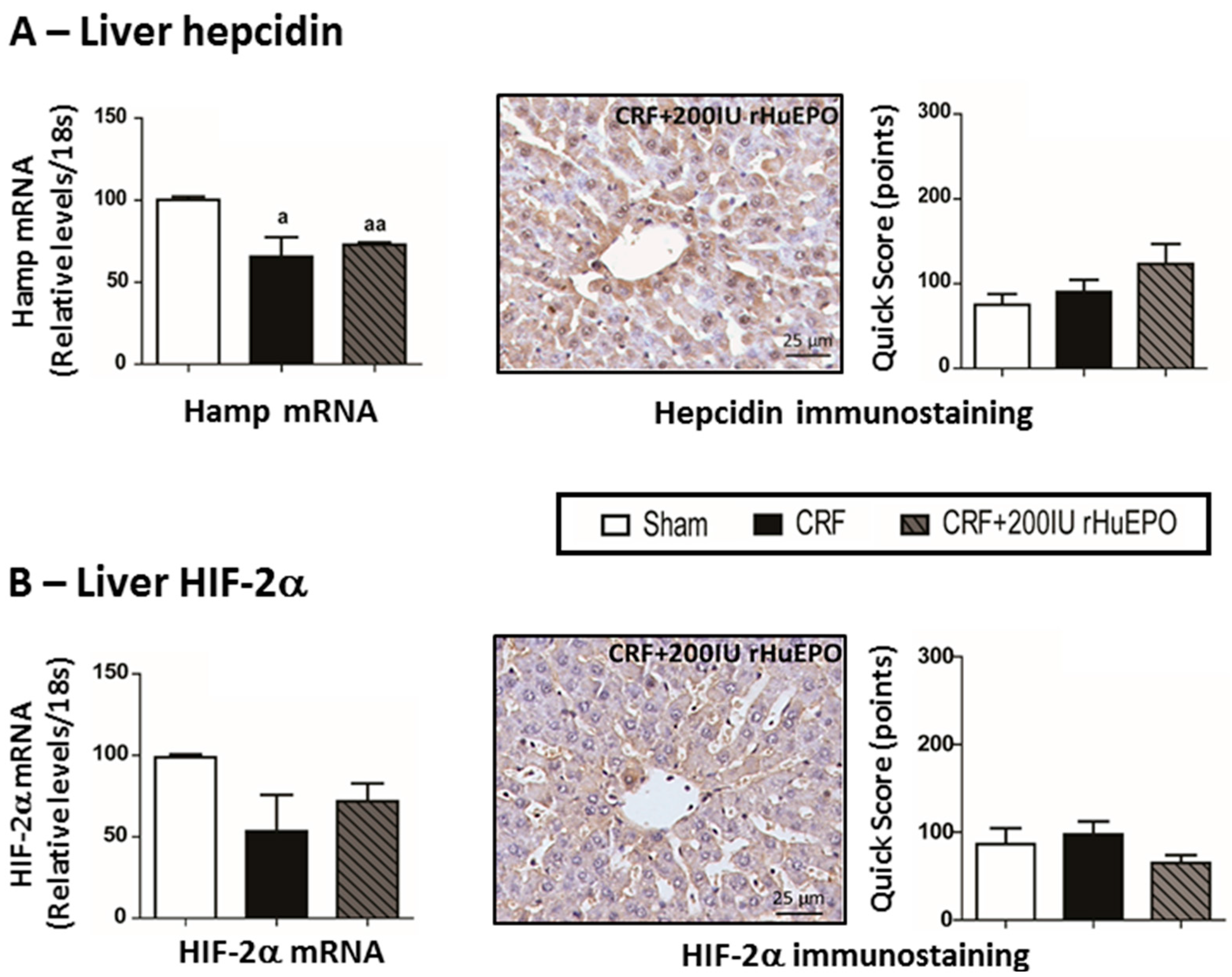
2.3. Serum EPO Concentration and EPO and EPOR mRNA Expression in the Liver and Kidney

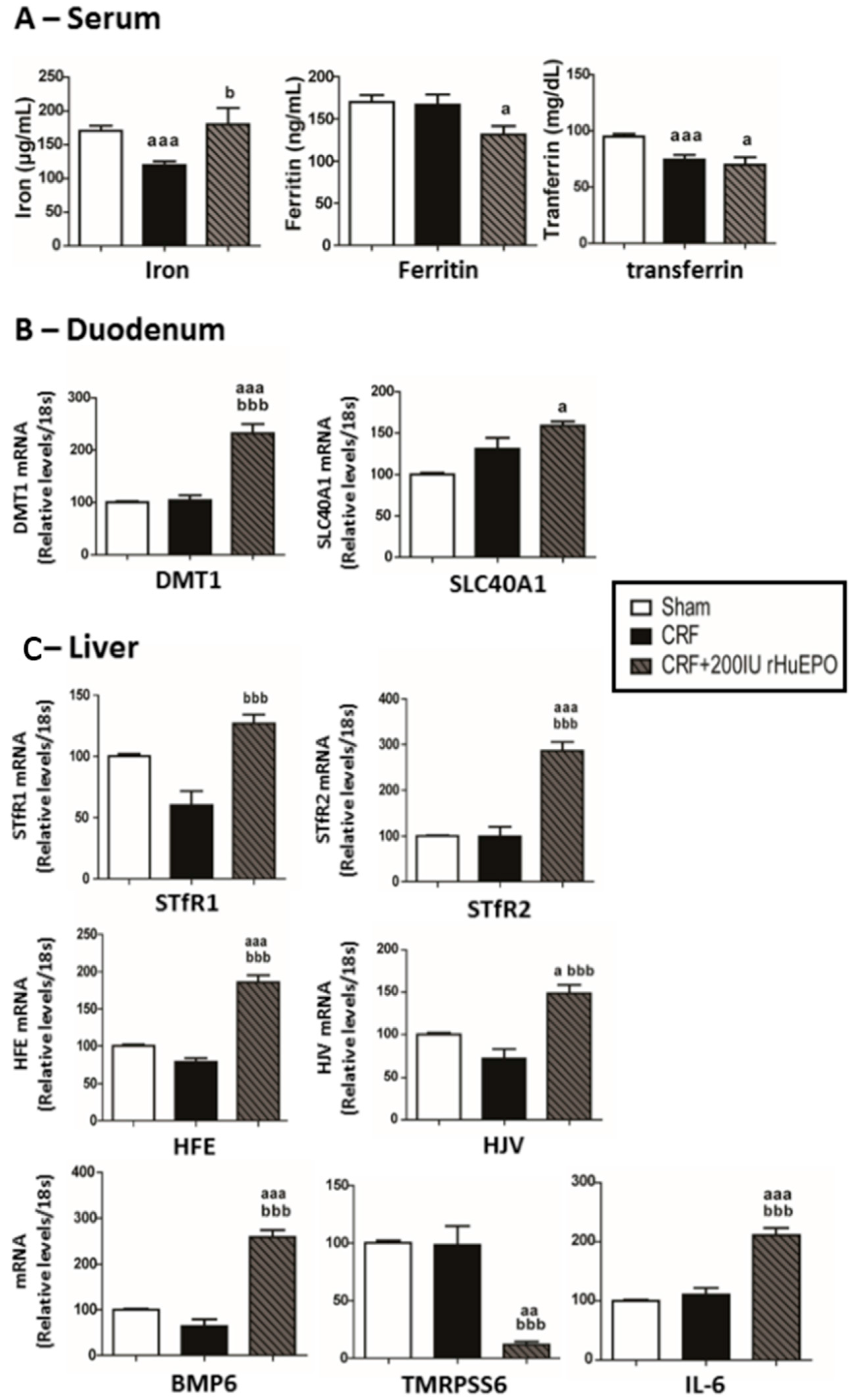
2.4. Iron Metabolism


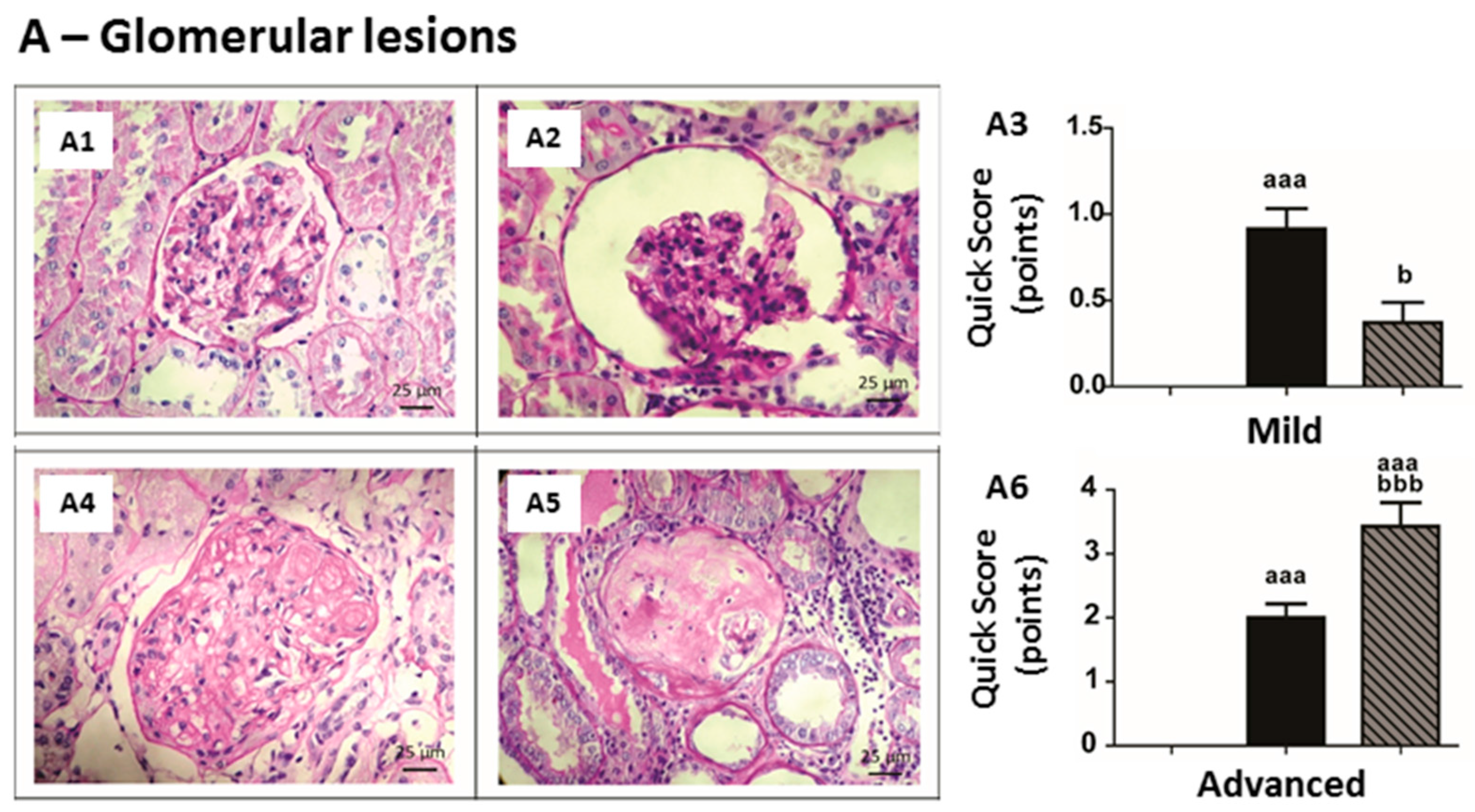
2.5. Kidney Lesions
| Mild Lesions | Group | Score (n, %) | ||||
|---|---|---|---|---|---|---|
| 0 (Absent) | 1 (<25%) | 2 (25%–50%) | 3 (>50%) | Total Score | ||
| Thickening of Bowman´s Capsule | Sham | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 |
| CRF | 1 (14.3%) | 3 (42.9%) | 1 (14.3%) | 2 (28.6%) | 1.57 ± 0.43 a,1 | |
| CRF + 200 IU rHuEPO | 3 (42.9%) | 3 (42.9%) | 1 (14.3%) | 0 | 0.71 ± 0.28 | |
| Hyalinosis of the Vascular Pole | Sham | 7 (100%) | 0 | 0 (0%) | 0 (0%) | 0.00 ± 0.00 |
| CRF | 1 (14.3%) | 6 (85.7%) | 0 | 0 | 0.86 ± 0.14 | |
| CRF + 200 IU rHuEPO | 5 (71.4%) | 1 (14.3%) | 0 | 1 (14.3%) | 0.57 ± 0.43 | |
| Glomerular Atrophy | Sham | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 |
| CRF | 1 (14.3%) | 6 (85.7%) | 0 | 0 | 0.86 ± 0.14 a | |
| CRF + 200 IU rHuEPO | 5 (71.4%) | 2 (28.6%) | 0 | 0 | 0.29 ± 0.18 | |
| Hypercellularity | Sham | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 |
| CRF | 0 | 7 (100%) | 0 | 0 | 1.00 ± 0.00 aa | |
| CRF + 200 IU rHuEPO | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 bb | |
| Dilatation of the Bowman’s Space | Sham | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 |
| CRF | 5 (71.4%) | 2 (28.6%) | 0 | 0 | 0.29 ± 0.18 | |
| CRF + 200 IU rHuEPO | 5 (71.4%) | 2 (28.6%) | 0 | 0 | 0.28 ± 0.18 | |
| Total Group Score | Sham | 0.00 ± 0.00 | ||||
| CRF | 0.91 ± 0.12 aaa | |||||
| CRF + 200 IU rHuEPO | 0.37 ± 0.12 b | |||||
| Group | Advanced Glomerular Lesions (n, %) | |||||
| None of the Previous Lesions (0) | Thickening of GBM (1) | Mesangial Expansion (2) | Nodular Sclerosis (3) | Global Glomerulosclerosis (4) | Total Group Score | |
| Sham | 7 (100%) | 0 | 0 | 0 | 0 | 0.00 ± 0.00 |
| CRF | 0 | 1 (14.3%) | 5 (71.4%) | 1 (14.3%) | 0 | 2.00 ± 0.22 aaa |
| CRF + 200 IU rHuEPO | 0 | 0 | 2 (28.6%) | 0 | 5 (71.4%) | 3.43 ± 0.37 aaa,bbb |
| Mild Lesions | Group | Score (n, %) | ||||
| 0 (Absent) | 1 (<25%) | 2 (25%–50%) | 3 (>50%) | Total Score | ||
| Tubular Hyaline Droplets | Sham | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 |
| CRF | 0 | 7 (100%) | 0 | 0 | 1.00 ± 0.00 aaa | |
| CRF + 200 IU rHuEPO | 0 | 7 (100%) | 0 | 0 | 1.00 ± 0.00 aaa | |
| TBM Irregularity | Sham | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 |
| CRF | 0 | 3 (42.9%) | 2 (28.5%) | 2 (28.5%) | 1.86 ± 0.34 aaa | |
| CRF + 200 IU rHuEPO | 0 | 5 (71.4%) | 2 (28.5%) | 0 | 1.29 ± 0.18 a | |
| Tubular Dilatation | Sham | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 |
| CRF | 2 (28.5%) | 1 (14.3%) | 2 (28.5%) | 2 (28.5%) | 1.57 ± 0.48 a | |
| CRF + 200 IU rHuEPO | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 bb | |
| Interstitial Inflammatory Infiltrate | Sham | 4 (57.2%) | 3 (42.9%) | 0 | 0 | 0.29 ± 0.18 |
| CRF | 0 | 0 | 7 (100%) | 0 | 2.00 ± 0.00 aaa | |
| CRF + 200 IU rHuEPO | 0 | 0 | 7 (100%) | 2.00 ± 0.00 aaa | ||
| Vacuolar Tubular Degeneration | Sham | 0 | 7 (100%) | 0 | 0 | 1.00 ± 0.00 |
| CRF | 3 (42.9%) | 3 (42.9%) | 1 (14.3%) | 0 | 0.71 ± 0.29 aaa | |
| CRF + 200 IU rHuEPO | 0 | 7 (100%) | 0 | 0 | 1.00 ± 0.00 bb | |
| Total Group Score | Sham | 0.26 ± 0.08 | ||||
| CRF | 1.43 ± 0.15 aaa | |||||
| CRF + 200 IU rHuEPO | 1.06 ± 0.12 aaa | |||||
| Advanced Lesions | Group | Score (n, %) | ||||
| 0 (Absent) | 1 (<25%) | 2 (25%–50%) | 3 (>50%) | Total Score | ||
| Hyaline Cylinders | Sham | 6 (85.7%) | 1 (14.3%) | 0 | 0 | 0.14 ± 0.14 |
| CRF | 0 | 1 (14.3%) | 6 (85.7%) | 0 | 1.86 ± 0.14 aa | |
| CRF + 200 IU rHuEPO | 0 | 0 | 4 (57.1%) | 3 (42.9%) | 2.43 ± 0.20 aaa | |
| Tubular Calcification | Sham | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 |
| CRF | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 | |
| CRF + 200 IU rHuEPO | 1 (14.3%) | 6 (85.7%) | 0 | 0 | 0.85 ± 0.14 aaa,bbb | |
| Necrosis | Sham | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 |
| CRF | 2 (28.6%) | 5 (71.4%) | 0 | 0 | 0.71 ± 0.18 | |
| CRF + 200 IU rHuEPO | 0 | 0 | 4 (57.1%) | 3 (42.9%) | 2.43±0.20 aaa,bbb | |
| IFTA | Sham | 7 (100%) | 0 | 0 | 0 | 0.00 ± 0.00 |
| CRF | 0 | 3 (42.9%) | 4 (57.1%) | 0 | 1.57 ± 0.20 aaa | |
| CRF + 200 IU rHuEPO | 0 | 0 | 0 | 7 (100%) | 3.00 ± 0.00 aaa,bbb | |
| Total Group Score | Sham | 0.04 ± 0.04 | ||||
| CRF | 1.04 ± 0.16 aaa | |||||
| CRF + 200 IU rHuEPO | 2.18 ± 0.17 aaa,bbb | |||||


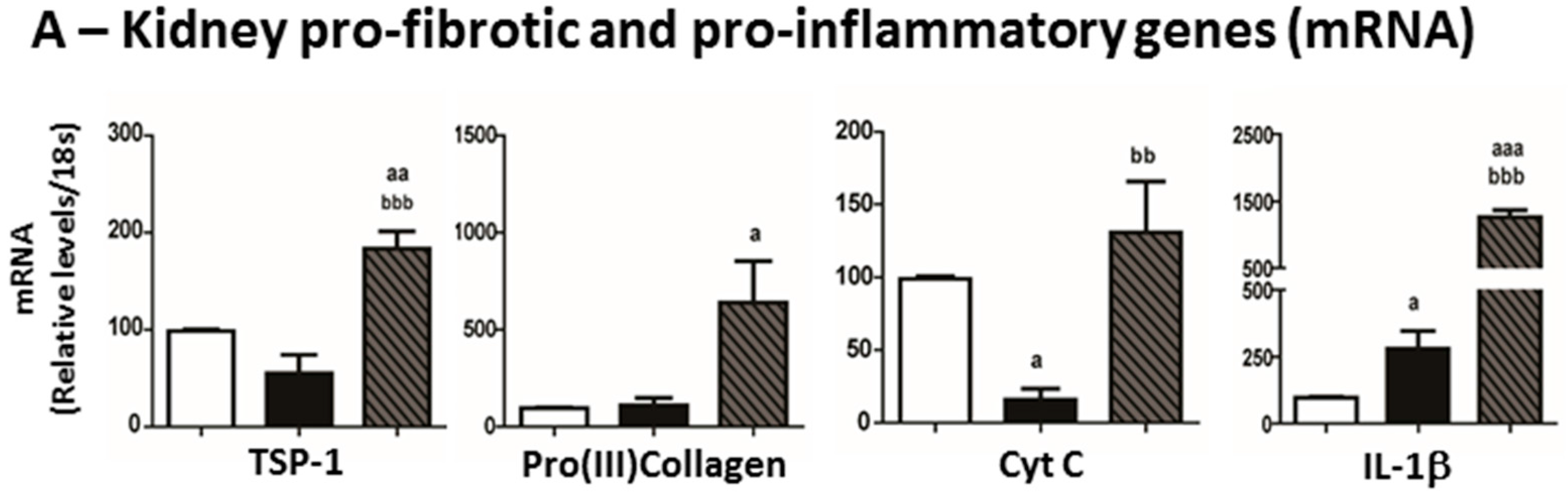
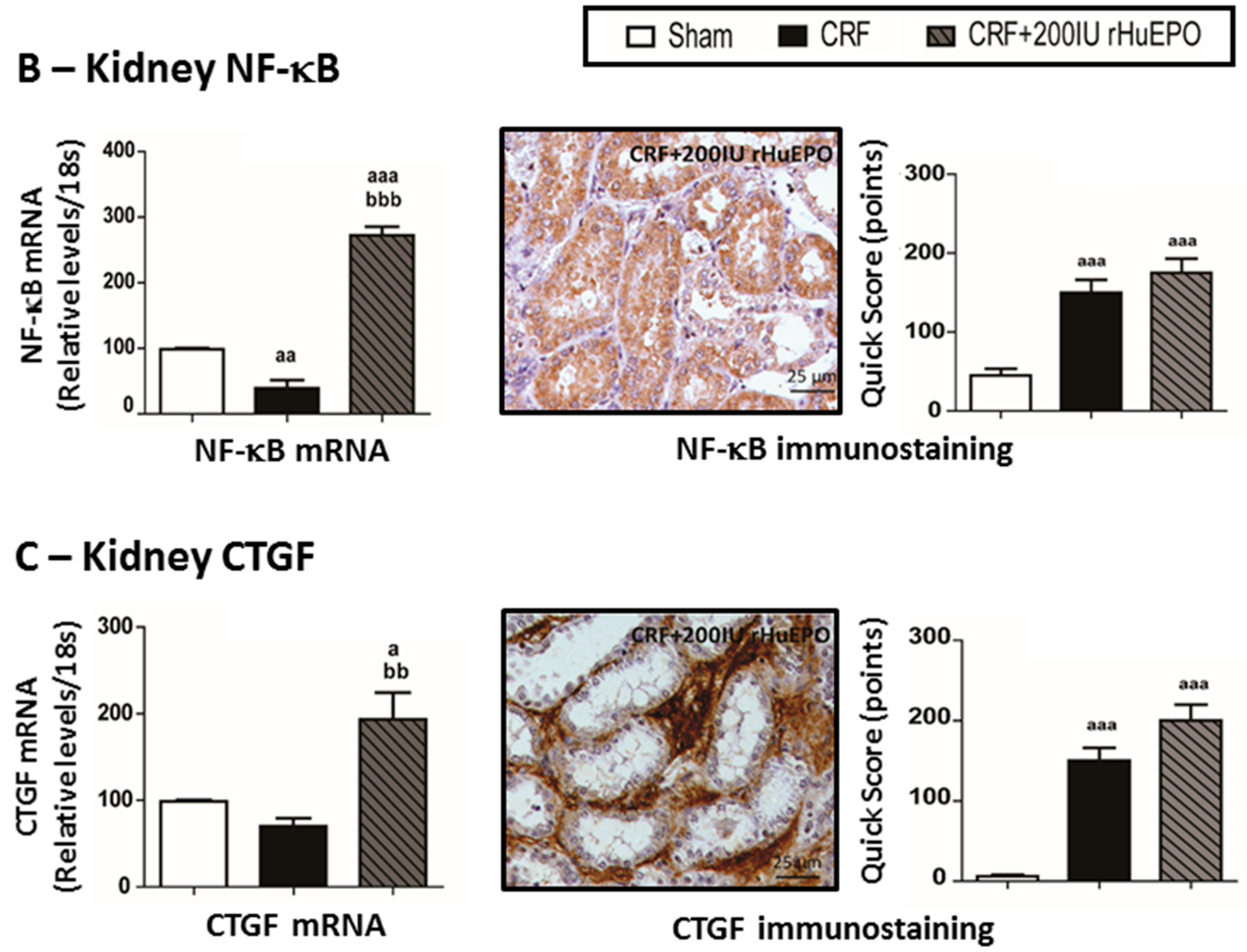
2.6. Mediators of Kidney Lesions


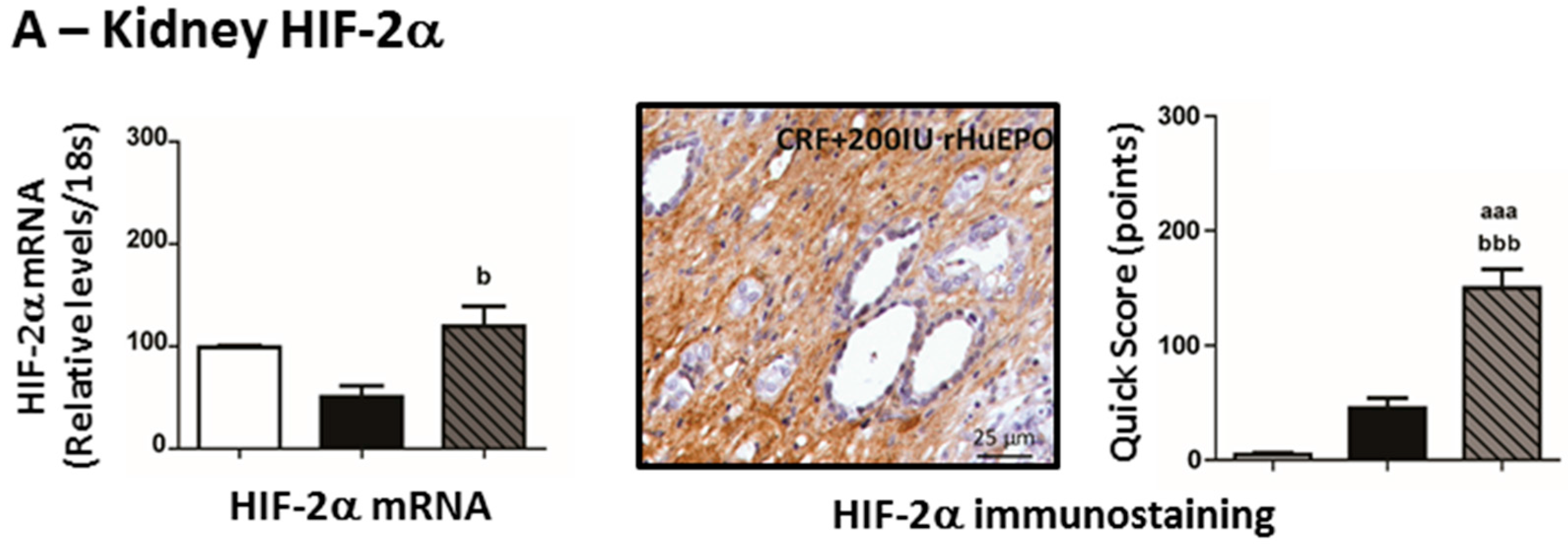
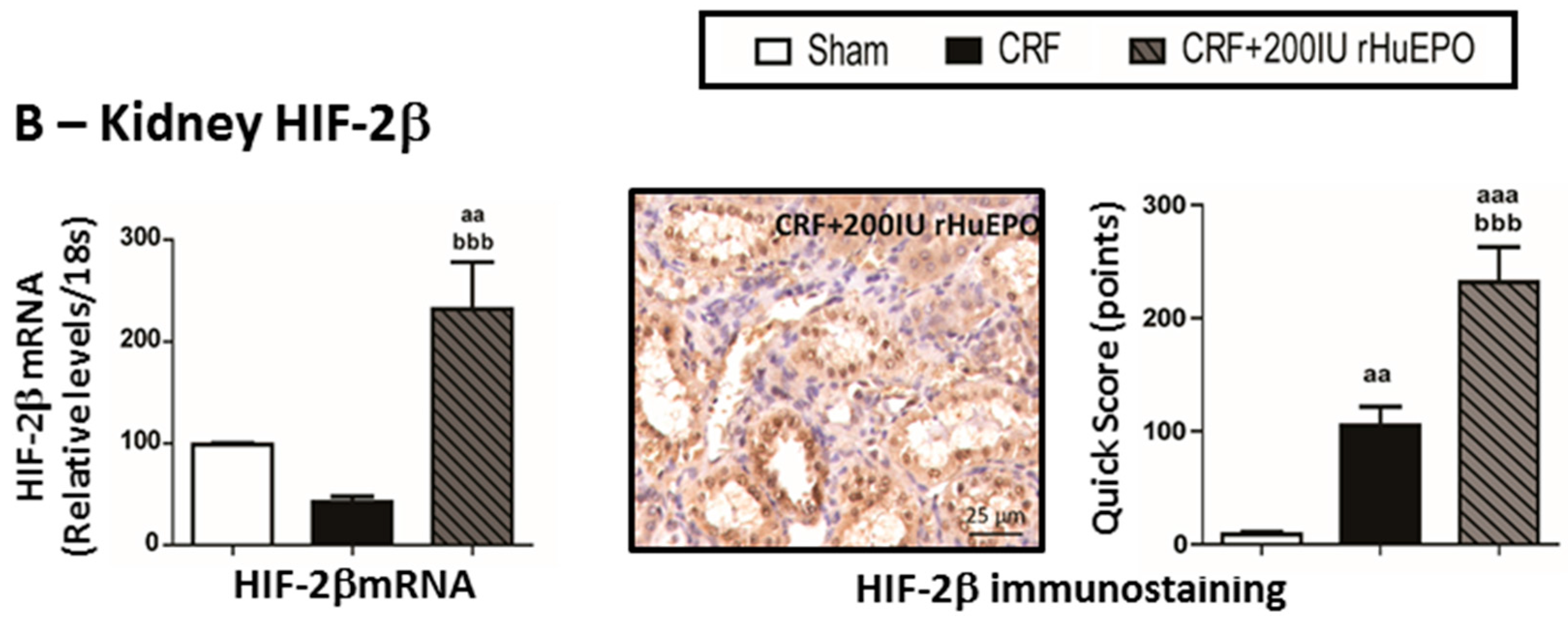
2.7. Kidney mRNA and Protein Expression of Hypoxia Inducible Factor 2α and 2β


3. Discussion

4. Experimental Section
4.1. Animals and Experimental Protocol
4.2. Sample Collection and Preparation
4.3. Biochemical and Hematological Assays
4.4. Detection of Anti-EPO Antibodies
4.5. Gene Expression Analysis
| Gene | Primer Sequences | Gene | Primer Sequences |
|---|---|---|---|
| EPO | F: 5’-AGG GTC ACG AAG CCA TGA AG-3’ | IL-6 | F: 5’-CGA GCC CAC CAG GAA CGA AAG TC-3’ |
| R: 5’-GAT TTC GGC TGT TGC CAG TG-3’ | R: 5’-CTG GCT GGA AGT CTC TTG CGG AG-3’ | ||
| EPOR | F: 5’-GCG ACT TGG ACC CTC TCA TC-3’ | IL-1β | F: 5’-CTC TGT GAC TCG TGG GAT GAT G-3’ |
| R: 5’-AGT TAC CCT TGT GGG TGG TG-3’ | R: 5’-CAC TTG TTG GCT TAT GTT CTG TCC-3’ | ||
| Hamp | F: 5’-GAA GGC AAG ATG GCA CTA AGC-3’ | CTGF | F: 5’-CGT AGA CGG TAA AGC AAT GG-3’ |
| R: 5’-CAG AGC CGT AGT CTG TCT CG-3’ | R: 5’-AGT CAA AGA AGC AGC AAA CAC-3’ | ||
| TfR2 | F: 5’-CAA GCT TCG CCC AGA AGG TA-3’ | NF-κβ | F: 5’-ACC TGA GTC TTC TGG ACC GCT G-3’ |
| R: 5’-CGT GTA AGG GTC CCC AGT TC-3’ | R: 5’-CCA GCC TTC TCC CAA GAG TCG T-3’ | ||
| SLC40A1 | F: 5’-CAG GCT TAG GGT CTA CTG CG-3’ | VEGF | F: 5’-GAA GTT CAT GGA CGT CTA CCA G -3’ |
| R: 5’-CCG AAA GAC CCC AAA GGA CA-3’ | R: 5’-CAT CTG CTA TGC TGC AGG AAG CT -3’ | ||
| HJV | F: 5’-GCC TAC TTC CAA TCC TGC GT-3’ | Pro (III) Collagen | F: 5’-CCA CCC TGA ACT CAA GAG TGG-3’ |
| R: 5’-GGT CAA GAA GAC TCG GGC AT-3’ | R: 5’-CCA TCC AGA ACT GTG TAA GTG-3’ | ||
| TF | F: 5’-GGC ATC AGA CTC CAG CAT CA-3’ | TSP-1 | F: 5’-CCG GTT TGA TCA GAG TGG T-3’ |
| R: 5’-GCA GGC CCA TAG GGA TGT T-3’ | R: 5’ GGT TTC GGA AGG TGC AAT-3’ | ||
| Hfe | F: 5’-CTG GAT CAG CCT CTC ACT GC-3’ | Cyt C | F: 5’- CTT GTC ATA AAG TGG ATA TGA TC-3’ |
| R: 5’-GTC ACC CAT GGT TCC TCC TG-3’ | R: 5’ CAA TAG GTT TGA GGC GAC ACC CTC-3’ | ||
| DMT1 | F: 5’-CAA CTC TAC CCT GGC TGT GG-3’ | HIF-2α | F: 5’-TGA CTT CAC TCA TCC TTG CGA CCA-3’ |
| R: 5’-GTC ATG GTG GAG CTC TGT CC-3’ | R: 5’-ATT CAT AGG CAG AGC GGC CAA GTA-3’ | ||
| TfR1 | F: 5’-GCT CGT GGA GAC TAC TTC CG-3’ | HIF-2β | F: 5’-TGA AAG AAG GAG AAG CCC AAT A-3’ |
| R: 5’-GCC CCA GAA GAT GTG TCG G-3’ | R: 5’-CAT CAG AGT TAT GCC GAG ACA G-3’ | ||
| TMPRSS6 | F: 5’-CCG AAT ATG AGG TGG ACC CG-3’ | 18S | F: 5’-CCA CTA AAG GGC ATC CTG GG-3’ |
| R: 5’-GGT TCA CGT AGC TGT AGC GG-3’ | R: 5’-CAT TGA GAG CAA TGC CAG CC-3’ | ||
| BMP6 | F: 5’-GCT GCC AAC TAT TGT GAC GG-3’ | Actb | F: 5’-GAG ATT ACT GCC CTG GCT CC-3’ |
| R: 5’-GGT TTG GGG ACG TAC TCG G-3’ | R: 5’-CGG ACT CAT CGT ACT CCT GC-3’ |
4.6. Histopathological Analysis
4.7. Immunohistochemistry Analysis
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, Q.L.; Rothenbacher, D. Prevalence of chronic kidney disease in population-based studies: Systematic review. BMC. Public Health 2008, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Coresh, J.; Selvin, E.; Stevens, L.A.; Manzi, J.; Kusek, J.W.; Eggers, P.; van Lente, F.; Levey, A.S. Prevalence of chronic kidney disease in the United States. JAMA 2007, 298, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.A.; Li, S.; Wang, C.; Huang, C.; Becker, B.N.; Bomback, A.S.; Brown, W.W.; Burrows, N.R.; Jurkovitz, C.T.; McFarlane, S.I.; et al. Prevalence of CKD and comorbid illness in elderly patients in the United States: Results from the Kidney Early Evaluation Program (KEEP). Am. J. Kidney Dis. 2010, 55, S23–S33. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, Y.; Miyazaki, S. Heart failure, chronic kidney disease, and biomarkers—An integrated viewpoint. Circ. J. 2010, 74, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Berl, T.; Henrich, W. Kidney-heart interactions: Epidemiology, pathogenesis, and treatment. Clin. J. Am. Soc. Nephrol. 2006, 1, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, T.; Kiyohara, Y.; Kubo, M.; Tanizaki, Y.; Doi, Y.; Okubo, K.; Wakugawa, Y.; Hata, J.; Oishi, Y.; Shikata, K.; et al. Chronic kidney disease and cardiovascular disease in a general Japanese population: The Hisayama Study. Kidney Int. 2005, 68, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Schiffrin, E.L.; Lipman, M.L.; Mann, J.F. Chronic kidney disease: Effects on the cardiovascular system. Circulation 2007, 116, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Cases-Amenos, A.; Martinez-Castelao, A.; Fort-Ros, J.; Bonal-Bastons, J.; Ruiz, M.P.; Valles-Prats, M.; Coll-Piera, E.; Galcerán-Gui, J.M. Investigadores del Estudio Micenas. Prevalence of anaemia and its clinical management in patients with stages 3-5 chronic kidney disease not on dialysis in Catalonia: MICENAS I study. Nefrologia 2014, 34, 189–198. [Google Scholar] [PubMed]
- Lankhorst, C.E.; Wish, J.B. Anemia in renal disease: Diagnosis and management. Blood Rev. 2010, 24, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G. Iron metabolism in the anemia of chronic disease. Biochim. Biophys. Acta 2009, 1790, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Guidi, G.C.; Lechi Santonastaso, C. Advancements in anemias related to chronic conditions. Clin. Chem. Lab. Med. 2010, 48, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Obrador, G.T.; Roberts, T.; St Peter, W.L.; Frazier, E.; Pereira, B.J.; Collins, A.J. Trends in anemia at initiation of dialysis in the United States. Kidney Int. 2001, 60, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.N.; Parfrey, P.S.; Harnett, J.D.; Kent, G.M.; Murray, D.C.; Barre, P.E. The impact of anemia on cardiomyopathy, morbidity, and and mortality in end-stage renal disease. Am. J. Kidney Dis. 1996, 28, 53–61. [Google Scholar] [CrossRef]
- Moreno, F.; Sanz-Guajardo, D.; Lopez-Gomez, J.M.; Jofre, R.; Valderrabano, F. Increasing the hematocrit has a beneficial effect on quality of life and is safe in selected hemodialysis patients. Spanish Cooperative Renal Patients Quality of Life Study Group of the Spanish Society of Nephrology. J. Am. Soc. Nephrol. 2000, 11, 335–342. [Google Scholar] [PubMed]
- Palmer, S.C.; Navaneethan, S.D.; Craig, J.C.; Johnson, D.W.; Tonelli, M.; Garg, A.X.; Pellegrini, F.; Ravani, P.; Jardine, M.; Perkovic, V.; et al. Meta-analysis: Erythropoiesis-stimulating agents in patients with chronic kidney disease. Ann. Intern. Med. 2010, 153, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Clement, F.M.; Klarenbach, S.; Tonelli, M.; Johnson, J.A.; Manns, B.J. The impact of selecting a high hemoglobin target level on health-related quality of life for patients with chronic kidney disease: A systematic review and meta-analysis. Arch. Intern. Med. 2009, 169, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Parfrey, P.S.; Lauve, M.; Latremouille-Viau, D.; Lefebvre, P. Erythropoietin therapy and left ventricular mass index in CKD and ESRD patients: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2009, 4, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W.; Pollock, C.A.; Macdougall, I.C. Erythropoiesis-stimulating agent hyporesponsiveness. Nephrology 2007, 12, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, R.D.; Critchlow, C.W.; Fishbane, S.; Besarab, A.; Stehman-Breen, C.; Krishnan, M.; Bradbury, B.D. Greater epoetin alfa responsiveness is associated with improved survival in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Fukuma, S.; Yamaguchi, T.; Hashimoto, S.; Nakai, S.; Iseki, K.; Tsubakihara, Y.; Fukuhara, S. Erythropoiesis-stimulating agent responsiveness and mortality in hemodialysis patients: Results from a cohort study from the dialysis registry in Japan. Am. J. Kidney Dis. 2012, 59, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Drueke, T.B.; Eckardt, K.U. Role of secondary hyperparathyroidism in erythropoietin resistance of chronic renal failure patients. Nephrol. Dial. Transplant. 2002, 17, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Smrzova, J.; Balla, J.; Barany, P. Inflammation and resistance to erythropoiesis-stimulating agents—What do we know and what needs to be clarified? Nephrol. Dial. Transplant. 2005, 20, viii2–viii7. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.; Rocha, S.; Rocha-Pereira, P.; Reis, F.; Castro, E.; Teixeira, F.; Miranda, V.; do Sameiro Faria, M.; Loureiro, A.; Quintanilha, A.; et al. DMT1 (NRAMP2/DCT1) genetic variability and resistance to recombinant human erythropoietin therapy in chronic kidney disease patients under haemodialysis. Acta Haematol. 2008, 120, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Do Sameiro-Faria, M.; Ribeiro, S.; Rocha-Pereira, P.; Fernandes, J.; Reis, F.; Bronze-da-Rocha, E.; Miranda, V.; Quintanilha, A.; Costa, E.; Belo, L.; et al. Body mass index and resistance to recombinant human erythropoietin therapy in maintenance hemodialysis patients. Ren. Fail. 2013, 35, 1392–1398. [Google Scholar] [CrossRef] [PubMed]
- Macdougall, I.C. Antibody-mediated pure red cell aplasia (PRCA): Epidemiology, immunogenicity and risks. Nephrol. Dial. Transplant. 2005, 20, iv9–iv15. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, N. Pure red cell aplasia and anti-erythropoietin antibodies in patients treated with epoetin. Nephrol. Dial. Transplant. 2003, 18, viii37–viii41. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, K.U.; Casadevall, N. Pure red-cell aplasia due to anti-erythropoietin antibodies. Nephrol. Dial. Transplant. 2003, 18, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.C.; Lin, Y.P.; Tarng, D.C. Erythropoiesis-stimulating agents in chronic kidney disease: What have we learned in 25 years? J. Formos. Med. Assoc. 2014, 113, 3–10. [Google Scholar] [PubMed]
- Garrido, P.; Ribeiro, S.; Fernandes, J.; Vala, H.; Bronze-da-Rocha, E.; Rocha-Pereira, P.; Belo, L.; Costa, E.; Santos-Silva, A.; Reis, F. Iron-hepcidin dysmetabolism, anemia and renal hypoxia, inflammation and fibrosis in the remnant kidney rat model. PLoS ONE 2015, 10, e0124048. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.; Pereira, B.J.; Rocha-Pereira, P.; Rocha, S.; Reis, F.; Castro, E.; Teixeira, F.; Miranda, V.; do Sameiro Faria, M.; Loureiro, A.; et al. Role of prohepcidin, inflammatory markers and iron status in resistance to rhEPO therapy in hemodialysis patients. Am. J. Nephrol. 2008, 28, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.C.; Garrido, P.; Ribeiro, S.; Rocha-Pereira, P.; Bronze-da-Rocha, E.; Belo, L.; Costa, E.; Reis, F.; Santos-Silva, A. Iron as the key modulator of hepcidin expression in erythroid antibody-mediated hypoplasia. Biomed. Res. Int. 2014, 2014, 421304. [Google Scholar] [CrossRef] [PubMed]
- Garrido, P.; Costa, E.; Santos-Silva, A.; Reis, F. Animal models of kidney disease-associated anemia. In Frontiers in Drug Discovery, Erythropoietic Stimulating Agents; Costa, E., Santos-Silva, A., Reis, F., Eds.; Bentham E-Books Publisher: Berlin, Germany, 2013; pp. 61–80. [Google Scholar]
- Strauch, M.; Gretz, N. Animal models to induce renal failure: A historical survey. Contrib. Nephrol. 1988, 60, 1–8. [Google Scholar] [PubMed]
- Morrison, A.B. Experimentally induced chronic renal insufficiency in the rat. Lab. Investig. 1962, 11, 321–332. [Google Scholar] [PubMed]
- Nankivell, B.J.; Chen, J.; Boadle, R.A.; Harris, D.C. The role of tubular iron accumulation in the remnant kidney. J. Am. Soc. Nephrol. 1994, 4, 1598–1607. [Google Scholar] [PubMed]
- Nankivell, B.J.; Boadle, R.A.; Harris, D.C. Iron accumulation in human chronic renal disease. Am. J. Kidney Dis. 1992, 20, 580–584. [Google Scholar] [CrossRef]
- Harris, D.C.; Tay, C.; Nankivell, B.J. Lysosomal iron accumulation and tubular damage in rat puromycin nephrosis and ageing. Clin. Exp. Pharmacol. Physiol. 1994, 21, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Izuhara, Y.; Nangaku, M.; Inagi, R.; Tominaga, N.; Aizawa, T.; Kurokawa, K.; van Ypersele de Strihou, C.; Miyata, T. Renoprotective properties of angiotensin receptor blockers beyond blood pressure lowering. J. Am. Soc. Nephrol. 2005, 16, 3631–3641. [Google Scholar] [CrossRef] [PubMed]
- Egrie, J.C.; Browne, J.K. Development and characterization of darbepoetin alfa. Oncology 2002, 16, 13–22. [Google Scholar] [PubMed]
- Higuchi, M.; Oh-eda, M.; Kuboniwa, H.; Tomonoh, K.; Shimonaka, Y.; Ochi, N. Role of sugar chains in the expression of the biological activity of human erythropoietin. J. Biol. Chem. 1992, 267, 7703–7709. [Google Scholar] [PubMed]
- Locatelli, F.; Aljama, P.; Barany, P.; Canaud, B.; Carrera, F.; Eckardt, K.U.; Macdougall, I.C.; Macleod, A.; Hörl, W.H.; Wiecek, A.; et al. Erythropoiesis-stimulating agents and antibody-mediated pure red-cell aplasia: Here are we now and where do we go from here? Nephrol. Dial. Transplant. 2004, 19, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, N. What is antibody-mediated pure red cell aplasia (PRCA)? Nephrol. Dial. Transplant. 2005, 20, iv3–iv8. [Google Scholar] [CrossRef] [PubMed]
- Lombardero, M.; Kovacs, K.; Scheithauer, B.W. Erythropoietin: A hormone with multiple functions. Pathobiology 2011, 78, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.N. Erythropoietin: Physiology and molecular mechanisms. Heart Fail. Rev. 2008, 13, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.W. Erythropoietin: Physiology and pharmacology update. Exp. Biol. Med. 2003, 228, 1–14. [Google Scholar]
- Sviglerova, J.; Kuncova, J.; Nalos, L.; Tonar, Z.; Rajdl, D.; Stengl, M. Cardiovascular parameters in rat model of chronic renal failure induced by subtotal nephrectomy. Physiol. Res. 2010, 59, S81–S88. [Google Scholar] [PubMed]
- Vaziri, N.D. Anemia and anemia correction: Surrogate markers or causes of morbidity in chronic kidney disease? Nat. Clin. Pract. Nephrol. 2008, 4, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Garimella, P.S.; Uhlig, K. Current issues in the management and monitoring of hypertension in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2013, 22, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Cardiovascular effects of erythropoietin and anemia correction. Curr. Opin. Nephrol. Hypertens. 2001, 10, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Hypoxia-inducible factors in the kidney. Am. J. Physiol. Ren. Physiol. 2006, 291, F271–F281. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. The sweet side of HIF. Kidney Int. 2010, 78, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Shoji, K.; Tanaka, T.; Nangaku, M. Role of hypoxia in progressive chronic kidney disease and implications for therapy. Curr. Opin. Nephrol. Hypertens. 2014, 23, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Donderski, R.; Szczepanek, J.; Domagalski, K.; Tretyn, A.; Korenkiewicz, J.; Marszalek, A.; Szymański, A.; Wolski, Z.; Odrowąż-Sypniewska, G.; Manitius, J. Analysis of relative expression level of VEGF (vascular endothelial growth factor), HIF-1alpha (hypoxia inducible factor 1alpha) and CTGF (connective tissue growth factor) genes in chronic glomerulonephritis (CGN) patients. Kidney Blood Press. Res. 2013, 38, 83–91. [Google Scholar]
- Rodriguez-Iturbe, B.; Garcia Garcia, G. The role of tubulointerstitial inflammation in the progression of chronic renal failure. Nephron Clin. Pract. 2010, 116, c81–c88. [Google Scholar] [CrossRef] [PubMed]
- Risdon, R.A.; Sloper, J.C.; De Wardener, H.E. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet 1968, 2, 363–366. [Google Scholar] [CrossRef]
- Eddy, A.A. Progression in chronic kidney disease. Adv. Chronic Kidney Dis. 2005, 12, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.; Swinkels, D.W.; Laarakkers, C.M.; Rocha-Pereira, P.; Rocha, S.; Reis, F.; Teixeira, F.; Miranda, V.; do Sameiro Faria, M.; Loureiro, A.; et al. Hepcidin serum levels and resistance to recombinant human erythropoietin therapy in haemodialysis patients. Acta Haematol. 2009, 122, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Preza, G.C.; Pinon, R.; Ganz, T.; Nemeth, E. Cellular catabolism of the iron-regulatory peptide hormone hepcidin. PLoS ONE 2013, 8, e58934. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.S. Control of systemic iron homeostasis by the hemojuvelin-hepcidin axis. Adv. Nutr. 2010, 1, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Finberg, K.E. Unraveling mechanisms regulating systemic iron homeostasis. Hematol. Am. Soc. Hematol. Educ. Program. 2011, 2011, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Canavesi, E.; Alfieri, C.; Pelusi, S.; Valenti, L. Hepcidin and HFE protein: Iron metabolism as a target for the anemia of chronic kidney disease. World J. Nephrol. 2012, 1, 166–176. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 997–1030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.S.; Yang, F.; Wang, J.; Tsukamoto, H.; Enns, C.A. Hemojuvelin-neogenin interaction is required for bone morphogenic protein-4-induced hepcidin expression. J. Biol. Chem. 2009, 284, 22580–22589. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Porayette, P.; Sripichai, O.; Noh, S.J.; Byrnes, C.; Bhupatiraju, A.; Lee, Y.T.; Goodnough, J.B.; Harandi, O.; Ganz, T.; et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009, 114, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Hanawa, H.; Jiao, S.; Ohno, Y.; Hayashi, Y.; Yoshida, K.; Kashimura, T.; Obata, H.; Minamino, T. Inappropriate expression of hepcidin by liver congestion contributes to anemia and relative iron deficiency. J. Card. Fail. 2014, 20, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Song, S.H.; Lee, J.H.; Kim, H.J.; Yang, H.R. Serum hepcidin levels and iron parameters in children with iron deficiency. Korean J. Hematol. 2012, 47, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Artunc, F.; Risler, T. Serum erythropoietin concentrations and responses to anaemia in patients with or without chronic kidney disease. Nephrol. Dial. Transplant. 2007, 22, 2900–2908. [Google Scholar] [CrossRef] [PubMed]
- Reis, F.; Rocha, L.; Ponte, L.; Alcobia, T.; Almeida, L.; Costa-Almeida, C.; Teixeira, F. Effect of preventive and regressive isosorbide 5-mononitrate treatment on catecholamine levels in plasma, platelets, adrenals, left ventricle and aorta in cyclosporin A-induced hypertensive rats. Life Sci. 2005, 77, 2514–2528. [Google Scholar] [CrossRef] [PubMed]
- Urra, J.M.; de la Torre, M.; Alcazar, R.; Peces, R. Rapid method for detection of anti-recombinant human erythropoietin antibodies as a new form of erythropoietin resistance. Clin. Chem. 1997, 43, 848–849. [Google Scholar] [PubMed]
- Teixeira, M.; Rodrigues-Santos, P.; Garrido, P.; Costa, E.; Parada, B.; Sereno, J.; Alves, R.; Belo, L.; Teixeira, F.; Santos-Silva, A.; et al. Cardiac antiapoptotic and proproliferative effect of recombinant human erythropoietin in a moderate stage of chronic renal failure in the rat. J. Pharm. Bioallied Sci. 2012, 4, 76–83. [Google Scholar] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Method Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Acs, G.; Zhang, P.J.; McGrath, C.M.; Acs, P.; McBroom, J.; Mohyeldin, A.; Liu, S.; Lu, H.; Verma, A. Hypoxia-inducible erythropoietin signaling in squamous dysplasia and squamous cell carcinoma of the uterine cervix and its potential role in cervical carcinogenesis and tumor progression. Am. J. Pathol. 2003, 162, 1789–1806. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/)
Share and Cite
Garrido, P.; Ribeiro, S.; Fernandes, J.; Vala, H.; Rocha-Pereira, P.; Bronze-da-Rocha, E.; Belo, L.; Costa, E.; Santos-Silva, A.; Reis, F. Resistance to Recombinant Human Erythropoietin Therapy in a Rat Model of Chronic Kidney Disease Associated Anemia. Int. J. Mol. Sci. 2016, 17, 28. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010028
Garrido P, Ribeiro S, Fernandes J, Vala H, Rocha-Pereira P, Bronze-da-Rocha E, Belo L, Costa E, Santos-Silva A, Reis F. Resistance to Recombinant Human Erythropoietin Therapy in a Rat Model of Chronic Kidney Disease Associated Anemia. International Journal of Molecular Sciences. 2016; 17(1):28. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010028
Chicago/Turabian StyleGarrido, Patrícia, Sandra Ribeiro, João Fernandes, Helena Vala, Petronila Rocha-Pereira, Elsa Bronze-da-Rocha, Luís Belo, Elísio Costa, Alice Santos-Silva, and Flávio Reis. 2016. "Resistance to Recombinant Human Erythropoietin Therapy in a Rat Model of Chronic Kidney Disease Associated Anemia" International Journal of Molecular Sciences 17, no. 1: 28. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010028