
Altered Pre-mRNA Splicing Caused by a Novel Intronic Mutation c.1443+5G>A in the Dihydropyrimidinase (DPYS) Gene

Abstract
:
1. Introduction
2. Results
2.1. Urinary Concentration of Pyrimidine Metabolites Determined by HPLC-MS/MS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Patient (Age) | Uracil | Thymine | DHU a | DHT b | NCβ-Alanine c | NCβ-AIB d |
|---|---|---|---|---|---|---|
| 1 (10 years) | 42 | 26 | 237 | 138 | <1 | <0.5 |
| 2 (1.8 years) | 115 | 85 | 539 | 309 | <1 | <1 |
| Control (<3 years, n = 104) | 11.8 ± 9.1 | 0.5 ± 0.6 | 6.3 ± 5.3 | 3.1 ± 2.1 | 11.0 ± 9.2 | 1.8 ± 2.3 |
| Control (>3 years, n = 111) | 7.0 ± 5.4 | 0.1 ± 0.3 | 2.1 ± 1.6 | 1.0 ± 0.7 | 2.8 ± 2.0 | 0.1 ± 0.4 |
2.2. Genotype
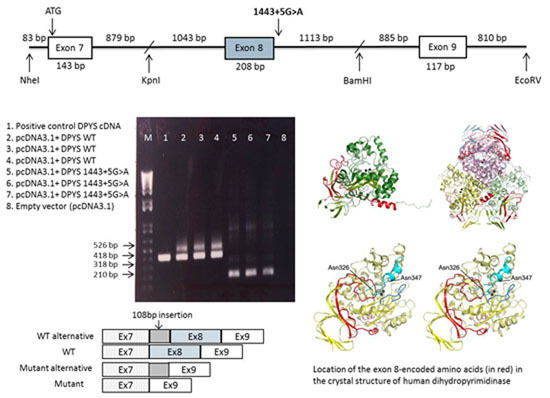
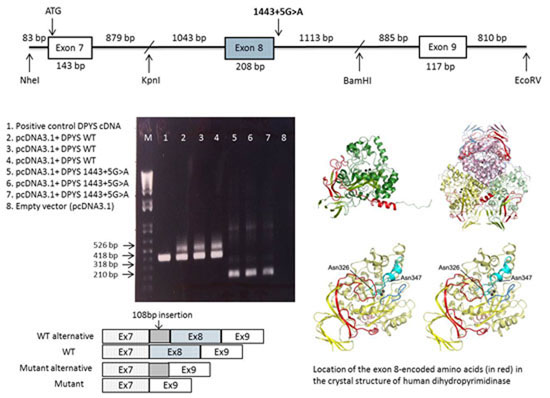
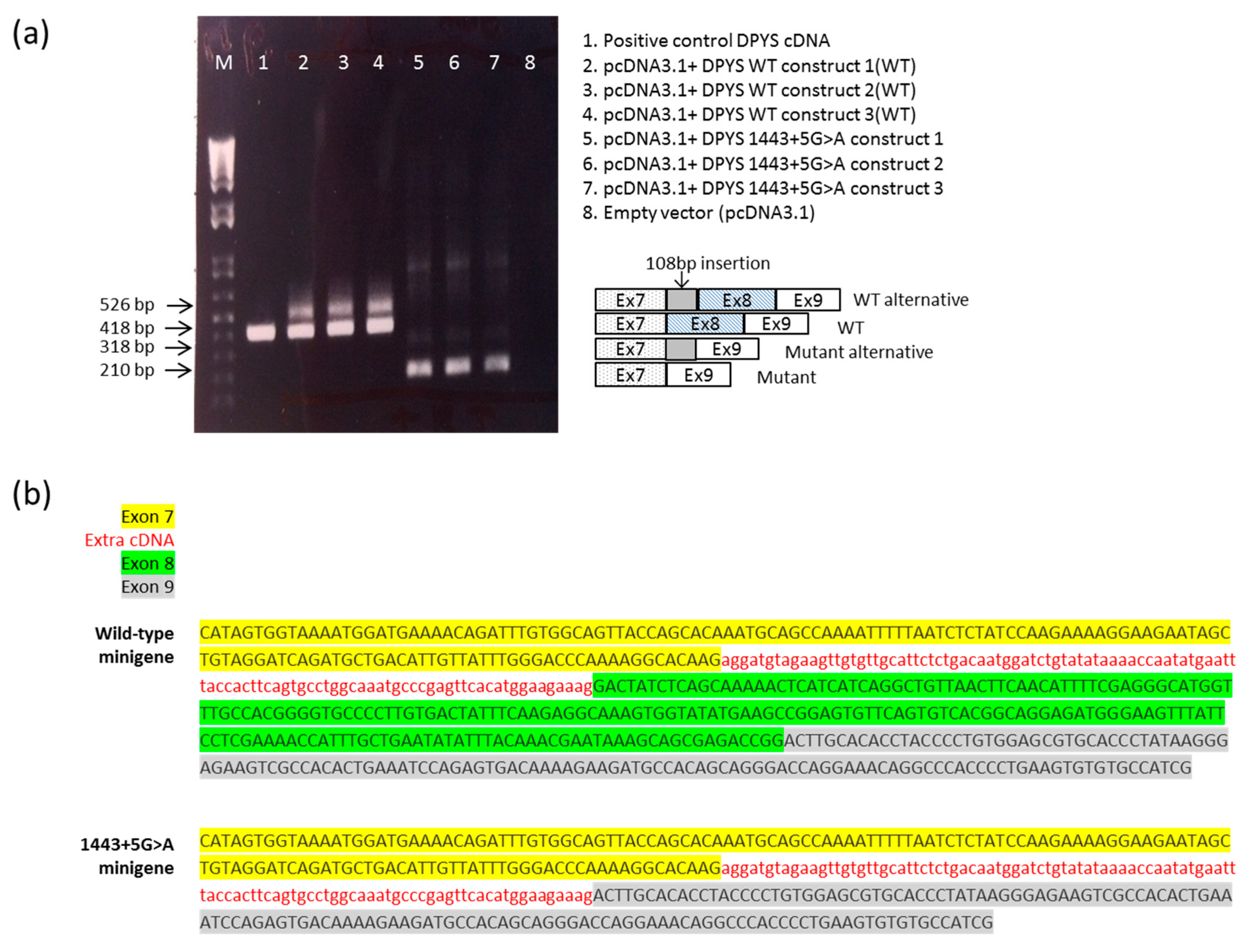
2.3. DPYS Minigene Expression


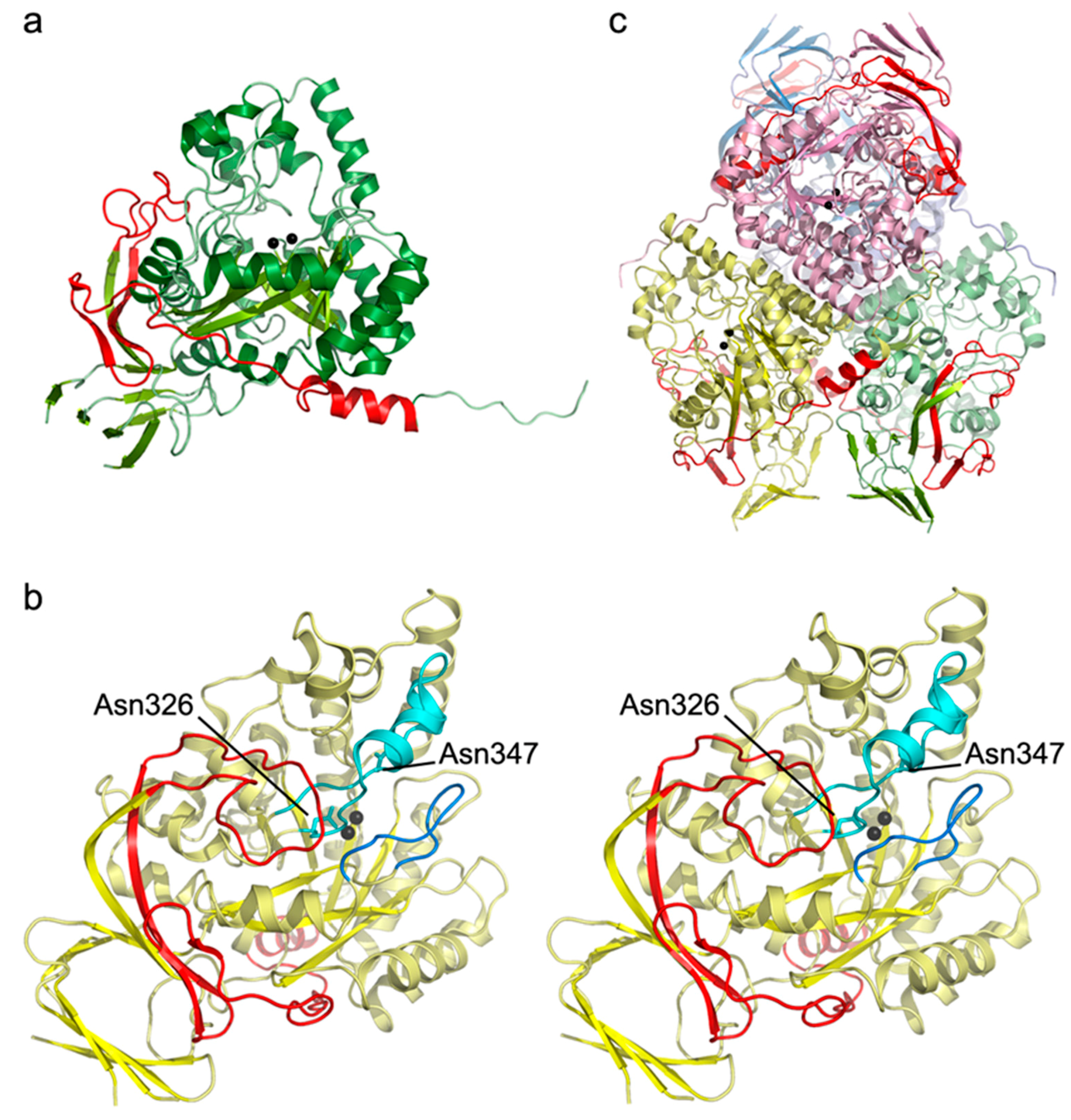
2.4. Analysis of the Crystal Structure of Human DHP

3. Discussion
4. Methods
4.1. Patients
4.2. Quantitative Analysis of Pyrimidines
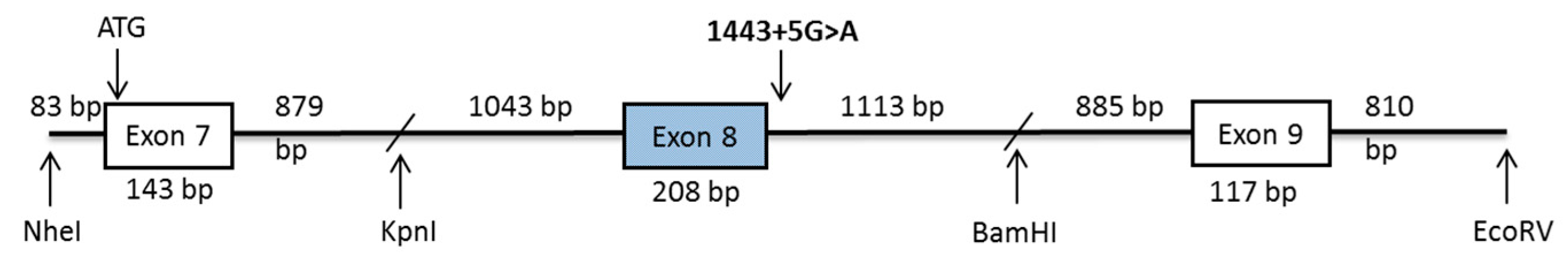
4.3. PCR Amplification of Coding Exons of DPYS
4.4. Construction of the Dihydropyrimidinaase Minigene
| PCR Product | Primer Sequence | Including Exon | Bp | Restriction Enzymes |
|---|---|---|---|---|
| Fragment A | F: 5′-tcatGCTAGCtgcaagtcttgtcattttatcg-3′ | Exon 7 | 1268 | NheI Acc65I |
| R: 5′-gaaggctgcttgcttgctat–3′ | ||||
| Fragment B | F: 5′-tcatGGTACCgtgggagcaaagctatgagg-3′ | Exon 8 | 2382 | Acc65I BamHI |
| R: 5′-tcatGGATCCcatcaaaaggggaaagcaaa-3′ | ||||
| Fragment C | F: 5′-tttcagatgtggtggtccaa-3′ | Exon 9 | 2493 | BamHI EcoRV |
| R: 5′-gcattgaatcgcattccttt-3′ |
4.5. Cell Culture and Transient Transfection
4.6. RNA Analysis of the Overexpressed pcDNA3.1Zeo-DPYS Minigene
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Van Kuilenburg, A.B.; Meijer, J.; Dobritzsch, D.; Meinsma, R.; Duran, M.; Lohkamp, B.; Zoetekouw, L.; Abeling, N.G.; van Tinteren, H.L.; Bosch, A.M. Clinical, biochemical and genetic findings in two siblings with a dihydropyrimidinase deficiency. Mol. Genet. Metab. 2007, 91, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, A. Inborn errors of purine and pyrimidine metabolism. J. Inherit. Metab Dis. 2009, 32, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Hamajima, N.; Kouwaki, M.; Vreken, P.; Matsuda, K.; Sumi, S.; Imaeda, M.; Ohba, S.; Kidouchi, K.; Nonaka, M.; Sasaki, M.; et al. Dihydropyrimidinase deficiency: Structural organization, chromosomal localization, and mutation analysis of the human dihydropyrimidinase gene. Am. J. Hum. Genet. 1998, 63, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Duran, M.; Rovers, P.; de Bree, P.K.; Schreuder, C.H.; Beukenhorst, H.; Dorland, L.; Berger, R. Dihydropyrimidinuria: A new inborn error of pyrimidine metabolism. J. Inherit. Metab. Dis. 1991, 14, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Assmann, B.; Hoffmann, G.F.; Wagner, L.; Brautigam, C.; Seyberth, H.W.; Duran, M.; van Kuilenburg, A.B.; Wevers, R.; van Gennip, A.H. Dihydropyrimidinase deficiency and congenital microvillous atrophy: Coincidence or genetic relation? J. Inherit. Metab. Dis. 1997, 20, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Duran, M.; Rovers, P.; de Bree, P.K.; Schreuder, C.H.; Beukenhorst, H.; Dorland, L.; Berger, R. Dihydropyrimidinuria. Lancet 1990, 336, 817–818. [Google Scholar] [CrossRef]
- Henderson, M.J.; Ward, K.; Simmonds, H.A.; Duley, J.A.; Davies, P.M. Dihydropyrimidinase deficiency presenting in infancy with severe developmental delay. J. Inherit. Metab. Dis. 1993, 16, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Putman, C.W.; Rotteveel, J.J.; Wevers, R.A.; van Gennip, A.H.; Bakkeren, J.A.; de Abreu, R.A. Dihydropyrimidinase deficiency, a progressive neurological disorder? Neuropediatrics 1997, 28, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Sumi, S.; Imaeda, M.; Kidouchi, K.; Ohba, S.; Hamajima, N.; Kodama, K.; Togari, H.; Wada, Y. Population and family studies of dihydropyrimidinuria: Prevalence, inheritance mode, and risk of fluorouracil toxicity. Am. J. Med. Genet. 1998, 78, 336–340. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.; Dobritzsch, D.; Meijer, J.; Meinsma, R.; Benoist, J.F.; Assmann, B.; Schubert, S.; Hoffmann, G.F.; Duran, M.; de Vries, M.C.; et al. Dihydropyrimidinase deficiency: Phenotype, genotype and structural consequences in 17 patients. Biochim. Biophys. Acta 2010, 1802, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.W.; Yau, M.M.; Ma, C.K.; Siu, T.S.; Tam, S.; Lam, C.W. Diagnosis of dihydropyrimidinase deficiency in a Chinese boy with dihydropyrimidinuria. Hong Kong Med. J. 2013, 19, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, M.; Yamashita, H.; Akai, F.; Hosono, H.; Hishinuma, E.; Hirasawa, N.; Mori, T. Genetic polymorphisms of dihydropyrimidinase in a Japanese patient with capecitabine-induced toxicity. PLoS ONE 2015, 10, e0124818. [Google Scholar] [CrossRef] [PubMed]
- Daguenet, E.; Dujardin, G.; Valcarcel, J. The pathogenicity of splicing defects: Mechanistic insights into pre-mRNA processing inform novel therapeutic approaches. EMBO Rep. 2015, 16, 1640–1655. [Google Scholar] [CrossRef] [PubMed]
- Gaildrat, P.; Killian, A.; Martins, A.; Tournier, I.; Frebourg, T.; Tosi, M. Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods Mol. Biol. 2010, 653, 249–257. [Google Scholar] [PubMed]
- Gamez-Pozo, A.; Palacios, I.; Kontic, M.; Menendez, I.; Camino, I.; Garcia-Miguel, P.; Abelairas, J.; Pestana, A.; Alonso, J. Pathogenic validation of unique germline intronic variants of RB1 in retinoblastoma patients using minigenes. Hum. Mutat. 2007, 28, 1245. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, G.; Casarin, A.; Trevisson, E.; Dona, M.; Cassina, M.; Graziano, C.; Picci, L.; Clementi, M.; Salviati, L. Validation of CFTR intronic variants identified during cystic fibrosis population screening by a minigene splicing assay. Clin. Chem. Lab. Med. 2015, 53, 1719–1723. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.B.; Sorensen, S.; Cartegni, L.; Corydon, T.J.; Doktor, T.K.; Schroeder, L.D.; Reinert, L.S.; Elpeleg, O.; Krainer, A.R.; Gregersen, N.; et al. Seemingly neutral polymorphic variants may confer immunity to splicing-inactivating mutations: a synonymous SNP in exon 5 of MCAD protects from deleterious mutations in a flanking exonic splicing enhancer. Am. J. Hum. Genet. 2007, 80, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Lohkamp, B.; Andersen, B.; Piskur, J.; Dobritzsch, D. The crystal structures of dihydropyrimidinases reaffirm the close relationship between cyclic amidohydrolases and explain their substrate specificity. J. Biol. Chem. 2006, 281, 13762–13776. [Google Scholar] [CrossRef] [PubMed]
- Cheon, Y.H.; Kim, H.S.; Han, K.H.; Abendroth, J.; Niefind, K.; Schomburg, D.; Wang, J.; Kim, Y. Crystal structure of d-hydantoinase from Bacillus stearothermophilus: insight into the stereochemistry of enantioselectivity. Biochemistry 2002, 41, 9410–9417. [Google Scholar] [CrossRef] [PubMed]
- Van Kuilenburg, A.B.; van Lenthe, H.; van Gennip, A.H. Activity of pyrimidine degradation enzymes in normal tissues. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1211–1214. [Google Scholar] [CrossRef] [PubMed]
- Van Kuilenburg, A.B.; van Lenthe, H.; van Cruchten, A.; Kulik, W. Quantification of 5,6-dihydrouracil by HPLC-electrospray tandem mass spectrometry. Clin Chem. 2004, 50, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Van Lenthe, H.; van Kuilenburg, A.B.; Ito, T.; Bootsma, A.H.; van Cruchten, A.; Wada, Y.; van Gennip, A.H. Defects in pyrimidine degradation identified by HPLC-electrospray tandem mass spectrometry of urine specimens or urine-soaked filter paper strips. Clin. Chem. 2000, 46, 1916–1922. [Google Scholar] [PubMed]
- Meijer, J.; Nakajima, Y.; Zhang, C.; Meinsma, R.; Ito, T.; van Kuilenburg, A.B. Identification of a novel synonymous mutation in the human beta-Ureidopropionase Gene UPB1 affecting pre-mRNA splicing. Nucleosides Nucleotides Nucleic Acids 2013, 32, 639–645. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakajima, Y.; Meijer, J.; Zhang, C.; Wang, X.; Kondo, T.; Ito, T.; Dobritzsch, D.; Van Kuilenburg, A.B.P. Altered Pre-mRNA Splicing Caused by a Novel Intronic Mutation c.1443+5G>A in the Dihydropyrimidinase (DPYS) Gene. Int. J. Mol. Sci. 2016, 17, 86. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010086
Nakajima Y, Meijer J, Zhang C, Wang X, Kondo T, Ito T, Dobritzsch D, Van Kuilenburg ABP. Altered Pre-mRNA Splicing Caused by a Novel Intronic Mutation c.1443+5G>A in the Dihydropyrimidinase (DPYS) Gene. International Journal of Molecular Sciences. 2016; 17(1):86. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010086
Chicago/Turabian StyleNakajima, Yoko, Judith Meijer, Chunhua Zhang, Xu Wang, Tomomi Kondo, Tetsuya Ito, Doreen Dobritzsch, and André B. P. Van Kuilenburg. 2016. "Altered Pre-mRNA Splicing Caused by a Novel Intronic Mutation c.1443+5G>A in the Dihydropyrimidinase (DPYS) Gene" International Journal of Molecular Sciences 17, no. 1: 86. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010086