
The Functional Analysis of Histone Acetyltransferase MOF in Tumorigenesis


Abstract
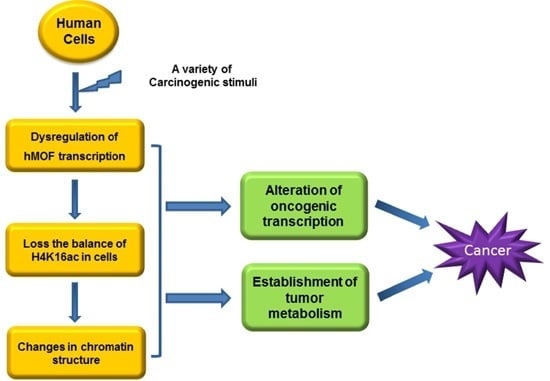
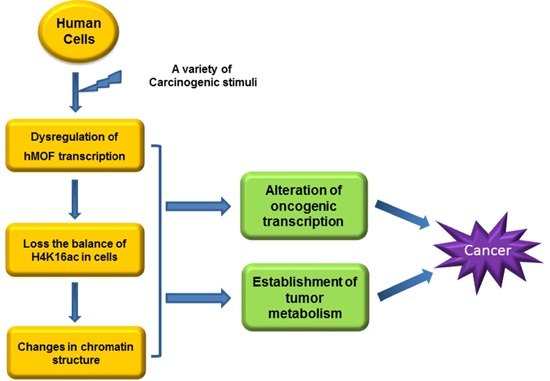
:
1. Introduction
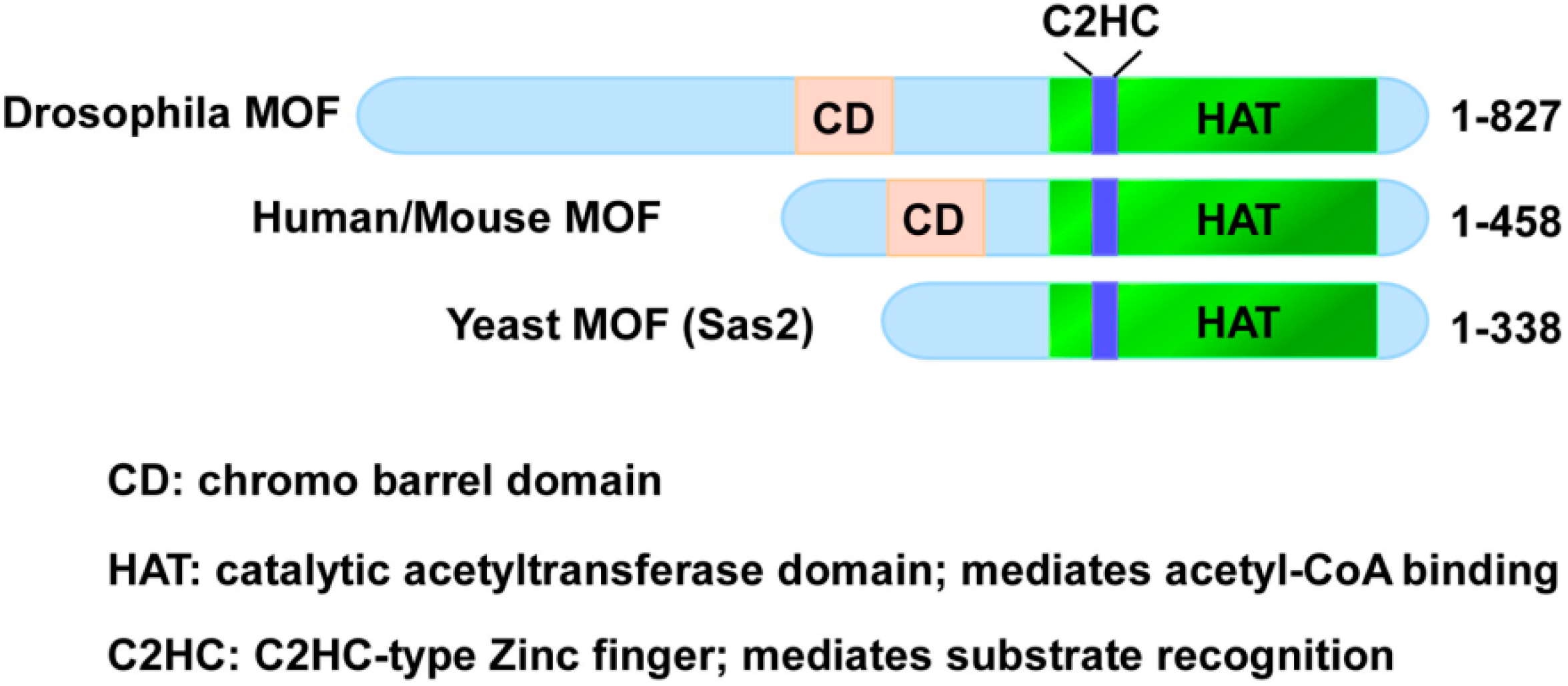
2. MOF Belongs to the MYST Family of HATs

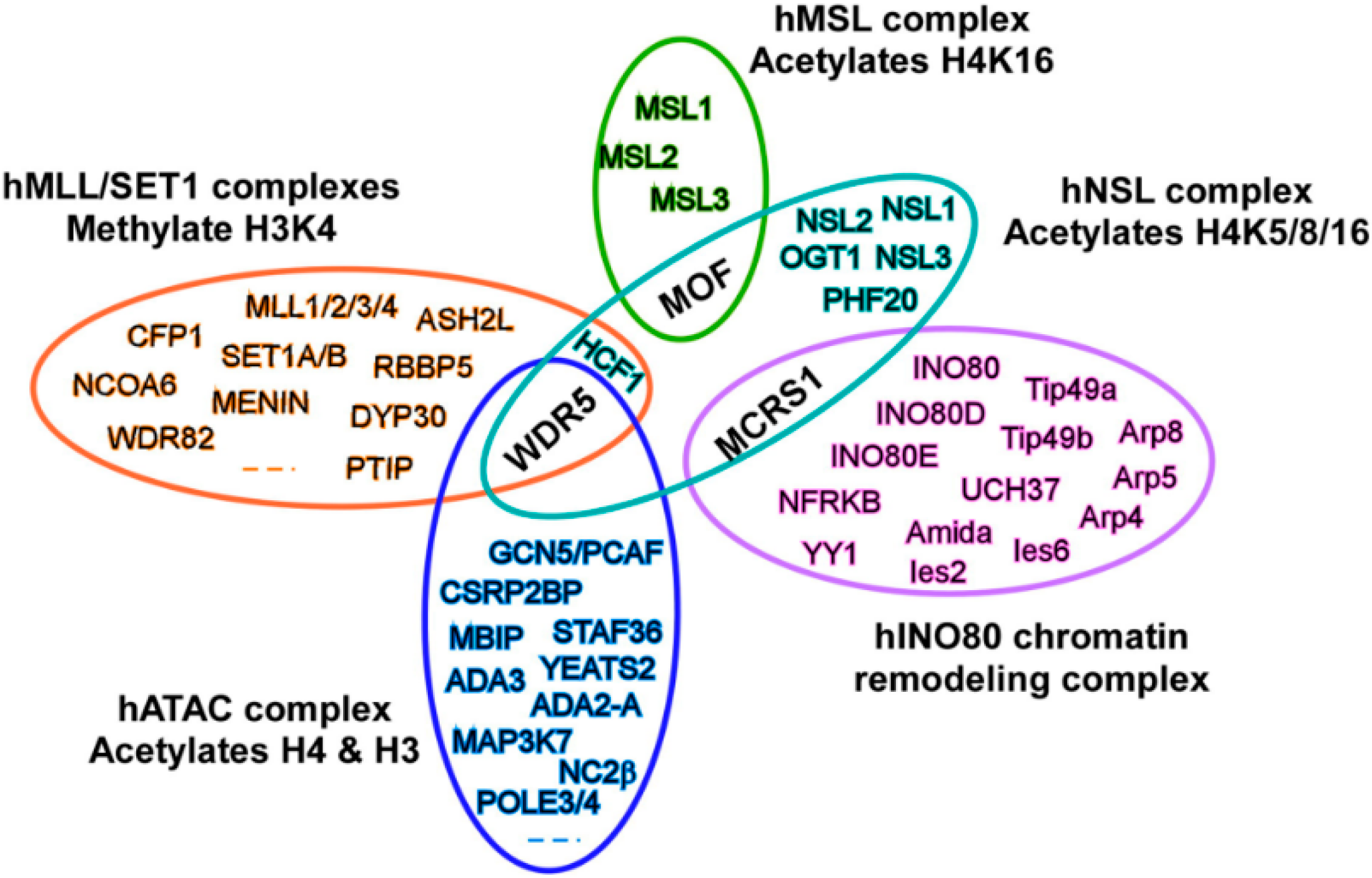
3. MOF Forms Two Distinct Multiprotein Complexes in Cells
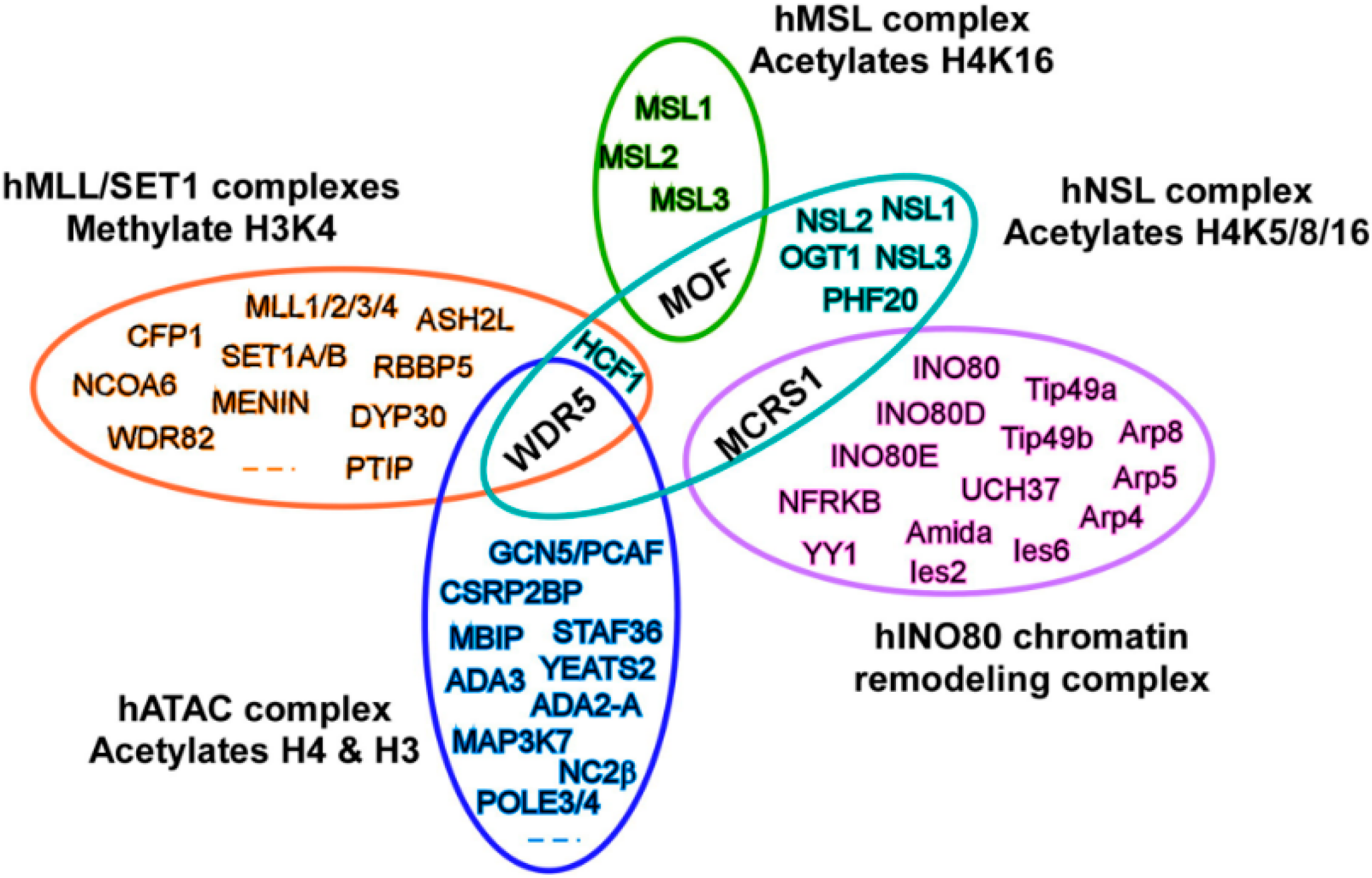
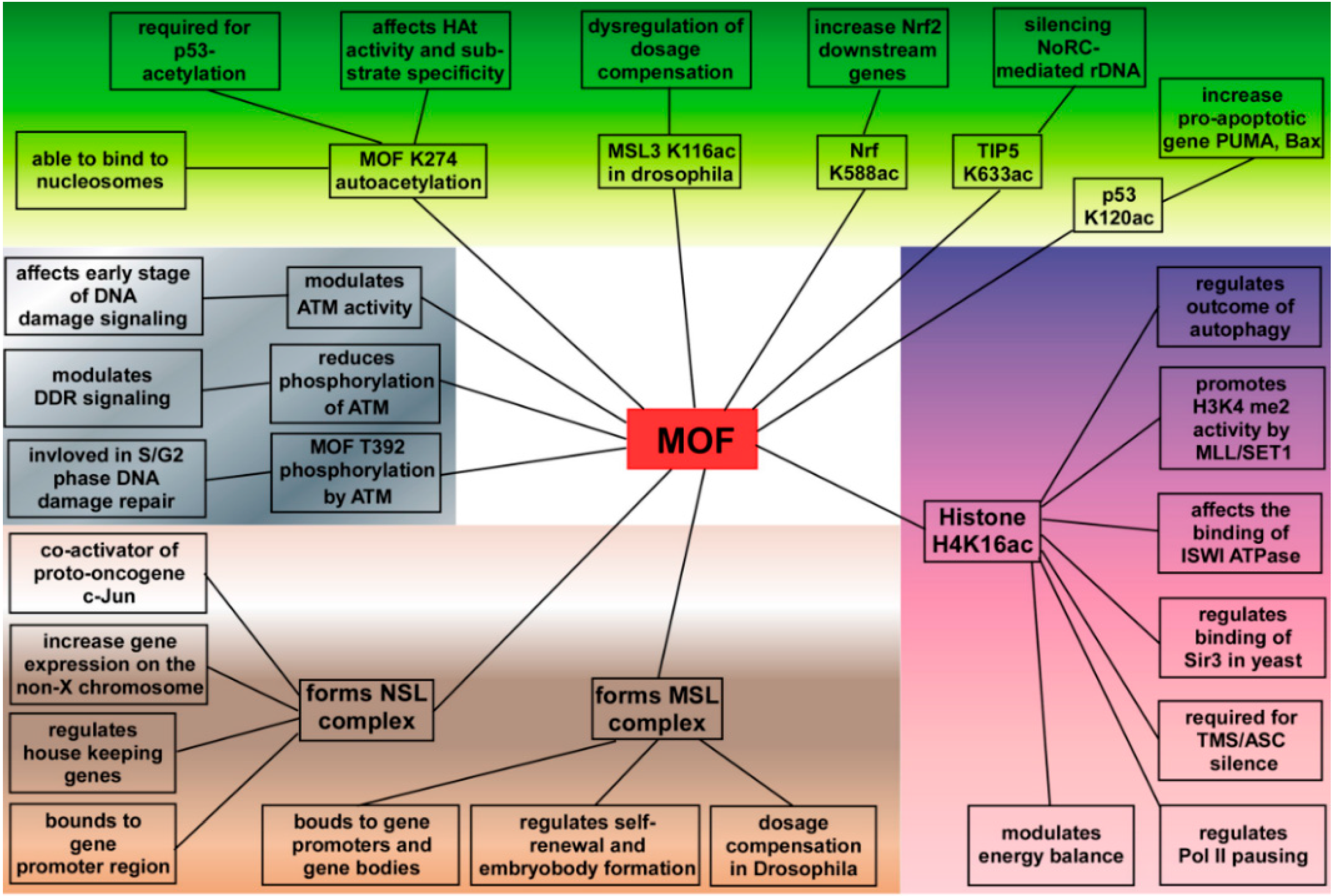
4. MOF Plays a Critical Role in Cells
4.1. MOF in Gene Transcriptional Regulation
4.2. Functions of MOF in DNA Damage Repair
4.3. Functions of MOF in ESCs
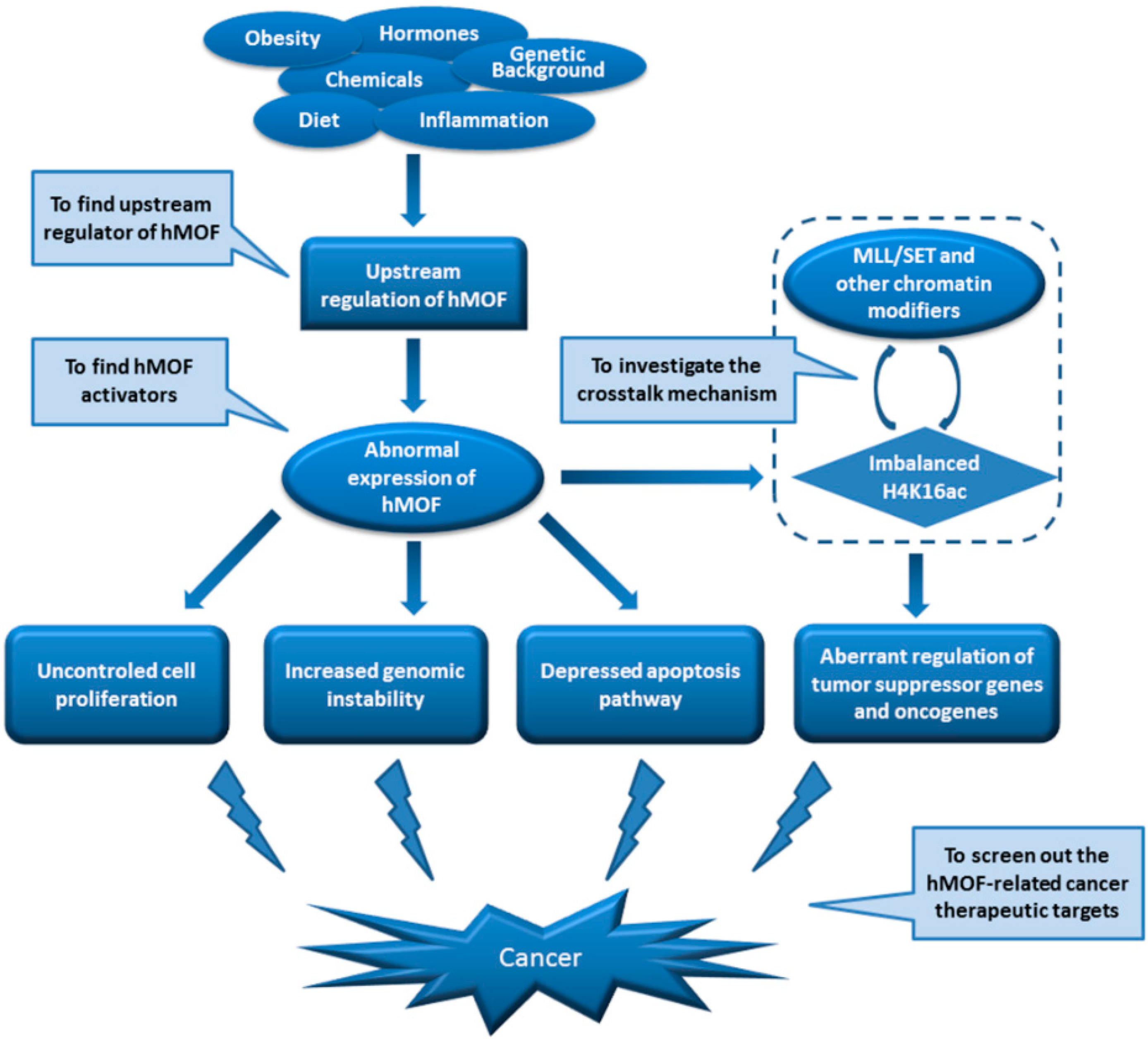
5. Roles of MOF in Tumorigenesis
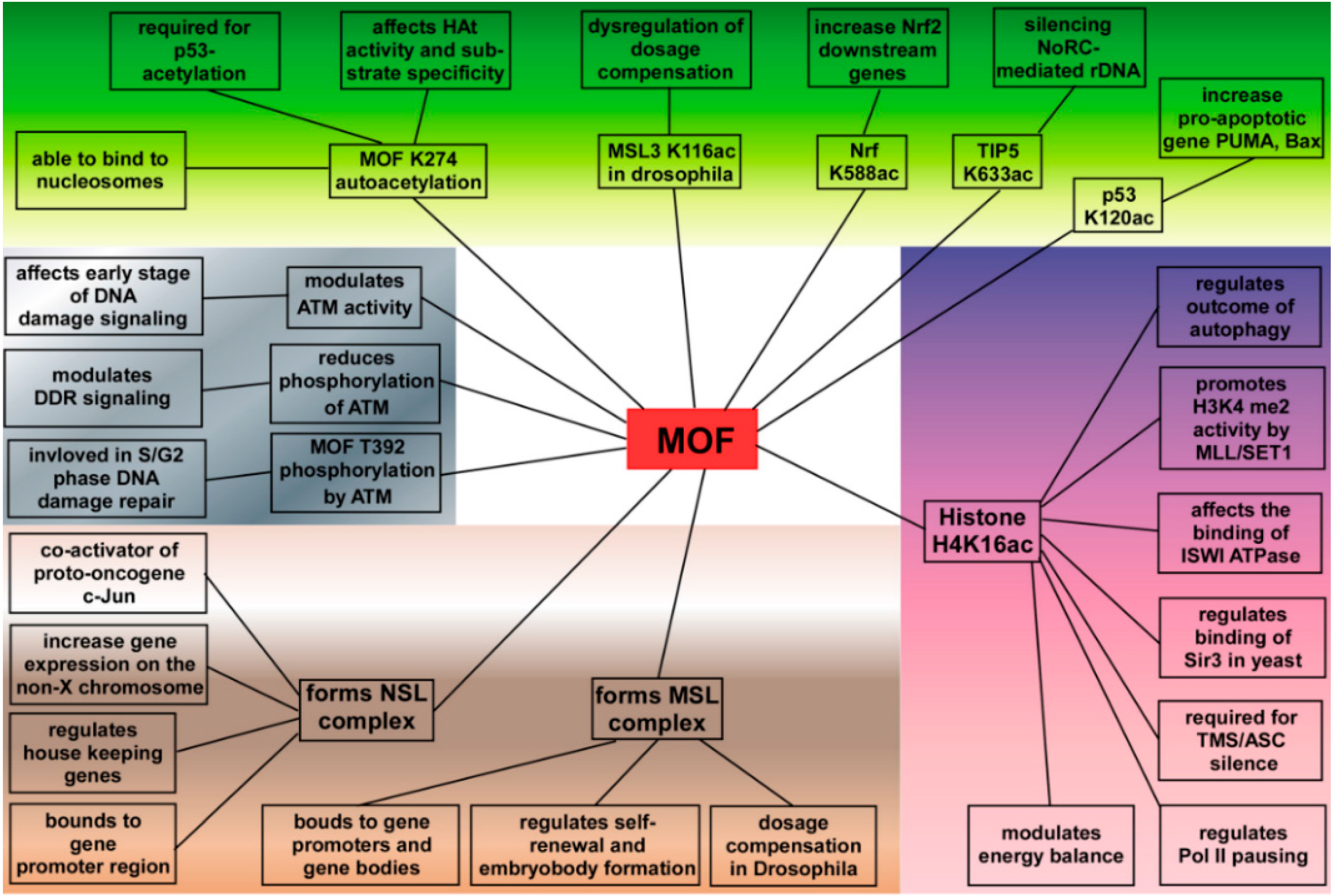
5.1. Histone Acetylation Modifiers in Cancer
5.2. MOF Expression in Cancer
5.3. Regulation of MOF in Cancer
5.4. Epigenetic Therapies Targeting Histone Acetylation in Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Samples | Method | Modifiers | Results | H4K16ac | Reference |
|---|---|---|---|---|---|---|
| Breast cancer | Tissue (98) | Gene profiling | MOF | Downregulated (40%) | ---- | [104] |
| Tissue (298) | IHC | MOF | Reduced or undetectable (18%) | H4K16ac ↓ | [104] | |
| Cell lines | RT-PCR, WB | MOF/SUV420H2 | Regulate Pol II pausing | ---- | [40] | |
| Cell lines | RT-PCR, WB | MOF | Silencing TMS1 gene | H4K16ac ↓ | [117] | |
| RCC | Tissue (47) | RT-qPCR | MOF | Reduced mRNA (74%) | H4K16ac ↓ | [108] |
| Tissue (21) | RT-PCR, WB, IHC | MOF | Reduced mRNA/protein (91%) | H4K16ac ↓ | [105] | |
| Medulloblastoma | Tissue (14) | RT-PCR | MOF | mRNA downregulated (79%) | ---- | [104] |
| Tissue (180) | IHC | MOF | Reduced or undetectable (40%) | H4K16ac ↓ | [104] | |
| Gastric cancer | Tissue (16) | RT-qPCR | MOF | mRNA downregulated (94%) | ---- | [108] |
| Tissue (52) | RT-qPCR | MOF | mRNA downregulated (81%) | ---- | [107] | |
| Cell lines | RT-qPCR, WB, IF | MOF/HDAC4 | Low MOF/high HDAC4 | H4K16ac ↓ | [107] | |
| Colorectal cancer | Tissue (44) | RT-qPCR, IHC | MOF | Reduced mRNA (57%)/protein | H4K16ac ↓ | [108] |
| Ovarian cancer | Tissue (57) | RT-qPCR, WB, IHC | MOF | Reduced mRNA/protein (65%) | H4K16ac ↓ | [54] |
| Tissue (30) | RT-PCR, WB | MOF | Reduced mRNA/protein | H4K16ac ↓ | [106] | |
| Hepatocellular | Tissue (70) | RT-qPCR, WB, IHC | MOF | Reduced mRNA/protein | ---- | [109] |
| Cell lines | RT-qPCR | MOF | Reduced mRNA | ---- | [109] | |
| NSCLC | Tissue (43) | IHC | MOF | Increase protein (14/43) | H4K16ac ↑ | [111] |
| Tissue (54) | RT-PCR, IHC | MOF | Increased mRNA/protein (>34) | ---- | [57] | |
| Tissue (20) | RT-PCR | MOF | Increased mRNA (50%) | ---- | [110] |
6. Conclusions and Perspectives

Acknowledgments
Authors Contributions
Conflicts of Interest
Abbreviations
References
- Van Holde, K.; Zlatanova, J. Chromatin fiber structure: Where is the problem now? Semin. Cell Dev. Biol. 2007, 18, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef] [PubMed]
- Gerhold, C.B.; Gasser, S.M. INO80 and SWR complexes: Relating structure to function in chromatin remodeling. Trends Cell Biol. 2014, 24, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Längst, G.; Manelyte, L. Chromatin remodelers: From function to dysfunction. Genes 2015, 6, 299–324. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Cai, Y.; Li, B.; Conaway, R.C.; Workman, J.L.; Conaway, J.W.; Kusch, T. In and out: Histone variant exchange in chromatin. Trends Biochem. Sci. 2005, 30, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.; Schneider, R. Targeting histone modifications-epigenetics in cancer. Curr. Opin. Cell Biol. 2013, 25, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Kind, J.; Vaquerizas, J.M.; Gebhardt, P.; Gentzel, M.; Luscombe, N.M.; Bertone, P.; Akhtar, A. Genome-wide analysis reveals MOF as a key regulator of dosage compensation and gene expression in Drosophila. Cell 2008, 133, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Guerin-Peyrou, T.G.; Sharma, G.G.; Park, C.; Agarwal, M.; Ganju, R.K.; Pandita, S.; Choi, K.; Sukumar, S.; Pandita, R.K.; et al. The mammalian ortholog of Drosophla MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Mol. Cell. Biol. 2008, 28, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.G.; So, S.; Gupta, A.; Kumar, R.; Cayrou, C.; Avvakumov, N.; Bhadra, U.; Pandita, R.K.; Porteus, M.H.; Chen, D.J.; et al. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol. Cell. Biol. 2010, 30, 3582–3595. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.; Xouri, G.; Akhtar, A. Males absent on the first (MOF): From flies to humans. Oncogene 2007, 26, 5385–5394. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Dyda, F.; Klein, D.C.; Hickman, A.B. GCN5-related N-acetyltransferases: A structural overview. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 81–103. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Pillus, L. MYSTs mark chromatin for chromosomal functions. Curr. Opin. Cell Biol. 2008, 20, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Awakumov, N.; Cöté, J. The MYST family of histone acetyltransferase and their intimate links to cancer. Oncogene 2007, 26, 5395–5407. [Google Scholar] [CrossRef] [PubMed]
- Belote, J.M.; Lucchesi, J.C. Male-specific lethal mutations of Drosophila melanogaster. Genetics 1980, 96, 165–186. [Google Scholar] [PubMed]
- Hilfiker, A.; Hilfiker-Kleiner, D.; Pannuti, A.; Lucchesi, J.C. Mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J. 1997, 16, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Bone, J.R.; Kuroda, M.I. Dosage compensation regulatory proteins and the evolution of sex chromosomes in Drosophila. Genetics 1996, 144, 705–713. [Google Scholar] [PubMed]
- Hilfiker, A.; Yang, Y.; Hayes, D.H.; Beard, C.A.; Manning, J.E.; Lucchesi, J.C. Dosage compensation in Drosophila: The X-chromosomal binding of MSL-1 and MLE is dependent on Sxl activity. EMBO J. 1994, 13, 3542–3550. [Google Scholar] [PubMed]
- Conrad, T.; Akhtar, A. Dosage compensation in Drosophila melanogaster: Epigenetic fine-tuning of chromosome-wide transcription. Nat. Rev. Genet. 2012, 13, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Morales, V.; Straub, T.; Neumann, M.F.; Mengus, G.; Akhtar, A.; Becker, P.B. Functional integration of the histone acetyltransferase MOF into the dosage compensation complex. EMBO J. 2004, 23, 2258–2268. [Google Scholar] [CrossRef] [PubMed]
- Neal, K.C.; Pannuti, A.; Smith, E.R.; Lucchesi, J.C. A new human member of the MYST family of histone acetyl transferases with high sequence similarity to Drosophila MOF. Biochim. Biophys. Acta 2000, 1490, 170–174. [Google Scholar] [CrossRef]
- Deng, X.; Berletch, J.B.; Ma, W.; Nguyen, D.K.; Hiatt, J.B.; Noble, W.S.; Shendure, J.; Disteche, C.M. Mammalian X upregulation is associated with enhanced transcription initiation, RNA half-life, and MOF-mediated H4K16 acetylation. Dev. Cell 2013, 25, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.; Akhtar, A. MSL proteins and the regulation of gene expression. Curr. Top. Microbiol. Immunol. 2006, 310, 117–140. [Google Scholar] [PubMed]
- Kelley, R.L.; Kuroda, M.I. Noncoding RNA genes in dosage compensation and imprinting. Cell 2000, 103, 9–12. [Google Scholar] [CrossRef]
- Mendjan, S.; Taipale, M.; Kind, J.; Holz, H.; Gebhardt, P.; Schelder, M.; Vermeulen, M.; Buscaino, A.; Duncan, K.; Mueller, J.; et al. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol. Cell 2006, 21, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Cayrou, C.; Huang, R.; Lane, W.S.; Côté, J.; Lucchesi, J.C. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol. Cell. Biol. 2005, 25, 9175–9188. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Rea, S.; Richter, K.; Vilar, A.; Lichter, P.; Imhof, A.; Akhtar, A. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol. Cell. Biol. 2005, 25, 6798–6810. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Milne, T.A.; Tackett, A.J.; Smith, E.R.; Fukuda, A.; Wysocka, J.; Allis, C.D.; Chait, B.T.; Hess, J.L.; Roeder, R.G. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4K16 acetyltransferase MOF. Cell 2005, 121, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Jin, J.; Swanson, S.K.; Cole, M.D.; Choi, S.H.; Florens, L.; Washburn, M.P.; Conaway, J.W.; Conaway, R.C. Subunit composition and substrate specificity of a MOF-containing histone acetyltransferase distinct from the male-specific lethal (MSL) complex. J. Biol. Chem. 2010, 285, 4268–4272. [Google Scholar] [CrossRef] [PubMed]
- Raja, S.J.; Charapitsa, I.; Conrad, T.; Vaquerizas, J.M.; Gebhardt, P.; Holz, H.; Kadlec, J.; Fraterman, S.; Luscombe, N.M.; Akhtar, A. The nonspecific lethal complex is a transcriptional regulator in Drosophila. Mol. Cell 2010, 38, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Feller, C.; Prestel, M.; Hartmann, H.; Straub, T.; Söding, J.; Becker, P.B. The MOF-containing NSL complex associates globally with housekeeping genes, but activates only a defined subset. Nucleic Acids Res. 2012, 40, 1509–1522. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.C.; Muhlpfordt, F.; Vaquerizas, J.M.; Raja, S.J.; Holz, H.; Luscombe, N.M.; Manke, T.; Akhtar, A. The NSL complex regulates housekeeping genes in Drosophila. PLoS Genet. 2012, 8, e1002736. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.W.; Hong, T.; Hong, S.; Guo, H.; Yu, H.; Kim, D.; Guszczynski, T.; Dressler, G.R.; Copeland, T.D.; Kalkum, M.; et al. PTIP associates with MLL3- and MLL4-containing histone H3 methyltransferase complex. J. Biol. Chem. 2007, 282, 20395–20406. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, T.; Gutiérrez, J.L.; Li, B.; Florens, L.; Swanson, S.K.; Washburn, M.P.; Abmayr, S.M.; Workman, J.L. ATAC is a bouble histone acetyltransferase complex that stimulates nucleosome sliding. Nat. Struct. Mol. Biol. 2008, 15, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Cai, Y.; Yao, T.; Gottschalk, A.J.; Florens, L.; Swanson, S.K.; Gutiérrez, J.L.; Coleman, M.K.; Workman, J.L.; Mushegian, A.; et al. A mammalian chromatin remodeling complex with similarities to the yeast INO80 complex. J. Biol. Chem. 2005, 280, 41207–41212. [Google Scholar] [CrossRef] [PubMed]
- Kapoor-Vazirani, P.; Kagey, J.D.; Vertino, P.M. SUV420H2-mediated H4K20 trimethylation enforces RNA polymerase II promoter-proximal pausing by blocking hMOF-dependent H4K16 acetylation. Mol. Cell. Biol. 2011, 31, 1594–1609. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Su, J.; Wang, F.; Liu, D.; Ding, J.; Yang, Y.; Conaway, J.W.; Conaway, R.C.; Cao, L.; Wu, D.; et al. Crosstalk between NSL histone acetyltransferase and MLL/SET complexes: NSL complex functions in promoting histone H3K4 di-methylation activity by MLL/SET complexes. PLoS Genet. 2013, 9, e1003940. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Long, Y.; Xing, Z.; Zhang, D. C-Jun recruite the NSL complex to regulate its target gene expression by modulating H4K16 acetylation and promoting the release of the repressive NuRD complex. Oncotarget 2015, 6, 14497–14506. [Google Scholar] [CrossRef] [PubMed]
- Hajji, N.; Wallenborg, K.; Vlachos, P.; Füllgrabe, J.; Hermanson, O.; Joseph, B. Opposing effects of hMOF and SIRT1 on H4K16 acetylation and the sensitivity to the popoisomerase II inhibitor etoposide. Oncogene 2010, 29, 2192–2204. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Ling, H.; Yuan, Z.; Fang, B.; Bloom, G.; Fulasawa, K.; Koomen, J.; Chen, J. SIRT1 negatively regulates the activities, functions, and protein levels of hMOF and TIP60. Mol. Cell. Biol. 2012, 32, 2823–2836. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.W.; Wei, H.M.; Wang, D.L.; Ni, J.Q.; Sun, F.L. Depletion of histone deacetylase 3 antagonizes PI3K-mediated overgrowth of Drosophila organs through the acetylation of histone H4K16. J. Cell Sci. 2012, 125, 5369–5378. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Costa, M.; Sun, H. Structure and function of histone acetyltransferase MOF. AIMS Biophys. 2015, 2, 555–569. [Google Scholar] [CrossRef]
- Carrozza, M.J.; Utley, R.T.; Workman, J.L.; Côté, J. The divers functions of histone acetyltransferase complexes. Trends Genet. 2003, 19, 321–329. [Google Scholar] [CrossRef]
- Suka, N.; Luo, K.; Grunstein, M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine 16 and spreading of heterochromatin. Nat. Genet. 2002, 32, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Corona, D.F.; Clapier, C.R.; Becker, P.B.; Tamkun, J.W. Modulation of ISWI function by site-specific histone acetylation. EMBO Rep. 2002, 3, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Becker, P.B. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol. Cell 2000, 5, 367–375. [Google Scholar] [CrossRef]
- Prestel, M.; Feller, C.; Straub, T.; Mitlöhner, H.; Becker, P.B. The activation potential of MOF is constrained for dosage compensation. Mol. Cell 2010, 38, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Füllgrabe, J.; Lynch-Day, M.A.; Heldring, N.; Li, W.; Struijk, R.B.; Ma, Q.; Hermanson, O.; Rosenfeld, M.G.; Klionsky, D.; Joseph, B. The histone H4 lysine 16 acetyltranferase hMOF regulates the outcome of autophagy. Nature 2013, 500, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, B.N.; Bechtel-Walz, W.; Lucci, J.; Karpiuk, O.; Hild, I.; Hartleben, B.; Vornweg, J.; Helmstadter, M.; Sahyoun, A.H.; Bhardway, V.; et al. MOF maintains transcriptional programs regulating cellular stress response. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, R.; Zhao, X.; Su, J.; Bian, X.; Ni, J.; Yue, Y.; Cai, Y.; Jin, J. A potential diagnostic marker for ovarian cancer: Involvement of the histone acetyltransferase, human males absent on the first. Oncol. Lett. 2013, 6, 393–400. [Google Scholar] [PubMed]
- Brenachot, X.; Rigault, C.; Nédélec, E.; Laderriére, A.; Khanam, T.; Gouazé, A.; Chaudy, S.; Lemoine, A.; Datiche, F.; Gascuel, J.; et al. The histone acetyltransferase MOF activates hypothalamic polysialylation to prevent diet-induced obesity in mice. Mol. Metab. 2014, 3, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ye, X.; Tang, N.; Shen, S.; Li, Z.; Niu, X.; Lu, S.; Xu, L. The histone acetyltransferase hMOF acetylates Nfr2 and regulates anti-drug response in human non-small cell lung cancer. Br. J. Phamacol. 2014, 171, 3196–3211. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Schmitz, K.M.; Mayer, C.; Yuan, X.; Akhtar, A.; Grummt, I. Reversible acetylation of chromatin remodeling complex NoRC is required for non-coding RNA-dependent silencing. Nat. Cell Biol. 2009, 11, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Buscaino, A.; Köcher, T.; Kind, J.H.; Holz, H.; Taipale, M.; Wagner, K.; Wilm, M.; Akhtar, A. MOF-regualted acetylation of MSL3 in the Drosophila dosage compensation complex. Mol. Cell 2003, 11, 1265–1277. [Google Scholar] [CrossRef]
- Kadlec, J.; Hallacli, E.; Lipp, M.; Holz, H.; Sanchez-Weatherby, J.; Cusack, S.; Akhtar, A. Structural basis for MOF and MSL3 recruitment into the dosage compensation complex by MSL1. Nat. Struct. Mol. Biol. 2011, 18, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Guo, S.; Tang, Q.; Li, C.; Zeng, R.; Xiong, Z.; Zhong, C.; Ding, J. Regulation of the histone acetyltransferase activity of hMOF via autoacetylation of Lys274. Cell Res. 2011, 21, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, D.; Zhao, L.; Yang, Y.; Ding, J.; Dong, L.; Hu, L.; Wang, F.; Zhao, X.; Cai, Y.; et al. Arsenic trioxide reduces global histone H4 acetylation at lysine 16 through direct binding to histone acetyltransferase hMOF in human cells. PLoS ONE 2015, 10, e0141014. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.W.; Yu, D.Y.; Pray-Grant, M.G.; Qiu, Q.; Harmon, K.E.; Megee, P.C.; Grant, P.A.; Smith, M.M.; Christman, M.F. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 2002, 419, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Squatrito, M.; Gorrini, C.; Amati, B. Tip60 in DNA damage response and growth control: Many tricks in one HAT. Trends Cell Biol. 2006, 16, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, M.P.; Horikoshi, N.; Pushpavallipvalli, S.N.; Sarkar, A.; Bag, I.; Krishnan, A.; Lucchesi, J.C.; Kumar, R.; Yang, Q.; Pandita, R.K.; et al. The role of MOF in the ionizing radiation response is conserved in Drosophila melanogaster. Chromosoma 2012, 121, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Van Attikum, H.; Gasser, S.M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009, 19, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Sharma, G.G.; Young, C.S.; Agarwal, M.; Smith, E.R.; Paull, T.T.; Lucchesi, J.C.; Khanna, K.K.; Ludwig, T.; Pandita, T.K. Involvement of human MOF in ATM function. Mol. Cell. Biol. 2005, 25, 5292–5305. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Hunt, C.R.; Hegde, M.L.; Chakraborty, S.; Chakraborty, S.; Udayakumar, D.; Horikoshi, N.; Singh, M.; Ramnarain, D.B.; Hittelman, W.N.; et al. MOF phosphorylation by ATM regulates 53BP1-mediated double-strand break repair pathway choice. Cell. Rep. 2014, 8, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Corsa, C.A.; Pan, P.W.; Wu, L.; Ferguson, D.; Yu, X.; Min, J.; Dou, Y. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol. Cell. Biol. 2010, 30, 5335–5347. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Srajer, G.; Evrard, Y.A.; Phan, H.M.; Furuta, Y.; Dent, S.Y. Developmental potential of Gcn5 (-/-) embryonic stem cells in vivo and in vitro. Dev. Dyn. 2007, 236, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Jin, Y. Critical roles of coactivator p300 in mouse embryonic stem cell differentiation and Nanog expression. J. Biol. Chem. 2009, 284, 9168–9175. [Google Scholar] [CrossRef]
- Fazzio, T.G.; Huff, J.T.; Panning, B. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell 2008, 134, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Dixon, M.P.; Kueh, A.J.; Voss, A.K. MOF (MYST1 or KAT8) is essential for progression of embryonic development past the blastocyst stage and required for normal chromatin architecture. Mol. Cell. Biol. 2008, 28, 5093–5105. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, L.; Pandey, R.; Byun, J.S.; Gardner, K.; Qin, Z.; Dou, Y. The histone acetyltransferase MOF is a key regulator of the embryonic stem cell core transcriptional network. Cell Stem Cell 2012, 11, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.H.; Wu, Q.; Chew, L.J.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J.; et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006, 38, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Ravens, S.; Fournie, M.; Ye, T.; Stierle, M.; Dembele, D.; Chavant, V.; Tora, L. Mof-associated complexes have overlapping and unique roles in regulating pluripotency in embryonic stem cells and during differentiation. Elife 2014, 3, e02104. [Google Scholar] [CrossRef] [PubMed]
- Chelmicki, T.; Dündar, F.; Turley, M.J.; Khanam, T.; Aktas, T.; Ramírez, F.; Gendrel, A.V.; Wright, P.R.; Videm, P.; Backofen, R.; et al. MOF-associated complexes ensure stem cell identity and Xist repression. Elife 2014, 3, e02024. [Google Scholar] [CrossRef] [PubMed]
- Pushpavalli, S.N.; Sarkar, A.; Ramaiah, M.J.; Chowdhury, D.R.; Bhadra, U.; Pal-Nhadra, M. Drosophila MOF controls checkpoint proteins and regulates genomic stability during early embryogenesis. BMC Mol. Biol. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Atadja, P.W.; Johnstone, R.W. Epigenetics in cancer: Targeting chromatin modifications. Mol. Cancer Ther. 2009, 8, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Mack, G.S. Epigenetic cancer therapy marks headway. J. Natl. Cancer Inst. 2006, 98, 1443–1444. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, K.; Yokomizo, K.; Shirahata, A.; Goto, T.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; Hibi, K. TIP60 as a potential marker for the malignancy of gastric cancer. Anticancer Res. 2011, 31, 77–79. [Google Scholar] [PubMed]
- Yang, Q.; Wang, B.; Gao, W.; Huang, S.; Liu, Z.; Li, W.; Jia, J. SIRT1 is downregulated in gastric cancer and leads to G1-phase arrest via NF-kB/Cycling D1 signaling. Mol. Cancer Res. 2013, 11, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Song, J.J.; Lee, J.; Kim, M.Y. Epigenetics: An emerging player in gastric cancer. World J. Gatroenterol. 2014, 20, 6433–6447. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, F.R.; Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Scholman, K.; Denkert, C.; Dietel, M.; et al. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004, 32, 959–976. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Iwasaki, H.; Krivtsov, A.; Febbo, P.G.; Thorner, A.R.; Ernst, P.; Anastasiadou, E.; Kutok, J.L.; Kogan, S.C.; Zinkel, S.S.; et al. Conditional MLL-CBP targets GMP and models therapy-related myeloproliferative disease. EMBO J. 2005, 24, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.G.; Ozdag, H.; Caldas, C. p300/CBP and cancer. Oncogenes 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Cole, P.A. Chemical probes for histone-modifying enzymes. Nat. Chem. Biol. 2008, 4, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.; Li, W.; Zhao, W.; Wang, R. Targeting epigenentic regulation in cancer. Acta Biochim. Biophys. Sin. 2016, 48, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Pasheva, E.; Sarov, M.; Bidjekov, K.; Ugrinova, I.; Sarg, B.; Lindner, H.; Pashev, I.G. In vitro acetylation of HMGB-1 and -2 proteins by CBP: The role of the acidic tail. Biochemistry 2004, 43, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Ugrinova, I.; Pasheva, I.G.; Pasheva, E.A. Post-synthetic acetylation of HMGB1 protein modulates its interactions with supercoiled DNA. Mol. Biol. Rep. 2009, 36, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Klune, J.R.; Dhupar, J.; Cardinal, J.; Billiar, T.R.; Tsung, A. HMGB1: Endogenous danger signaling. Mol. Med. 2008, 14, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Travers, A.A. Priming the nucleosome: A role for HMGB proteins? EMBO Rep. 2003, 4, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Barreiro-Alonso, A.; Lamas-Maceiras, M.; Rodríguez-Belmonte, E.; Vazoso-Vázquez, Á.; Quindós, M.; Cerdán, E. High mobility group B proteins, their partners, and other redox sensors in ovarian and prostate cancer. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Topalova, D.; Ugrinova, I.; Pashev, I.G.; Pasheva, E.A. HMGB1 protein inhibits DNA replication in vitro: A role of the acetylation and the acidic tail. Int. J. Biochem. Cell Biol. 2008, 40, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.S.; Mitchell, D.L.; Vasquez, K.M. High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 10320–10325. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Pfister, S.; Rea, S.; Taipale, M.; Mendrzyk, F.; Straub, B.; Ittrich, C.; Thuerigen, O.; Sinn, H.P.; Akhtar, A.; Lichter, P. The histone acetyltransferase hMOF is frequently downregulated in primary breast carcinoma and medulloblastoma and constitutes a biomarker for clinical outcome in medulloblastoma. Int. J. Cancer 2008, 122, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, R.; Wu, D.; Lu, Z.; Sun, W.; Cai, Y.; Wang, C.; Jin, J. Epigenetic change in kidney tumor: Downregulation of histone acetyltransferase MYST1 in human renal cell carcinoma. J. Exp. Clin. Cancer Res. 2013, 32. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Hu, Z.; Liu, J.; Gao, J.; Tan, M.; Zhang, D.; Zhu, L.; Liu, S.; Hou, R.; Lin, B. Expression of hMOF in different ovarian tissues and its effects on ovarian cancer prognosis. Oncol. Rep. 2015, 33, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yang, J.; Zhao, L.; Yu, X.; Wang, L.; Wang, F.; Cai, Y.; Jin, J. Expression of hMOF, but not HDAC4, is responsible for the global histone H4K16 acetylation in gastric cancer. Int. J. Oncol. 2015, 46, 2535–2545. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zhu, L.; Yang, J.; Su, J.; Ni, J.; Du, Y.; Liu, D.; Wang, Y.; Wang, F.; Jin, J.; et al. Correlation of low expression of hMOF with clinicopathological features of colorectal carcinoma, gastric cancer and renal cell carcinoma. Int. J. Oncol. 2014, 44, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, H.; Pan, H.; Yang, Y.; Huang, G.; Yang, Y.; Zhou, W.P.; Pan, Z.Y. The histone acetyltransferase hMOF suppresses hepatocellular carcinoma growth. Biochem. Biophys. Res. Commun. 2014, 452, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Song, J.S.; Chun, S.M.; Lee, J.Y.; Kim, D.K.; Kim, Y.H.; Jang, S.J. The histone acetyltransferase hMOF is overexpressed in non-small cell lung carcinoma. Korean J. Pathol. 2011, 45, 386–396. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, D.L.; Liu, Y.; Chen, S.; Sun, F.L. Histone acetyltransferase hMOF promotes S phase entry and tumorigenesis in lung cancer. Cell Signal. 2013, 25, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Orpinell, M.; Fournier, M.; Riss, A.; Nagy, Z.; Krebs, A.R.; Frontini, M.; Tora, L. The ATAC acetyl transferase complex controls mitotic progression by targeting non-histone substrates. EMBO J. 2010, 29, 2381–2394. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, P.; Yang, F.; Di Stefano, L.; Ji, J.Y.; Ouyang, J.; Nishikawa, J.L.; Toiber, D.; Kulkarni, M.; Wang, Q.; Najafi-Shoushtari, S.H.; et al. A SIRT1-LSD1 corepressor complex regulates Notch target gene expression and development. Mol. Cell 2011, 42, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Schultz, R.M. Histone deacetylase 2 (HDAC2) regulates chromosome segregation and kinetochore function via H4K16 deacetylation during oocyte maturation in mouse. PLoS Genet. 2013, 9, e1003377. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.D.; Lowary, P.T.; Widom, J. Effects of histone acetylation on the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 2001, 307, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4K16 acetylation controls chrmatin structure and protein interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Kapoor-Vazirani, P.; Kagey, J.D.; Powell, D.R.; Vertino, P.M. Role of hMOF-dependent histone H4 lysine 16 acetylation in the maintenance of TMS1/ASC gene activity. Cancer Res. 2008, 68, 6810–6821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hao, C.; Wang, L.; Liu, P.; Zhao, L.; Zhu, C.; Tian, X. Inhibition of leukemic cells by valproic acid, an HDAC inhibitor, in xenograft tumors. Onco. Targets Ther. 2013, 6, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.; Cardoso, B.A.; Belo, H.; Almeida, A.M. Vorinostat induces apoptosis and differentiation in myeloid malignancies: Genetic and molecular mechanisms. PLoS ONE 2013, 8, e53766. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Shi, W.; Jankowski, K.; Kan, C.Y.; Cluse, L.; Martin, B.P.; MacKenzie, K.L.; Smyth, G.K.; Johnstone, R.W. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Federico, M.; Bagella, L. Histone deacetylase inhibitors in the treatment of hematological malignancies and solid tumors. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIb multicenter trial of vornostat in patients with persistant, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.L.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, O.A.; Horwitz, S.; Masszi, T.; van Hoof, A.; Brown, P.; Doorduijin, J.; Hess, G.; Jurczak, W.; Knoblauch, P.; Chawla, S.; et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIFE (CLI-19) study. J. Clin. Oncol. 2015, 33, 2492–2499. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new fromtier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Rajak, H.; Singh, A.; Raghuwanshi, K.; Kumar, R.; Dewangan, P.K.; Veerasamy, R.; Sharma, P.C.; Dixit, A.; Mishra, P. A structural insight into hydroxamic acid based histone deacetylase inhibitors for the presence of anticancer activity. Curr. Med. Chem. 2014, 21, 2642–2664. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, J.; Wang, F.; Cai, Y.; Jin, J. The Functional Analysis of Histone Acetyltransferase MOF in Tumorigenesis. Int. J. Mol. Sci. 2016, 17, 99. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010099
Su J, Wang F, Cai Y, Jin J. The Functional Analysis of Histone Acetyltransferase MOF in Tumorigenesis. International Journal of Molecular Sciences. 2016; 17(1):99. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010099
Chicago/Turabian StyleSu, Jiaming, Fei Wang, Yong Cai, and Jingji Jin. 2016. "The Functional Analysis of Histone Acetyltransferase MOF in Tumorigenesis" International Journal of Molecular Sciences 17, no. 1: 99. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010099