
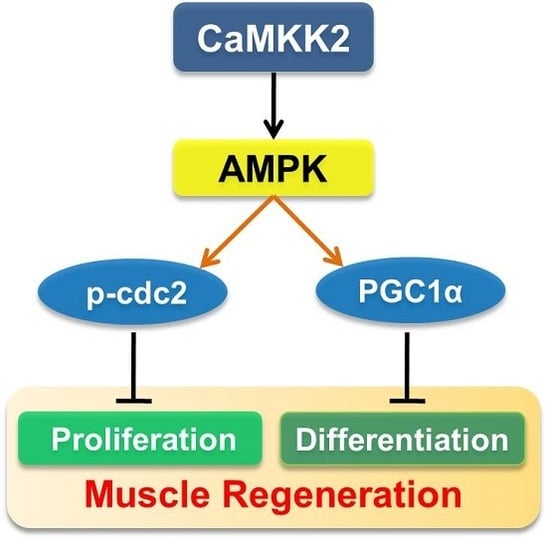
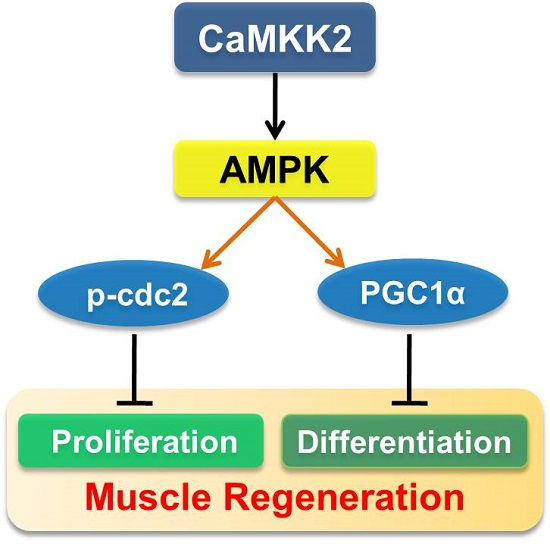
CaMKK2 Suppresses Muscle Regeneration through the Inhibition of Myoblast Proliferation and Differentiation

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
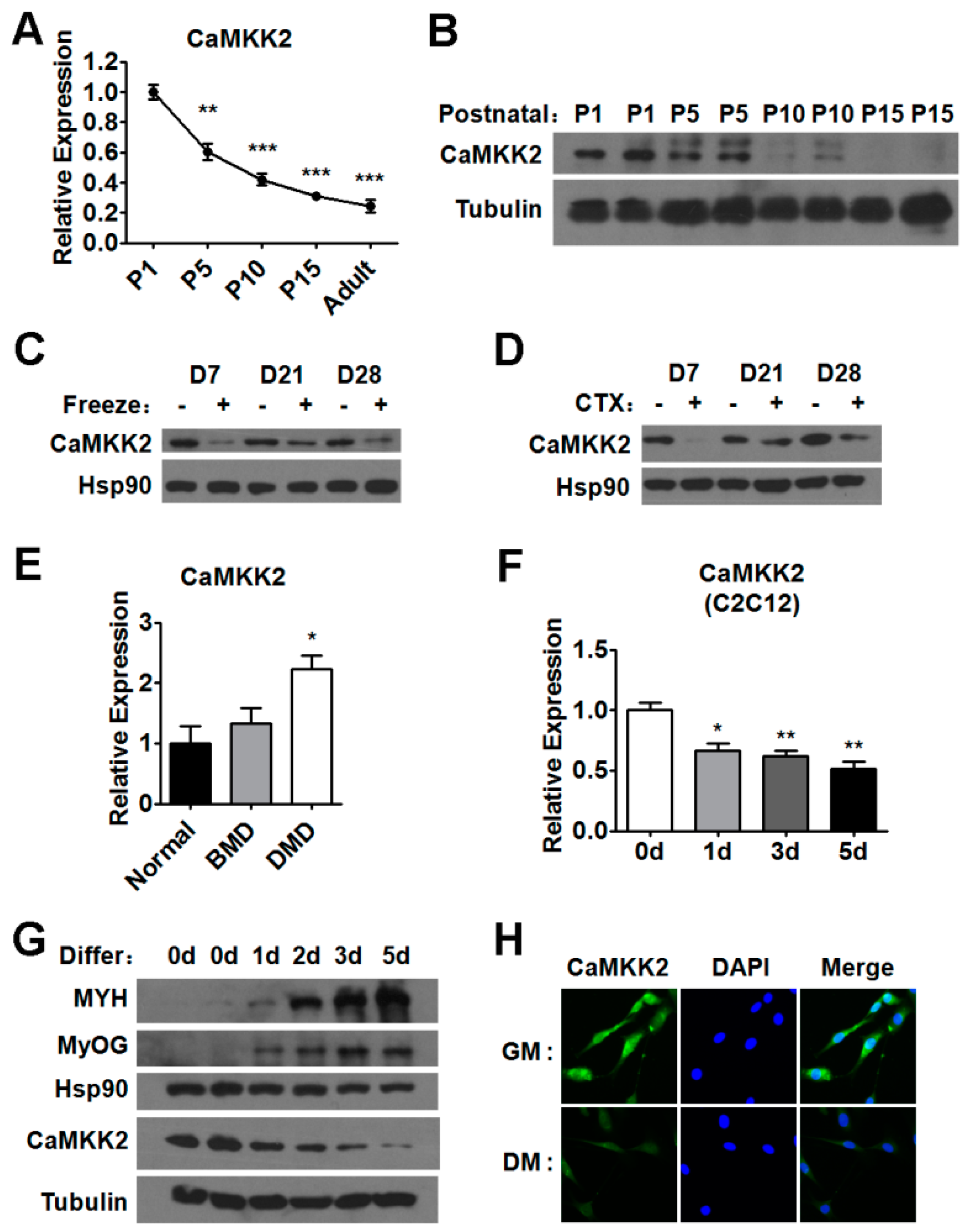
2.1. CaMKK2 Expression Levels during Postnatal Myogenesis and Muscle Regeneration
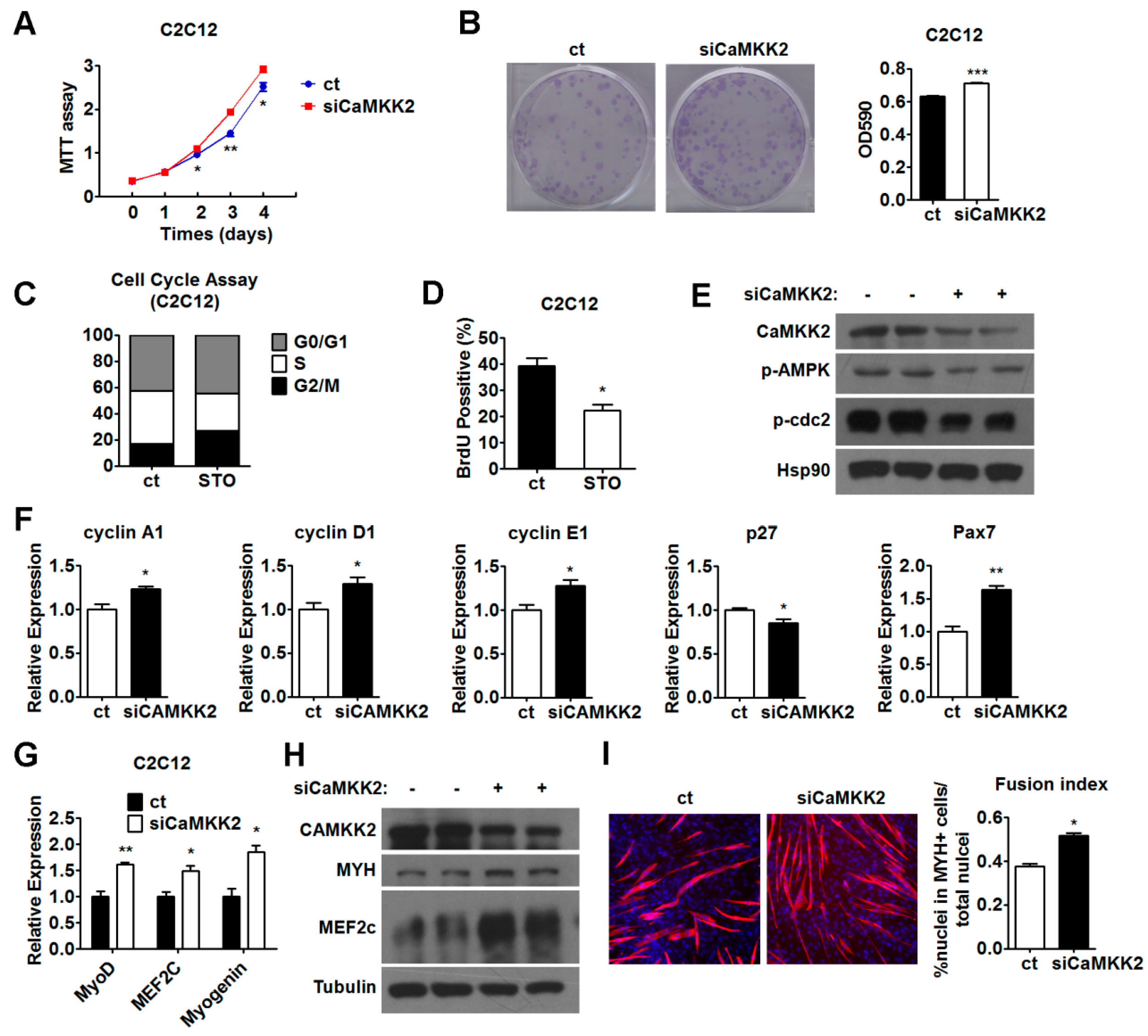
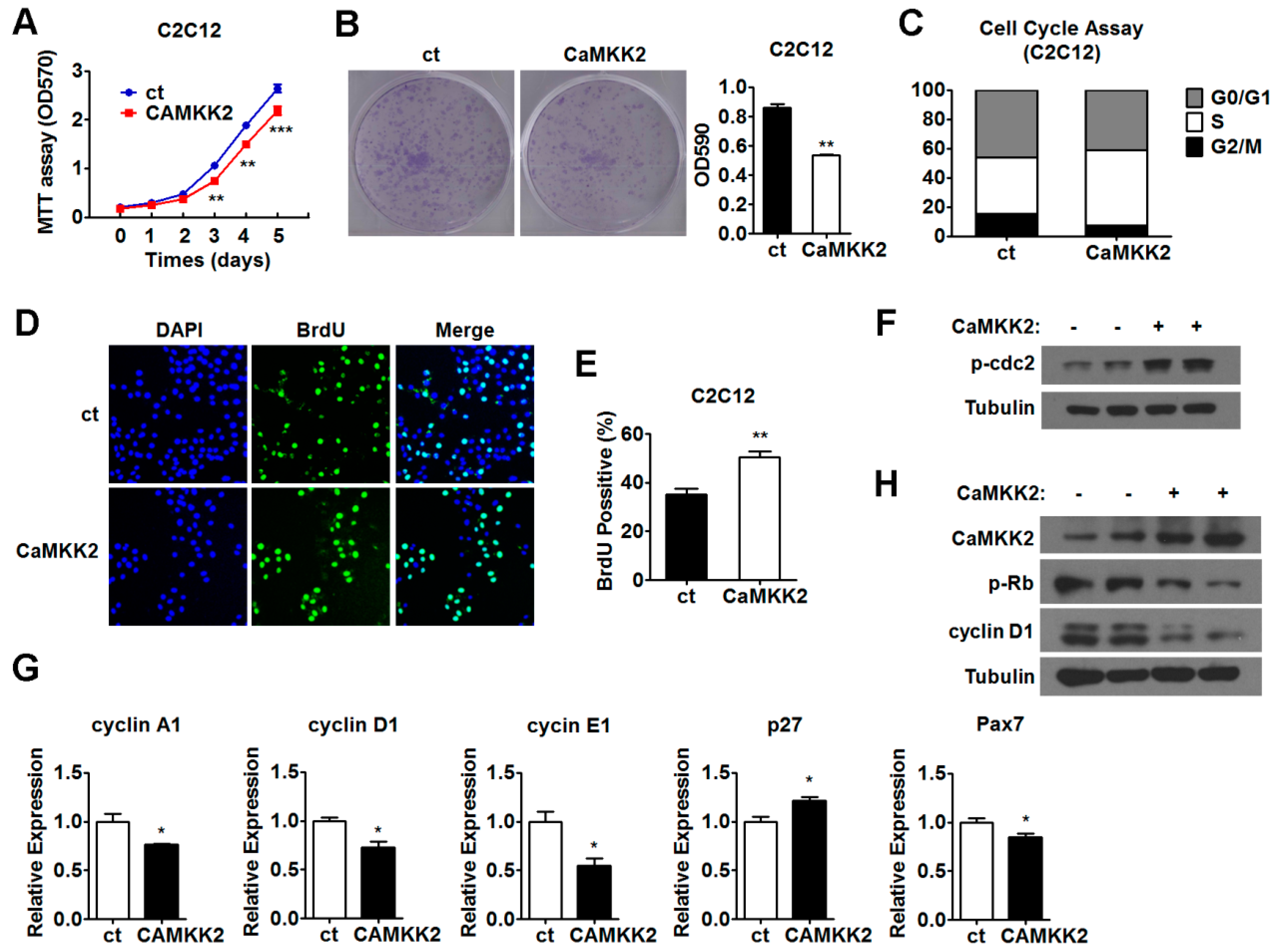
2.2. CaMKK2 Inhibits C2C12 Myoblasts Proliferation
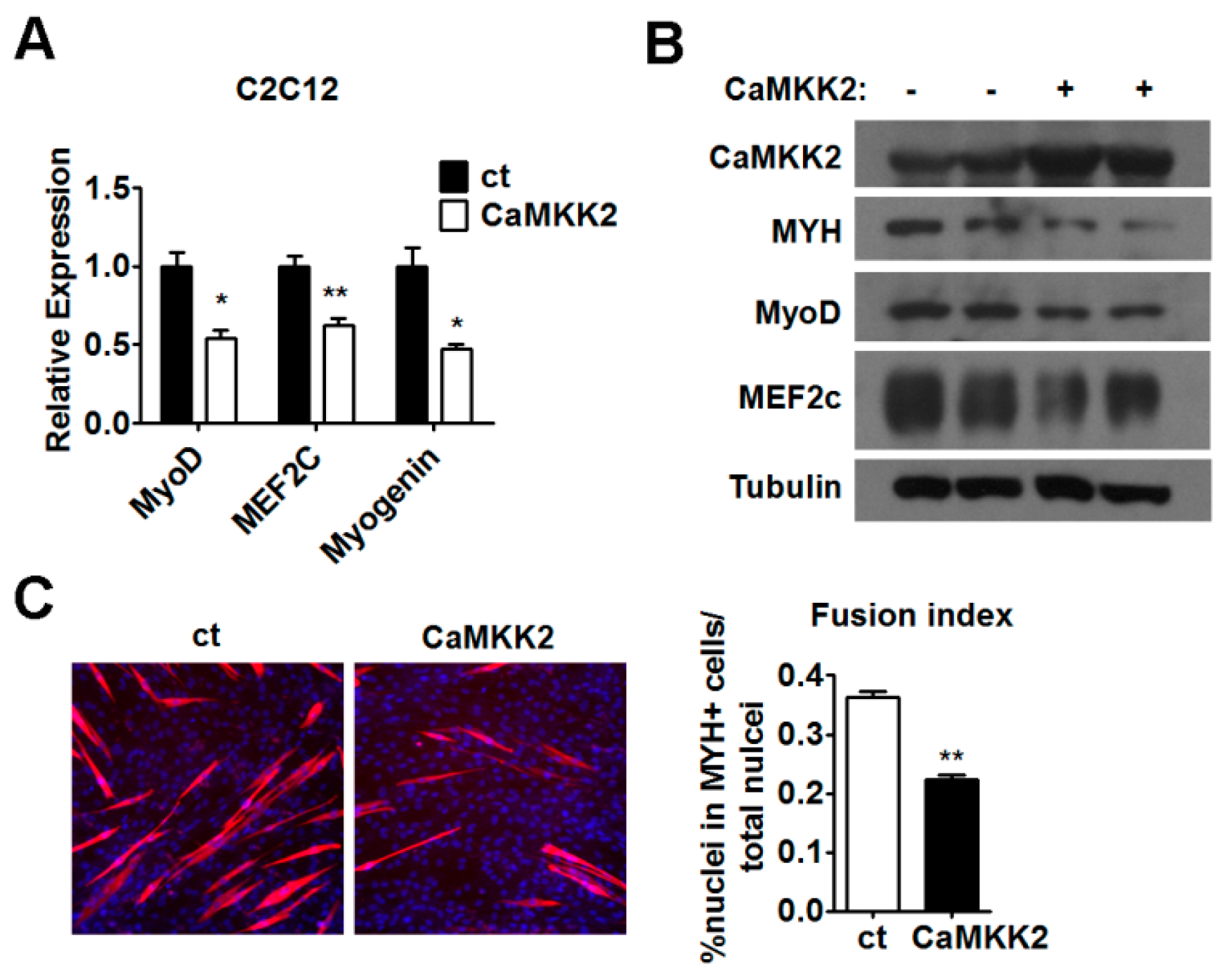
2.3. CaMKK2 Inhibits C2C12 Myoblasts Differentiation
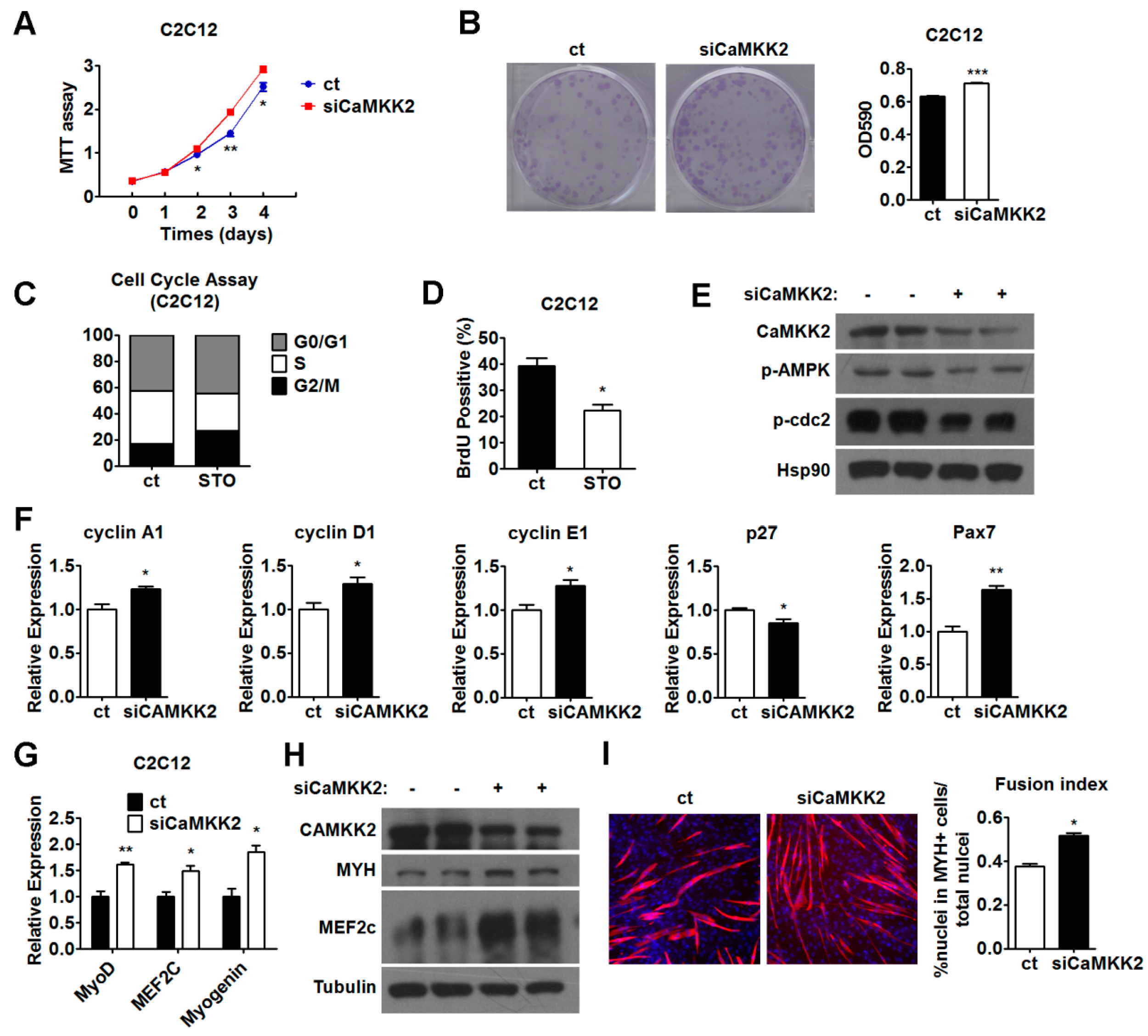
2.4. Inhibition of CaMKK2 Promotes Cell Proliferation and Differentiation
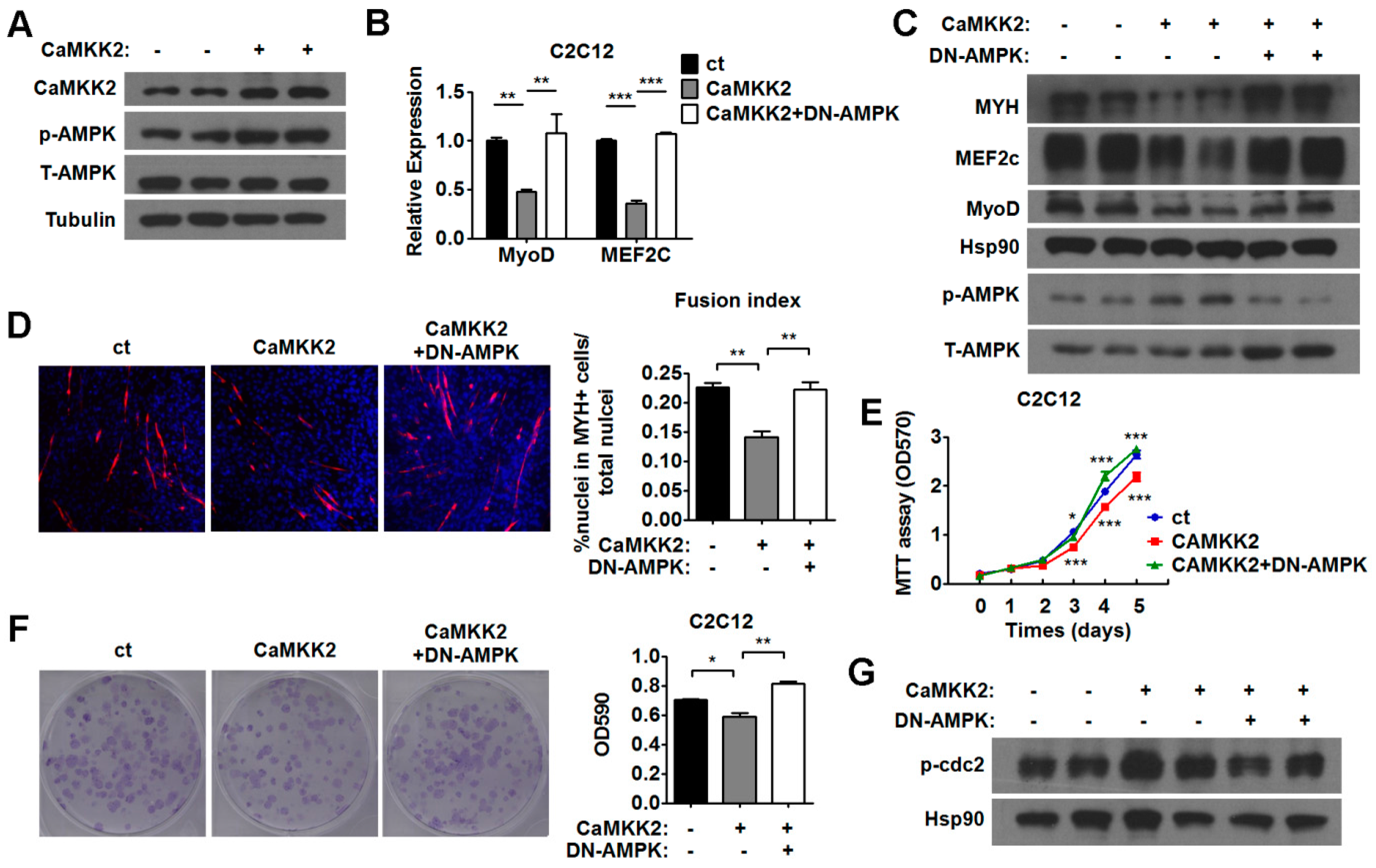
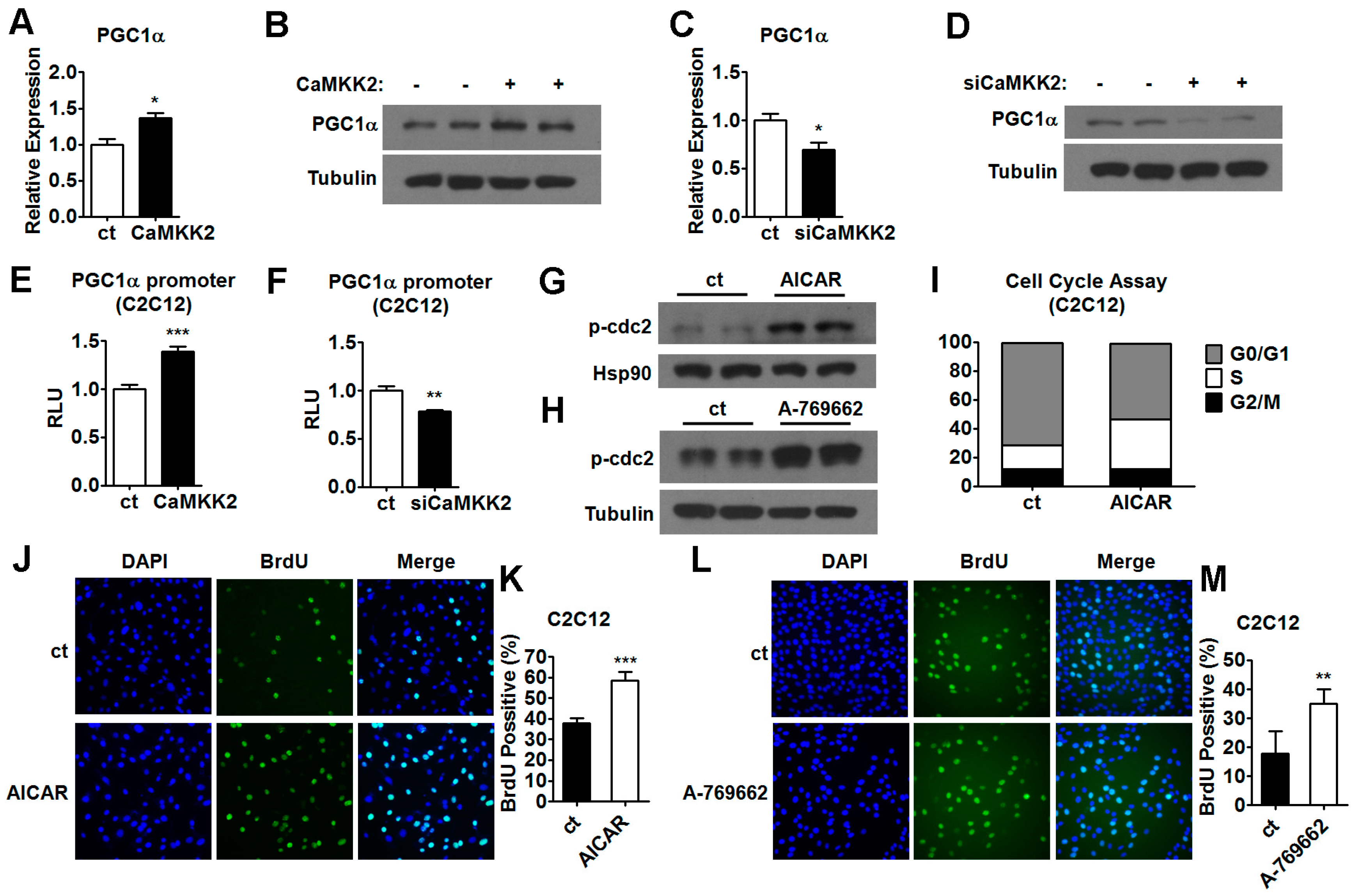
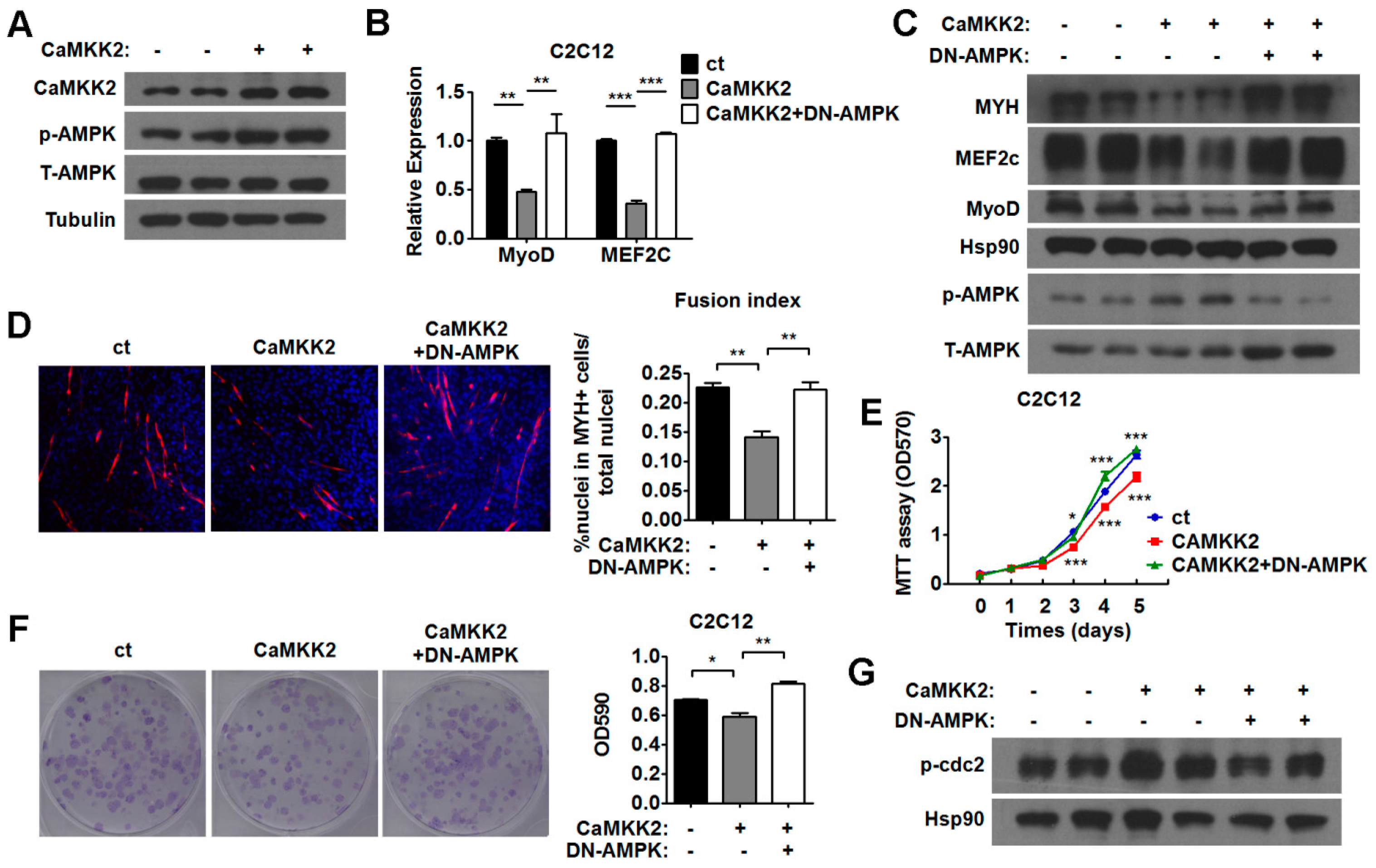
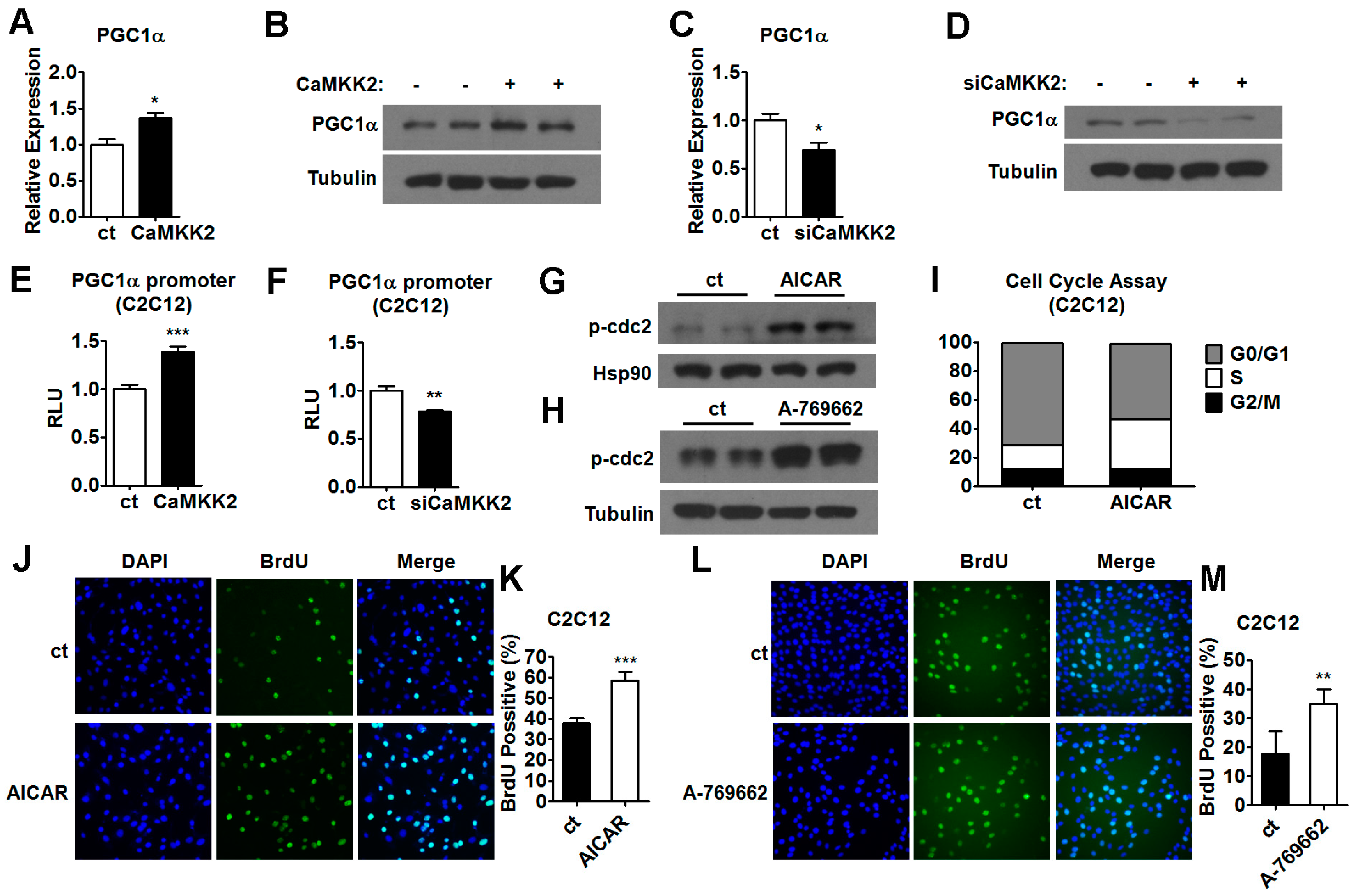
2.5. CaMKK2 Inhibits C2C12 Myoblasts Proliferation and Differentiation through AMPK
2.6. AMPK Pathways Mediate the Effect of CaMKK2 on Myoblasts Proliferation and Differentiation
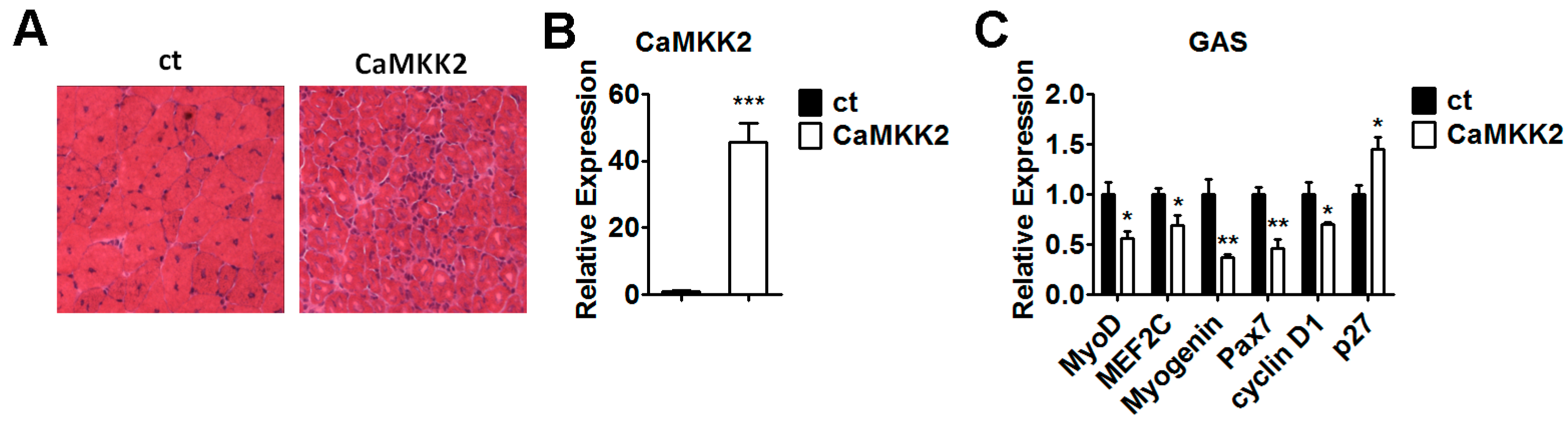
2.7. Overexpression of CaMKK2 Inhibits Muscle Regeneration in Vivo
3. Discussion
4. Methods
4.1. Cell Culture and Differentiation
4.2. CaMKK2 Expression Plasmid Construction
4.3. DNA and siRNA Transfection
4.4. Luciferase Reporter Assay
4.5. RNA Isolation and Real-Time PCR Analysis
4.6. Western Blotting
4.7. Immunocytochemistry
4.8. Cell Proliferation Assay
4.9. Cell Cycle Analysis
4.10. Muscle Regeneration
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AICAR | 5-Aminoimidazole-4-carboxamide1-β-d-ribofuranoside |
| AMPK | adenosine monophosphate activated protein kinase |
| BMD | Becker muscular dystrophy |
| BrdU | 5-Bromo-2-deoxyUridine |
| CaMK | Ca2+/calmodulin-dependent protein kinase |
| CaMKK2 | Calcium/calmodulin-dependent protein kinase kinase 2 |
| cdc2 | cell division cycle protein 2 homolog |
| cDNA | complementary deoxyribonucleic acid |
| CREB | cAMP response element-binding protein |
| CTX | cardiotoxin |
| DAPI | 4’,6-diamidino-2-phenylindole |
| DMD | duchenne muscular dystrophy |
| DMEM | Dulbecco minimum essential medium |
| GAS | gastrocnemius; HDAC5: histone deacetylase 5 |
| Hsp90 | heat shock protein 90 |
| MEF2C | myocyte-specific enhancer factor 2C |
| mTOR | mammalian target of rapamycin |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| MYH | myosin heavy chain |
| MyoD | myogenic differentiation protein |
| MyoG | myogenin; PBS: phosphate-buffered saline |
| PCR | polymerase chain reaction |
| PGC1α | peroxisome proliferator-activated receptor-γ coactivator-1α |
| Rb | retinoblastoma protein; S6K: ribosomal protein S6 kinase |
References
- Guller, I.; Russell, A.P. Micrornas in skeletal muscle: Their role and regulation in development, disease and function. J. Physiol. 2010, 588, 4075–4087. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.M.; Li, L.; Rozo, M.E.; Lepper, C. Making skeletal muscle from progenitor and stem cells: Development versus regeneration. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Nie, Q.; Zhang, X. Micrornas involved in skeletal muscle differentiation. J. Genet. Genom. 2013, 40, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Means, A.R. Regulation of the cell cycle by calcium and calmodulin. Endocr. Rev. 1993, 14, 40–58. [Google Scholar] [CrossRef] [PubMed]
- Colomer, J.; Means, A.R. Physiological roles of the Ca2+/cam-dependent protein kinase cascade in health and disease. Sub-Cell. Biochem. 2007, 45, 169–214. [Google Scholar]
- Means, A.R. The year in basic science: Calmodulin kinase cascades. Mol. Endocrinol. 2008, 22, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Ribar, T.J.; Lin, F.; Noeldner, P.K.; Green, M.F.; Muehlbauer, M.J.; Witters, L.A.; Kemp, B.E.; Means, A.R. Hypothalamic CAMKK2 contributes to the regulation of energy balance. Cell Metab. 2008, 7, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Mairet-Coello, G.; Courchet, J.; Pieraut, S.; Courchet, V.; Maximov, A.; Polleux, F. The camkk2-ampk kinase pathway mediates the synaptotoxic effects of abeta oligomers through TAU phosphorylation. Neuron 2013, 78, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Antunes-Martins, A.; Ris, L.; Peters, M.; Godaux, E.; Giese, K.P. Calcium/calmodulin kinase kinase β has a male-specific role in memory formation. Neuroscience 2007, 145, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, M.; Nishio, M.; Ribar, T.J.; Anderson, K.A.; West, A.E.; Means, A.R. BDNF-mediated cerebellar granule cell development is impaired in mice null for CAMKK2 or CAMKIV. J. Neurosci. 2009, 29, 8901–8913. [Google Scholar] [CrossRef] [PubMed]
- Cary, R.L.; Waddell, S.; Racioppi, L.; Long, F.; Novack, D.V.; Voor, M.J.; Sankar, U. Inhibition of Ca(2)(+)/calmodulin-dependent protein kinase kinase 2 stimulates osteoblast formation and inhibits osteoclast differentiation. J. Bone Miner. Res. 2013, 28, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Ribar, T.J.; Means, A.R. The Ca2+/calmodulin-dependent protein kinase kinase, CAMKK2, inhibits preadipocyte differentiation. Endocrinology 2011, 152, 3668–3679. [Google Scholar] [CrossRef] [PubMed]
- Teng, E.C.; Racioppi, L.; Means, A.R. A cell-intrinsic role for CAMKK2 in granulocyte lineage commitment and differentiation. J. Leukoc. Biol. 2011, 90, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, S.; Ross, F.A.; Vara Ciruelos, D.; Gray, A.; Gowans, G.J.; Hardie, D.G. AMPK causes cell cycle arrest in LKB1-deficient cells via activation of CAMKK2. Mol. Cancer Res. MCR 2016, 14, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, Z.; Ji, Y.; Sun, K.; Dai, Z.; Wu, G. l-glutamine enhances tight junction integrity by activating camk kinase 2-amp-activated protein kinase signaling in intestinal porcine epithelial cells. J. Nutr. 2016, 146, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Subbannayya, Y.; Syed, N.; Barbhuiya, M.A.; Raja, R.; Marimuthu, A.; Sahasrabuddhe, N.; Pinto, S.M.; Manda, S.S.; Renuse, S.; Manju, H.C.; et al. Calcium calmodulin dependent kinase kinase 2—A novel therapeutic target for gastric adenocarcinoma. Cancer Biol. Ther. 2015, 16, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Zhang, L.H.; Xu, W.P.; Jing, P.; Zhan, P.Y. Microrna-9 attenuates amyloid β-induced synaptotoxicity by targeting calcium/calmodulin-dependent protein kinase kinase 2. Mol. Med. Rep. 2014, 9, 1917–1922. [Google Scholar] [PubMed]
- Racioppi, L.; Means, A.R. Calcium/calmodulin-dependent protein kinase kinase 2: Roles in signaling and pathophysiology. J. Biol. Chem. 2012, 287, 31658–31665. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.L.; Butler, D.C.; Alway, S.E. Ampk inhibits myoblast differentiation through a PGC-1α-dependent mechanism. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E304–E314. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, M.S.; Scotton, C.; Passarelli, C.; Ferlini, A. Duchenne muscular dystrophy: From diagnosis to therapy. Molecules 2015, 20, 18168–18184. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, H.; Nishitani, H.; Seki, T.; Nishimoto, T. A dual-specificity phosphatase cdc25b is an unstable protein and triggers p34(cdc2)/cyclin b activation in hamster BHK21 cells arrested with hydroxyurea. J. Cell Biol. 1997, 138, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Singh, R.P.; Agarwal, C.; Siriwardana, S.; Sclafani, R.A.; Agarwal, R. Resveratrol causes cdc2-Tyr15 phosphorylation via ATM/ATR-chk1/2-cdc25c pathway as a central mechanism for s phase arrest in human ovarian carcinoma ovcar-3 cells. Carcinogenesis 2005, 26, 1978–1987. [Google Scholar] [CrossRef] [PubMed]
- Baldin, V.; Lukas, J.; Marcote, M.J.; Pagano, M.; Draetta, G. Cyclin d1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993, 7, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Korenjak, M.; Brehm, A. E2F-Rb complexes regulating transcription of genes important for differentiation and development. Curr. Opin. Genet. Dev. 2005, 15, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Inuzuka, H.; Ishikawa, Y.; Ikeda, M.; Saji, I.; Kobayashi, R. Sto-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J. Biol. Chem. 2002, 277, 15813–15818. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Frederich, M.; He, H.; Balschi, J.A. Relationship between 5-aminoimidazole-4-carboxamide-ribotide and AMP-activated protein kinase activity in the perfused mouse heart. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1235–H1243. [Google Scholar] [CrossRef] [PubMed]
- Anders, M.J.; Ali, Z.S.; Hegarty, B.D.; Heath, R.; Snowden, M.A.; Carling, D. Defining the mechanism of activation of amp-activated protein kinase by the small molecule a-769662, a member of the thienopyridone family. J. Biol. Chem. 2007, 282, 32539–32548. [Google Scholar] [CrossRef] [PubMed]
- Goransson, O.; McBride, A.; Hawley, S.A.; Ross, F.A.; Shpiro, N.; Foretz, M.; Viollet, B.; Hardie, D.G.; Sakamoto, K. Mechanism of action of a-769662, a valuable tool for activation of AMP-activated protein kinase. J. Biol. Chem. 2007, 282, 32549–32560. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulou, S.; Kolovou, P.E.; Morizane, Y.; Kayama, M.; Nicolaou, F.; Miller, J.W.; Gragoudas, E.; Ksander, B.R.; Vavvas, D.G. Retinoblastoma cells are inhibited by aminoimidazole carboxamide ribonucleotide (AICAR) partially through activation of AMP-dependent kinase. FASEB J. 2010, 24, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Irrcher, I.; Ljubicic, V.; Kirwan, A.F.; Hood, D.A. Amp-activated protein kinase-regulated activation of the PGC-α promoter in skeletal muscle cells. PLoS ONE 2008, 3, e3614. [Google Scholar] [CrossRef] [PubMed]
- Green, M.F.; Anderson, K.A.; Means, A.R. Characterization of the CAMKKβ-AMPK signaling complex. Cell Signal. 2011, 23, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; He, H.C.; Han, Z.D.; Wan, Y.P.; Luo, H.W.; Huang, Y.Q.; Cai, C.; Liang, Y.X.; Dai, Q.S.; Jiang, F.N.; et al. Microrna-224 and its target CAMKK2 synergistically influence tumor progression and patient prognosis in prostate cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Frigo, D.E.; Howe, M.K.; Wittmann, B.M.; Brunner, A.M.; Cushman, I.; Wang, Q.; Brown, M.; Means, A.R.; McDonnell, D.P. Cam kinase kinase β-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011, 71, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Shima, T.; Mizokami, A.; Miyagi, T.; Kawai, K.; Izumi, K.; Kumaki, M.; Ofude, M.; Zhang, J.; Keller, E.T.; Namiki, M. Down-regulation of calcium/calmodulin-dependent protein kinase kinase 2 by androgen deprivation induces castration-resistant prostate cancer. Prostate 2012, 72, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Marcelo, K.L.; Rajapakshe, K.; Coarfa, C.; Dean, A.; Wilganowski, N.; Robinson, H.; Sevick, E.; Bissig, K.D.; Goldie, L.C.; et al. The CAMKK2/CAMKIV relay is an essential regulator of hepatic cancer. Hepatology 2015, 62, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed]
- Karacosta, L.G.; Foster, B.A.; Azabdaftari, G.; Feliciano, D.M.; Edelman, A.M. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CAMKK2) and the androgen receptor in prostate cancer progression. J. Biol. Chem. 2012, 287, 24832–24843. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.J.; Kiens, B.; Richter, E.A. Ca2+-calmodulin-dependent protein kinase expression and signalling in skeletal muscle during exercise. J. Physiol. 2006, 574, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.R. The role of calcium and calcium/calmodulin-dependent kinases in skeletal muscle plasticity and mitochondrial biogenesis. Proc. Nutr. Soc. 2004, 63, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhu, M.J.; Dodson, M.V.; Du, M. AMP-activated protein kinase stimulates warburg-like glycolysis and activation of satellite cells during muscle regeneration. J. Biol. Chem. 2015, 290, 26445–26456. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhao, J.X.; Liang, J.; Zhu, M.J.; Foretz, M.; Viollet, B.; Du, M. AMP-activated protein kinase mediates myogenin expression and myogenesis via histone deacetylase 5. Am. J. Physiol. Cell Physiol. 2013, 305, C887–C895. [Google Scholar] [CrossRef] [PubMed]
- LaBarge, S.; McDonald, M.; Smith-Powell, L.; Auwerx, J.; Huss, J.M. Estrogen-related receptor-α (ERR-α) deficiency in skeletal muscle impairs regeneration in response to injury. FASEB J. 2014, 28, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chhipa, R.R.; Pooya, S.; Wortman, M.; Yachyshin, S.; Chow, L.M.; Kumar, A.; Zhou, X.; Sun, Y.; Quinn, B.; et al. Discrete mechanisms of mtor and cell cycle regulation by AMPK agonists independent of AMPK. Proc. Natl. Acad. Sci. USA 2014, 111, E435–E444. [Google Scholar] [CrossRef] [PubMed]
- Cisneros-Barroso, E.; Yance-Chavez, T.; Kito, A.; Sugiura, R.; Gomez-Hierro, A.; Gimenez-Zaragoza, D.; Aligue, R. Negative feedback regulation of calcineurin-dependent Prz1 transcription factor by the CAMKK-CAMK1 axis in fission yeast. Nucleic Acids Res. 2014, 42, 9573–9587. [Google Scholar] [CrossRef] [PubMed]
- Chazaud, B. Inflammation during skeletal muscle regeneration and tissue remodeling: Application to exercise-induced muscle damage management. Immunol. Cell Biol. 2016, 94, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Kharraz, Y.; Guerra, J.; Mann, C.J.; Serrano, A.L.; Munoz-Canoves, P. Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediat. Inflamm. 2013. [Google Scholar] [CrossRef] [PubMed]
- Racioppi, L.; Noeldner, P.K.; Lin, F.; Arvai, S.; Means, A.R. Calcium/calmodulin-dependent protein kinase kinase 2 regulates macrophage-mediated inflammatory responses. J. Biol. Chem. 2012, 287, 11579–11591. [Google Scholar] [CrossRef] [PubMed]
- Saini, J.; McPhee, J.S.; Al-Dabbagh, S.; Stewart, C.E.; Al-Shanti, N. Regenerative function of immune system: Modulation of muscle stem cells. Age. Res. Rev. 2016, 27, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, X.; Li, Y.; Zhao, L.; Lu, M.; Yao, X.; Xia, H.; Wang, Y.C.; Liu, M.F.; Jiang, J.; et al. Thyroid hormone regulates muscle fiber type conversion via miR-133a1. J. Cell Biol. 2014, 207, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, X.; Chen, C.; Li, Y.; Zhao, L.; Jing, Y.; Liu, W.; Wang, X.; Zhang, Y.; Xia, H.; et al. Attenuation of p38-mediated miR-1/133 expression facilitates myoblast proliferation during the early stage of muscle regeneration. PLoS ONE 2012, 7, e41478. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, C.; Zhang, D.; Zhao, L.; Li, Y.; Yao, X.; Wang, H.; Zhang, S.; Liu, W.; Cao, H.; Yu, S.; et al. CaMKK2 Suppresses Muscle Regeneration through the Inhibition of Myoblast Proliferation and Differentiation. Int. J. Mol. Sci. 2016, 17, 1695. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101695
Ye C, Zhang D, Zhao L, Li Y, Yao X, Wang H, Zhang S, Liu W, Cao H, Yu S, et al. CaMKK2 Suppresses Muscle Regeneration through the Inhibition of Myoblast Proliferation and Differentiation. International Journal of Molecular Sciences. 2016; 17(10):1695. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101695
Chicago/Turabian StyleYe, Cheng, Duo Zhang, Lei Zhao, Yan Li, Xiaohan Yao, Hui Wang, Shengjie Zhang, Wei Liu, Hongchao Cao, Shuxian Yu, and et al. 2016. "CaMKK2 Suppresses Muscle Regeneration through the Inhibition of Myoblast Proliferation and Differentiation" International Journal of Molecular Sciences 17, no. 10: 1695. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101695