
The Importance of Patient-Specific Factors for Hepatic Drug Response and Toxicity


Abstract
:1. Introduction
2. Socioeconomical Aspects of Drug Hepatotoxicity
3. Impact of Genetic Factors on Drug Metabolism
4. The Importance of Rare Variant Alleles for Pharmacogenetics
5. Mechanisms of Drug-Induced Hepatotoxicity
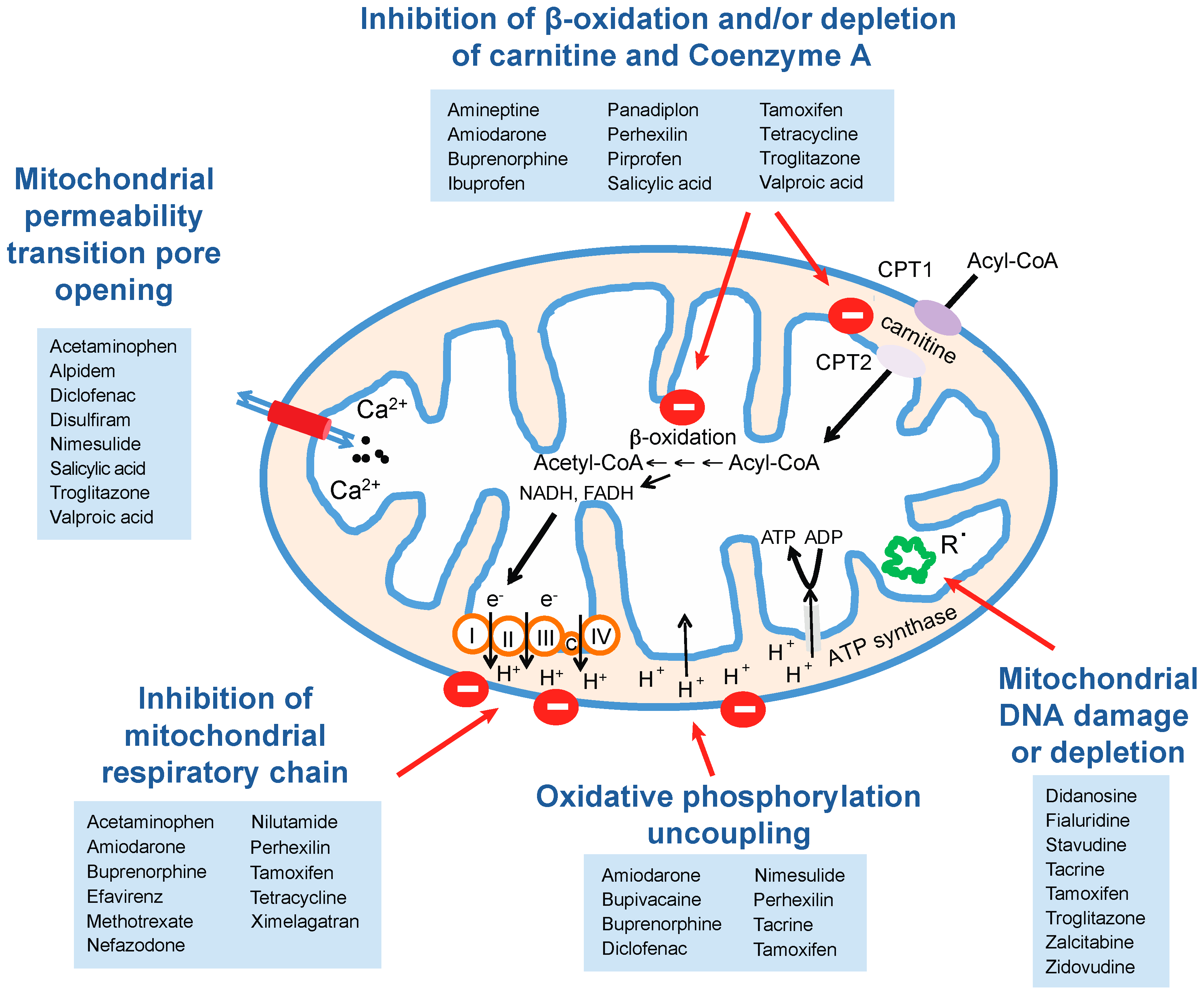
5.1. Mitochondrial Perturbations
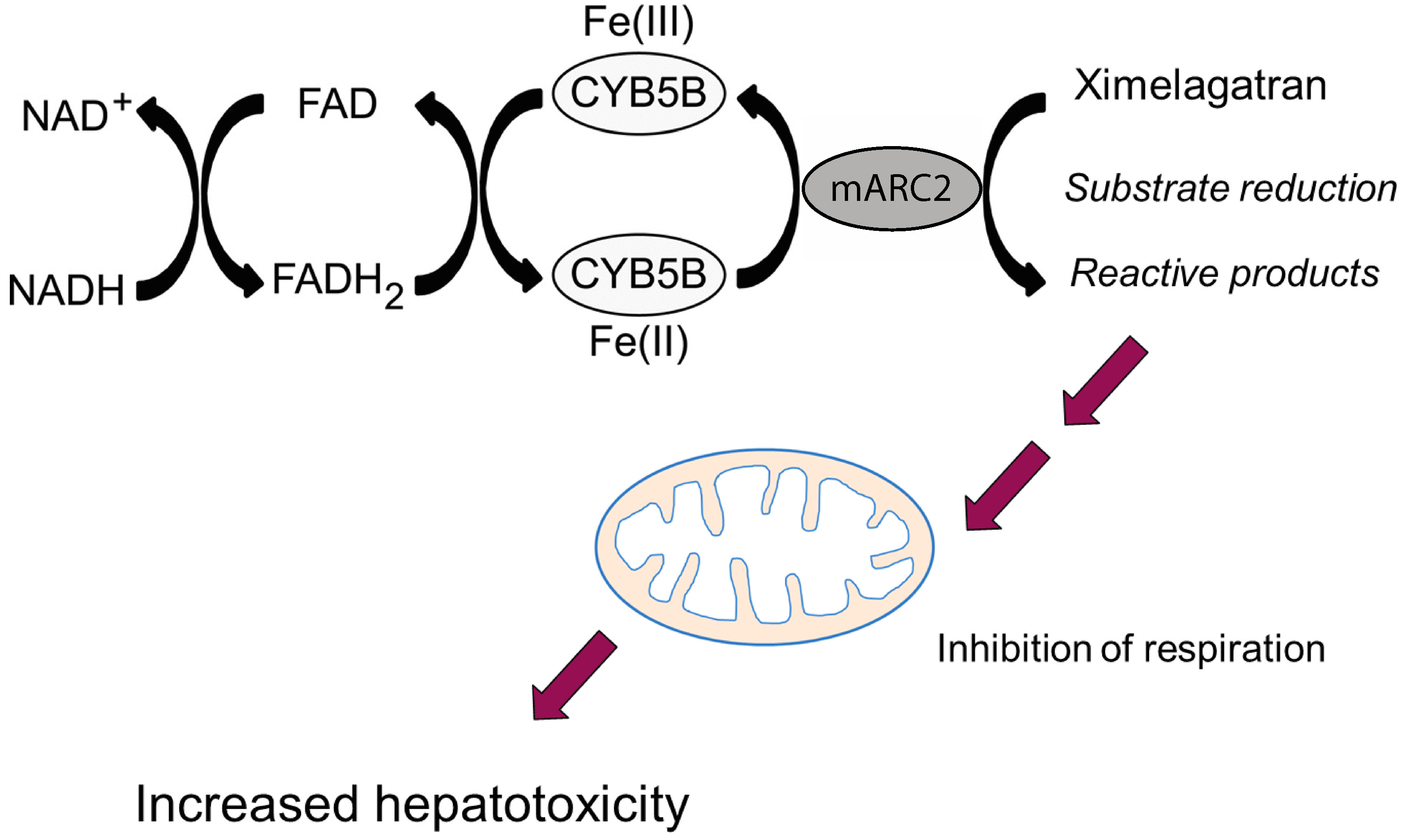
5.1.1. Inhibition of Mitochondrial Respiration
5.1.2. Effects on Mitochondrial Lipid Metabolism
5.1.3. Mitochondrial DNA Damage and Inhibition of Mitochondrial Gene Expression
5.2. Immune-Mediated Toxicity
5.2.1. Abacavir Hypersensitivity Syndrome (HSS)
5.2.2. Systemic Lupus Erythematosus (SLE)
5.2.3. Steven Johnson Syndrome (SJS) and Toxic Epidermal Necrolysis (TEN)
5.2.4. Clozapine-Induced Agranulocytosis
5.2.5. Immune-Related Drug-Induced Liver Injury (DILI)
6. The Impact of Liver Diseases on Drug Response
7. Epigenetics and Inter-Individual Differences
In Vitro Toxicity Models That Reflect Patient-Specific Factors
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Spear, B.B.; Heath-Chiozzi, M.; Huff, J. Clinical application of pharmacogenetics. Trends Mol. Med. 2001, 7, 201–204. [Google Scholar] [CrossRef]
- Sim, S.C.; Kacevska, M.; Ingelman-Sundberg, M. Pharmacogenomics of drug-metabolizing enzymes: A recent update on clinical implications and endogenous effects. Pharmacogenom. J. 2012, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Frueh, F.W.; Amur, S.; Mummaneni, P.; Epstein, R.S.; Aubert, R.E.; DeLuca, T.M.; Verbrugge, R.R.; Burckart, G.J.; Lesko, L.J. Pharmacogenomic Biomarker Information in Drug Labels Approved by the United States Food and Drug Administration: Prevalence of Related Drug Use. Pharmacotherapy 2008, 28, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, F.; Caneva, L.; Prasad, K.; Paulmichl, M.; Maliepaard, M.; Llerena, A.; Ingelman-Sundberg, M.; Papaluca-Amati, M. Pharmacogenomic information in drug labels: European Medicines Agency perspective. Pharmacogenom. J. 2015, 15, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Haga, S.B.; Mills, R.; Moaddeb, J. Pharmacogenetic information for patients on drug labels. Pharmacogenom. Pers. Med. 2014, 7, 297–305. [Google Scholar]
- Carr, D.; Alfirevic, A.; Pirmohamed, M. Pharmacogenomics: Current State-of-the-Art. Genes 2014, 5, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Lauschke, V.M.; Ingelman-Sundberg, M. Requirements for comprehensive pharmacogenetic genotyping platforms. Pharmacogenomics 2016, 17, 917–924. [Google Scholar] [CrossRef] [PubMed]
- CPIC Guidelines for Gene-Drug Interactions. Available online: https://cpicpgx.org/genes-drugs (accessed on 12 July 2016).
- Fujikura, K.; Ingelman-Sundberg, M.; Lauschke, V.M. Genetic variation in the human cytochrome P450 supergene family. Pharmacogenet. Genom. 2015, 25, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.S.; Tabor, H.K.; Johnson, A.D.; Snively, B.M.; Assimes, T.L.; Auer, P.L.; Ioannidis, J.P.A.; Peters, U.; Robinson, J.G.; Sucheston, L.E.; et al. Quantifying rare, deleterious variation in 12 human cytochrome P450 drug-metabolism genes in a large-scale exome dataset. Hum. Mol. Genet. 2014, 23, 1957–1963. [Google Scholar] [CrossRef] [PubMed]
- Kozyra, M.; Ingelman-Sundberg, M.; Lauschke, V.M. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lauschke, V.M.; Ingelman-Sundberg, M. Precision Medicine and Rare Genetic Variants. Trends Pharmacol. Sci. 2016, 37, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Pirmohamed, M.; James, S.; Meakin, S.; Green, C.; Scott, A.K.; Walley, T.J.; Farrar, K.; Park, B.K.; Breckenridge, A.M. Adverse drug reactions as cause of admission to hospital: Prospective analysis of 18 820 patients. Br. Med. J. 2004, 329, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Impicciatore, P.; Choonara, I.; Clarkson, A.; Provasi, D.; Pandolfini, C.; Bonati, M. Incidence of adverse drug reactions in paediatric in/out-patients: A systematic review and meta-analysis of prospective studies. Br. J. Clin. Pharmacol. 2001, 52, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Routledge, P.A.; O’Mahony, M.S.; Woodhouse, K.W. Adverse drug reactions in elderly patients. Br. J. Clin. Pharmacol. 2003, 57, 121–126. [Google Scholar] [CrossRef]
- Budnitz, D.S.; Shehab, N.; Kegler, S.R.; Richards, C.L. Medication use leading to emergency department visits for adverse drug events in older adults. Ann. Intern. Med. 2007, 147, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.W.; Spell, N.; Cullen, D.J.; Burdick, E.; Laird, N.; Petersen, L.A.; Small, S.D.; Sweitzer, B.J.; Leape, L.L. The costs of adverse drug events in hospitalized patients. Adverse drug events prevention study group. J. Am. Med. Assoc. 1997, 277, 307–311. [Google Scholar] [CrossRef]
- Gautier, S.; Bachelet, H.; Bordet, R.; Caron, J. The cost of adverse drug reactions. Exp. Opin. Pharmacother. 2005, 4, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Hug, B.L.; Keohane, C.; Seger, D.L.; Yoon, C.; Bates, D.W. The costs of adverse drug events in community hospitals. Jt. Comm. J. Qual. Patient Saf. 2012, 38, 120–126. [Google Scholar] [PubMed]
- Johnson, J.A.; Bootman, J.L. Drug-related morbidity and mortality and the economic impact of pharmaceutical care. Am. J. Health Syst. Pharm. 1997, 54, 554–558. [Google Scholar] [PubMed]
- Faich, G.A.; Knapp, D.; Dreis, M.; Turner, W. National adverse drug reaction surveillance: 1985. J. Am. Med. Assoc. 1987, 257, 2068–2070. [Google Scholar] [CrossRef]
- Goettler, M.; Schneeweiss, S.; Hasford, J. Adverse drug reaction monitoring-cost and benefit considerations. Part II: Cost and preventability of adverse drug reactions leading to hospital admission. Pharmacoepidemiol. Drug Saf. 1997, 6, S79–S90. [Google Scholar] [CrossRef]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 2014, 13, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Manning, F.J.; Swartz, M. Review of the Fialuridine (FIAU) Clinical Trials; National Academies Press: Atlanta, GA, USA, 1995. [Google Scholar]
- Kaku, K.; Enya, K.; Nakaya, R.; Ohira, T.; Matsuno, R. Efficacy and safety of fasiglifam (TAK-875), a G protein-coupled receptor 40 agonist, in Japanese patients with type 2 diabetes inadequately controlled by diet and exercise: A randomized, double-blind, placebo-controlled, phase III trial. Diabetes Obes. Metab. 2015, 17, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Lasser, K.E.; Allen, P.D.; Woolhandler, S.J.; Himmelstein, D.U.; Wolfe, S.M.; Bor, D.H. Timing of new black box warnings and withdrawals for prescription medications. J. Am. Med. Assoc. 2002, 287, 2215–2220. [Google Scholar] [CrossRef]
- Amstutz, U.; Froehlich, T.K.; Largiadèr, C.R. Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5-fluorouracil toxicity. Pharmacogenomics 2011, 12, 1321–1336. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.E.; Hon, Y.Y.; Bomgaars, L.; Coutre, S.; Holdsworth, M.; Janco, R.; Kalwinsky, D.; Keller, F.; Khatib, Z.; Margolin, J.; et al. Preponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprine. J. Clin. Oncol. 2001, 19, 2293–2301. [Google Scholar] [PubMed]
- Lennard, L. TPMT in the treatment of Crohn’s disease with azathioprine. Gut 2002, 51, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Gasche, Y.; Daali, Y.; Fathi, M.; Chiappe, A.; Cottini, S.; Dayer, P.; Desmeules, J. Codeine intoxication associated with ultrarapid CYP2D6 metabolism. N. Engl. J. Med. 2004, 351, 2827–2831. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, F.; Undevia, S.D.; Iyer, L.; Chen, P.X.; Das, S.; Kocherginsky, M.; Karrison, T.; Janisch, L.; Ramírez, J.; Rudin, C.M.; et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J. Clin. Oncol. 2004, 22, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Lei, H.-P.; Li, Z.; Tan, Z.-R.; Guo, D.; Fan, L.; Chen, Y.; Hu, D.-L.; Wang, D.; Zhou, H.-H. The CYP2C19 ultra-rapid metabolizer genotype influences the pharmacokinetics of voriconazole in healthy male volunteers. Eur. J. Clin. Pharmacol. 2008, 65, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.V.; Perel, P.; Shah, T.; Hingorani, A.D.; Casas, J.P. CYP2C19 Genotype, Clopidogrel Metabolism, Platelet Function, and Cardiovascular Events A Systematic Review and Meta-analysis. J. Am. Med. Assoc. 2011, 306, 2704–2714. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Gong, L.; Whirl-Carrillo, M.; Gage, B.F.; Scott, S.A.; Stein, C.M.; Anderson, J.L.; Kimmel, S.E.; Lee, M.T.M.; Pirmohamed, M.; et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 Genotypes and Warfarin Dosing. Clin. Pharmacol. Ther. 2011, 90, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Ohashi, K.; Kamata, T.; Takashima, M.; Kosuge, K.; Kawasaki, T.; Hanai, H.; Kubota, T.; Ishizaki, T.; Kaneko, E. Effect of genetic differences in omeprazole metabolism on cure rates for Helicobacter pylori infection and peptic ulcer. Ann. Intern. Med. 1998, 129, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Shirai, N.; Takashima, M.; Xiao, F.; Hanai, H.; Sugimura, H.; Ohashi, K.; Ishizaki, T.; Kaneko, E. Effect of genotypic differences in CYP2C19 on cure rates for Helicobacter pylori infection by triple therapy with a proton pump inhibitor, amoxicillin, and clarithromycin. Clin. Pharmacol. Ther. 2001, 69, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 variants and statin-induced myopathy—A genomewide study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [PubMed]
- Wang, L.; McLeod, H.L.; Weinshilboum, R.M. Genomics and drug response. N. Engl. J. Med. 2011, 364, 1144–1153. [Google Scholar] [PubMed]
- Hertz, D.L.; Rae, J. Pharmacogenetics of Cancer Drugs. Annu. Rev. Med. 2015, 66, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Evans, W.E. Pharmacogenomics in the clinic. Nature 2015, 526, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Abecasis, G.R.; Chakravarti, A.; Donnelly, P.; Eichler, E.E.; Gabriel, S.B.; Hurles, M.E.; Mardis, E.R.; Nickerson, D.A.; Gibbs, R.A.; Boerwinkle, E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [Green Version]
- Tennessen, J.A.; Bigham, A.W.; O’Connor, T.D.; Fu, W.; Kenny, E.E.; Gravel, S.; McGee, S.; Do, R.; Liu, X.; Jun, G.; et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012, 337, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.R.B.; Xu, C.; Lawson, D.; Hendricks, A.E.; Barroso, I.; Hurles, M.E.; Greenwood, C.M.T.; Bala, S.; Clapham, P.; Coates, G.; et al. The UK10K project identifies rare variants in health and disease. Nature 2015, 526, 82–90. [Google Scholar] [Green Version]
- Ragoussis, J. Genotyping Technologies for Genetic Research. Annu. Rev. Genom. Hum. Genet. 2009, 10, 117–133. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A. Bringing genome-wide association findings into clinical use. Nat. Rev. Genet. 2013, 14, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, J.; Brockmöller, J.; Tzvetkov, M.V.; Sehrt, D.; Sachse-Seeboth, C.; Hjelmborg, J.B.; Möller, S.; Halekoh, U.; Hofmann, U.; Schwab, M.; et al. Heritability of metoprolol and torsemide pharmacokinetics. Clin. Pharmacol. Ther. 2015, 98, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Kaplowitz, N. Idiosyncratic drug hepatotoxicity. Nat. Rev. Drug Discov. 2005, 4, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Dobo, K.L.; Obach, R.S.; Luffer-Atlas, D.; Bercu, J.P. A Strategy for the Risk Assessment of Human Genotoxic Metabolites. Chem. Res. Toxicol. 2009, 22, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Orr, S.T.M.; Ripp, S.L.; Ballard, T.E.; Henderson, J.L.; Scott, D.O.; Obach, R.S.; Sun, H.; Kalgutkar, A.S. Mechanism-Based Inactivation (MBI) of Cytochrome P450 Enzymes: Structure-Activity Relationships and Discovery Strategies To Mitigate Drug-Drug Interaction Risks. J. Med. Chem. 2012, 55, 4896–4933. [Google Scholar] [CrossRef] [PubMed]
- Kitteringham, N.R.; Kenna, J.G.; Park, B.K. Detection of autoantibodies directed against human hepatic endoplasmic reticulum in sera from patients with halothane-associated hepatitis. Br. J. Clin. Pharmacol. 1995, 40, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; You, Q.; Yin, H.; Holt, M.P.; Ju, C. Involvement of natural killer T-cells in halothane-induced liver injury in mice. Biochem. Pharmacol. 2010, 80, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Boelsterli, U.A. Xenobiotic acyl glucuronides and acyl CoA thioesters as protein-reactive metabolites with the potential to cause idiosyncratic drug reactions. Curr. Drug Metab. 2002, 3, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P.; Ramsay, L.; Daly, A.K.; Sonchit, N.; Leathart, J.B.S.; Alexander, G.; Kenna, J.G.; Caldwell, J.; Day, C.P. Hepatic adducts, circulating antibodies, and cytokine polymorphisms in patients with diclofenac hepatotoxicity. Hepatology 2004, 39, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S.; Dalvie, D. Predicting Toxicities of Reactive Metabolite-Positive Drug Candidates. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.A.; Roberts, D.W.; James, L.P. Adverse Drug Reactions (Handbook of Experimental Pharmacology), 1st ed.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 196, pp. 369–405. [Google Scholar]
- Koen, Y.M.; Sarma, D.; Williams, T.D.; Galeva, N.A.; Obach, R.S.; Hanzlik, R.P. Identification of Protein Targets of Reactive Metabolites of Tienilic Acid in Human Hepatocytes. Chem. Res. Toxicol. 2012, 25, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Fromenty, B.; Berson, A.; Robin, M.-A.; Lettéron, P.; Moreau, R.; Mansouri, A. Central role of mitochondria in drug-induced liver injury. Drug Metab. Rev. 2012, 44, 34–87. [Google Scholar] [CrossRef] [PubMed]
- Watmough, N.J.; Bindoff, L.A.; Birch-Machin, M.A.; Jackson, S.; Bartlett, K.; Ragan, C.I.; Poulton, J.; Gardiner, R.M.; Sherratt, H.S.; Turnbull, D.M. Impaired mitochondrial β-oxidation in a patient with an abnormality of the respiratory chain. Studies in skeletal muscle mitochondria. J. Clin. Investig. 1990, 85, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Blas-García, A.; Apostolova, N.; Ballesteros, D.; Monleón, D.; Morales, J.M.; Rocha, M.; Victor, V.M.; Esplugues, J.V. Inhibition of mitochondrial function by efavirenz increases lipid content in hepatic cells. Hepatology 2010, 52, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Dykens, J.A.; Jamieson, J.D.; Marroquin, L.D.; Nadanaciva, S.; Xu, J.J.; Dunn, M.C.; Smith, A.R.; Will, Y. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol. Sci. 2008, 103, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Schmets, L.; Fisch, C.; Fau, D.; Wolf, C.; Fromenty, B.; Deschamps, D.; Pessayre, D. Inhibition by nilutamide of the mitochondrial respiratory chain and ATP formation. Possible contribution to the adverse effects of this antiandrogen. J. Pharmacol. Exp. Ther. 1994, 270, 167–176. [Google Scholar] [PubMed]
- Fromenty, B.; Fisch, C.; Berson, A.; Letteron, P.; Larrey, D.; Pessayre, D. Dual effect of amiodarone on mitochondrial respiration. Initial protonophoric uncoupling effect followed by inhibition of the respiratory chain at the levels of complex I and complex II. J. Pharmacol. Exp. Ther. 1990, 255, 1377–1384. [Google Scholar] [PubMed]
- Neve, E.P.A.; Köfeler, H.; Hendriks, D.F.G.; Nordling, Å.; Gogvadze, V.; Mkrtchian, S.; Näslund, E.; Ingelman-Sundberg, M. Expression and Function of mARC: Roles in Lipogenesis and Metabolic Activation of Ximelagatran. PLoS ONE 2015, 10, e0138487. [Google Scholar] [CrossRef] [PubMed]
- Kon, K.; Kim, J.-S.; Jaeschke, H.; Lemasters, J.J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004, 40, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Descatoire, V.; Sutton, A.; Fau, D.; Maulny, B.; Vadrot, N.; Feldmann, G.; Berthon, B.; Tordjmann, T.; Pessayre, D. Toxicity of alpidem, a peripheral benzodiazepine receptor ligand, but not zolpidem, in rat hepatocytes: Role of mitochondrial permeability transition and metabolic activation. J. Pharmacol. Exp. Ther. 2001, 299, 793–800. [Google Scholar] [PubMed]
- Masubuchi, Y.; Nakayama, S.; Horie, T. Role of mitochondrial permeability transition in diclofenac-induced hepatocyte injury in rats. Hepatology 2002, 35, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Balakirev, M.Y.; Zimmer, G. Mitochondrial injury by disulfiram: Two different mechanisms of the mitochondrial permeability transition. Chem. Biol. Interact. 2001, 138, 299–311. [Google Scholar] [CrossRef]
- Mingatto, F.E.; dos Santos, A.C.; Rodrigues, T.; Pigoso, A.A.; Uyemura, S.A.; Curti, C. Effects of nimesulide and its reduced metabolite on mitochondria. Br. J. Pharmacol. 2000, 131, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Trost, L.C.; Lemasters, J.J. The mitochondrial permeability transition: A new pathophysiological mechanism for Reye’s syndrome and toxic liver injury. J. Pharmacol. Exp. Ther. 1996, 278, 1000–1005. [Google Scholar] [PubMed]
- Tirmenstein, M.A.; Hu, C.X.; Gales, T.L.; Maleeff, B.E.; Narayanan, P.K.; Kurali, E.; Hart, T.K.; Thomas, H.C.; Schwartz, L.W. Effects of troglitazone on HepG2 viability and mitochondrial function. Toxicol. Sci. 2002, 69, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.L.K.; Liu, J.; Go, M.L.; Boelsterli, U.A. The mitochondrial superoxide/thioredoxin-2/Ask1 signaling pathway is critically involved in troglitazone-induced cell injury to human hepatocytes. Toxicol. Sci. 2008, 101, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Meyers, L.L.; Beierschmitt, W.P.; Khairallah, E.A.; Cohen, S.D. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol. Appl. Pharmacol. 1988, 93, 378–387. [Google Scholar] [CrossRef]
- Donnelly, P.J.; Walker, R.M.; Racz, W.J. Inhibition of mitochondrial respiration in vivo is an early event in acetaminophen-induced hepatotoxicity. Arch. Toxicol. 1994, 68, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Imaizumi, N.; Chamberland, S.R.; Alder, N.N.; Boelsterli, U.A. Targeting mitochondria with methylene blue protects mice against acetaminophen-induced liver injury. Hepatology 2015, 61, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Fau, D.; Fornacciari, R.; Degove-Goddard, P.; Sutton, A.; Descatoire, V.; Haouzi, D.; Letteron, P.; Moreau, A.; Feldmann, G.; et al. Mechanisms for experimental buprenorphine hepatotoxicity: Major role of mitochondrial dysfunction versus metabolic activation. J. Hepatol. 2001, 34, 261–269. [Google Scholar] [CrossRef]
- Yamamoto, N.; Oliveira, M.B.; Campello Ade, P.; Lopes, L.C.; Klüppel, M.L. Methotrexate: Studies on the cellular metabolism. I. Effect on mitochondrial oxygen uptake and oxidative phosphorylation. Cell Biochem. Funct. 1988, 6, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, D.; DeBeco, V.; Fisch, C.; Fromenty, B.; Guillouzo, A.; Pessayre, D. Inhibition by perhexiline of oxidative phosphorylation and the beta-oxidation of fatty acids: Possible role in pseudoalcoholic liver lesions. Hepatology 1994, 19, 948–961. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, C.M.P.; Custódio, J.B.A.; Almeida, L.M.; Moreno, A.J.M. Mechanisms of the Deleterious Effects of Tamoxifen on Mitochondrial Respiration Rate and Phosphorylation Efficiency. Toxicol. Appl. Pharmacol. 2001, 176, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Larosche, I.; Lettéron, P.; Fromenty, B.; Vadrot, N.; Abbey-Toby, A.; Feldmann, G.; Pessayre, D.; Mansouri, A. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J. Pharmacol. Exp. Ther. 2007, 321, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Pious, D.A.; Hawley, P. Effect of antibiotics on respiration in human cells. Pediatr. Res. 1972, 6, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Dabadie, P.; Bendriss, P.; Erny, P.; Mazat, J.P. Uncoupling effects of local anesthetics on rat liver mitochondria. FEBS Lett. 1987, 226, 77–82. [Google Scholar] [CrossRef]
- Ponsoda, X.; Bort, R.; Jover, R.; Gómez-Lechón, M.J.; Castell, J.V. Molecular mechanism of diclofenac hepatotoxicity: Association of cell injury with oxidative metabolism and decrease in ATP levels. Toxicol. in Vitro 1995, 9, 439–444. [Google Scholar] [CrossRef]
- Syed, M.; Skonberg, C.; Hansen, S.H. Mitochondrial toxicity of diclofenac and its metabolites via inhibition of oxidative phosphorylation (ATP synthesis) in rat liver mitochondria: Possible role in drug induced liver injury (DILI). Toxicol. in Vitro 2016, 31, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Mingatto, F.E.; Rodrigues, T.; Pigoso, A.A.; Uyemura, S.A.; Curti, C.; Santos, A.C. The critical role of mitochondrial energetic impairment in the toxicity of nimesulide to hepatocytes. J. Pharmacol. Exp. Ther. 2002, 303, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Renault, S.; Letteron, P.; Robin, M.A.; Fromenty, B.; Fau, D.; Le Bot, M.A.; Riché, C.; Durand-Schneider, A.M.; Feldmann, G.; et al. Uncoupling of rat and human mitochondria: A possible explanation for tacrine-induced liver dysfunction. Gastroenterology 1996, 110, 1878–1890. [Google Scholar] [CrossRef] [PubMed]
- Walker, U.A.; Bäuerle, J.; Laguno, M.; Murillas, J.; Mauss, S.; Schmutz, G.; Setzer, B.; Miquel, R.; Gatell, J.M.; Mallolas, J. Depletion of mitochondrial DNA in liver under antiretroviral therapy with didanosine, stavudine, or zalcitabine. Hepatology 2004, 39, 311–317. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, R.; Fried, M.W.; Sallie, R.; Conjeevaram, H.; Di Bisceglie, A.M.; Park, Y.; Savarese, B.; Kleiner, D.; Tsokos, M.; Luciano, C. Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N. Engl. J. Med. 1995, 333, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Haouzi, D.; Descatoire, V.; Demeilliers, C.; Sutton, A.; Vadrot, N.; Fromenty, B.; Feldmann, G.; Pessayre, D.; Berson, A. Tacrine inhibits topoisomerases and DNA synthesis to cause mitochondrial DNA depletion and apoptosis in mouse liver. Hepatology 2003, 38, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Rachek, L.I.; Yuzefovych, L.V.; LeDoux, S.P.; Julie, N.L.; Wilson, G.L. Troglitazone, but not rosiglitazone, damages mitochondrial DNA and induces mitochondrial dysfunction and cell death in human hepatocytes. Toxicol. Appl. Pharmacol. 2009, 240, 348–354. [Google Scholar] [CrossRef] [PubMed]
- De la Asunción, J.G.; del Olmo, M.L.; Sastre, J.; Pallardó, F.V.; Viña, J. Zidovudine (AZT) causes an oxidation of mitochondrial DNA in mouse liver. Hepatology 1999, 29, 985–987. [Google Scholar] [CrossRef] [PubMed]
- Le Dinh, T.; Fréneaux, E.; Labbe, G.; Letteron, P.; Degott, C.; Genève, J.; Berson, A.; Larrey, D.; Pessayre, D. Amineptine, a tricyclic antidepressant, inhibits the mitochondrial oxidation of fatty acids and produces microvesicular steatosis of the liver in mice. J. Pharmacol. Exp. Ther. 1988, 247, 745–750. [Google Scholar] [PubMed]
- Kennedy, J.A.; Unger, S.A.; Horowitz, J.D. Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem. Pharmacol. 1996, 52, 273–280. [Google Scholar] [CrossRef]
- Fréneaux, E.; Fromenty, B.; Berson, A.; Labbe, G.; Degott, C.; Letteron, P.; Larrey, D.; Pessayre, D. Stereoselective and nonstereoselective effects of ibuprofen enantiomers on mitochondrial beta-oxidation of fatty acids. J. Pharmacol. Exp. Ther. 1990, 255, 529–535. [Google Scholar] [PubMed]
- Baldwin, G.S.; Murphy, V.J.; Yang, Z.; Hashimoto, T. Binding of nonsteroidal antiinflammatory drugs to the alpha-subunit of the trifunctional protein of long chain fatty acid oxidation. J. Pharmacol. Exp. Ther. 1998, 286, 1110–1114. [Google Scholar] [PubMed]
- Ulrich, R.G.; Bacon, J.A.; Cramer, C.T.; Petrella, D.K.; Sun, E.L.; Meglasson, M.D.; Holmuhamedov, E. Disruption of mitochondrial activities in rabbit and human hepatocytes by a quinoxalinone anxiolytic and its carboxylic acid metabolite. Toxicology 1998, 131, 33–47. [Google Scholar] [CrossRef]
- Genève, J.; Hayat-Bonan, B.; Labbe, G.; Degott, C.; Letteron, P.; Fréneaux, E.; Le Dinh, T.; Larrey, D.; Pessayre, D. Inhibition of mitochondrial β-oxidation of fatty acids by pirprofen. Role in microvesicular steatosis due to this nonsteroidal anti-inflammatory drug. J. Pharmacol. Exp. Ther. 1987, 242, 1133–1137. [Google Scholar] [PubMed]
- Deschamps, D.; Fisch, C.; Fromenty, B.; Berson, A.; Degott, C.; Pessayre, D. Inhibition by salicylic acid of the activation and thus oxidation of long chain fatty acids: Possible role in the development of Reye’s syndrome. J. Pharmacol. Exp. Ther. 1991, 259, 894–904. [Google Scholar] [PubMed]
- Fréneaux, E.; Labbe, G.; Letteron, P.; Dinh, T.L.; Degott, C.; Genève, J.; Larrey, D.; Pessayre, D. Inhibition of the mitochondrial oxidation of fatty acids by tetracycline in mice and in man: Possible role in microvesicular steatosis induced by this antibiotic. Hepatology 1988, 8, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Fulgencio, J.-P.; Kohl, C.; Girard, J.; Pégorier, J.-P. Troglitazone Inhibits Fatty Acid Oxidation and Esterification, and Gluconeogenesis in Isolated Hepatocytes from Starved Rats. Diabetes 1996, 45, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Aires, C.C.P.; Ijlst, L.; Stet, F.; Prip-Buus, C.; de Almeida, I.T.; Duran, M.; Wanders, R.J.A.; Silva, M.F.B. Inhibition of hepatic carnitine palmitoyl-transferase I (CPT IA) by valproyl-CoA as a possible mechanism of valproate-induced steatosis. Biochem. Pharmacol. 2010, 79, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Derks, T.G.J.; van Dijk, T.H.; Grefhorst, A.; Rake, J.-P.; Smit, G.P.A.; Kuipers, F.; Reijngoud, D.-J. Inhibition of mitochondrial fatty acid oxidation in vivo only slightly suppresses gluconeogenesis but enhances clearance of glucose in mice. Hepatology 2008, 47, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Khungar, V.; Han, S.-H. A Systematic Review of Side Effects of Nucleoside and Nucleotide Drugs Used for Treatment of Chronic Hepatitis B. Curr. Hepat. Rep. 2010, 9, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hanes, J.; Johnson, K.A. Toxicity of Nucleoside Analogues Used to Treat AIDS and the Selectivity of the Mitochondrial DNA Polymerase. Biochemistry 2003, 42, 14711–14719. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.; Copeland, W.C.; Day, B.J. Mitochondrial dna depletion, oxidative stress, and mutation: Mechanisms of dysfunction from nucleoside reverse transcriptase inhibitors. Lab. Investig. 2001, 81, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Moullan, N.; Mouchiroud, L.; Wang, X.; Ryu, D.; Williams, E.G.; Mottis, A.; Jovaisaite, V.; Frochaux, M.V.; Quiros, P.M.; Deplancke, B.; et al. Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep. 2015, 10, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.C.; Adamson, J.S.; Workman, W.W.; Norman, T.D. Fatal Liver Disease after Intravenous Administration of Tetracycline in High Dosage. N. Engl. J. Med. 1963, 269, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.H.; Ju, C.; Ramaiah, S.K.; Uetrecht, J.; Jaeschke, H. Mechanisms of Immune-Mediated Liver Injury. Toxicol. Sci. 2010, 115, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Holt, M.P.; Cheng, L.; Ju, C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J. Leukoc. Biol. 2008, 84, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Reilly, T.P.; Bourdi, M.; Radonovich, M.F.; Brady, J.N.; George, J.W.; Pohl, L.R. Protective Role of Kupffer Cells in Acetaminophen-Induced Hepatic Injury in Mice. Chem. Res. Toxicol. 2002, 15, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Michael, S.L.; Pumford, N.R.; Mayeux, P.R.; Niesman, M.R.; Hinson, J.A. Pretreatment of mice with macrophage inactivators decreases acetaminophen hepatotoxicity and the formation of reactive oxygen and nitrogen species. Hepatology 1999, 30, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Mallal, S.; Nolan, D.; Witt, C.; Masel, G.; Martin, A.M.; Moore, C.; Sayer, D.; Castley, A.; Mamotte, C.; Maxwell, D.; et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 2002, 359, 727–732. [Google Scholar] [CrossRef]
- Illing, P.T.; Vivian, J.P.; Dudek, N.L.; Kostenko, L.; Chen, Z.; Bharadwaj, M.; Miles, J.J.; Kjer-Nielsen, L.; Gras, S.; Williamson, N.A.; et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 2012, 486, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Chessman, D.; Kostenko, L.; Lethborg, T.; Purcell, A.W.; Williamson, N.A.; Chen, Z.; Kjer-Nielsen, L.; Mifsud, N.A.; Tait, B.D.; Holdsworth, R.; et al. Human Leukocyte Antigen Class I-Restricted Activation of CD8+ T-Cells Provides the Immunogenetic Basis of a Systemic Drug Hypersensitivity. Immunity 2008, 28, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, J.R.; Welsh, K.I.; Tinoco, R.M.; Dollery, C.T.; Hughes, G.R.; Bernstein, R.; Ryan, P.; Naish, P.F.; Aber, G.M.; Bing, R.F.; et al. Hydralazine-induced systemic lupus erythematosus: Influence of HLA-DR and sex on susceptibility. Lancet 1980, 1, 1107–1109. [Google Scholar] [CrossRef]
- Dunphy, J.; Oliver, M.; Rands, A.L.; Lovell, C.R.; McHugh, N.J. Antineutrophil cytoplasmic antibodies and HLA class II alleles in minocycline-induced lupus-like syndrome. Br. J. Dermatol. 2000, 142, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.-H.; Hung, S.-I.; Hong, H.-S.; Hsih, M.-S.; Yang, L.-C.; Ho, H.-C.; Wu, J.-Y.; Chen, Y.-T. Medical genetics: A marker for Stevens-Johnson syndrome. Nature 2004, 428, 486. [Google Scholar] [CrossRef] [PubMed]
- Tangamornsuksan, W.; Chaiyakunapruk, N.; Somkrua, R.; Lohitnavy, M.; Tassaneeyakul, W. Relationship Between the HLA-B*1502Allele and Carbamazepine-Induced Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis. JAMA Dermatol. 2013, 149, 1025–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormack, M.; Alfirevic, A.; Bourgeois, S.; Farrell, J.J.; Kasperavičiūtė, D.; Carrington, M.; Sills, G.J.; Marson, T.; Jia, X.; de Bakker, P.I.W.; et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N. Engl. J. Med. 2011, 364, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, T.; Mushiroda, T.; Yowang, A.; Takahashi, A.; Kubo, M.; Shirakata, Y.; Ikezawa, Z.; Iijima, M.; Shiohara, T.; Hashimoto, K.; et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum. Mol. Genet. 2011, 20, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Locharernkul, C.; Loplumlert, J.; Limotai, C.; Korkij, W.; Desudchit, T.; Tongkobpetch, S.; Kangwanshiratada, O.; Hirankarn, N.; Suphapeetiporn, K.; Shotelersuk, V. Carbamazepine and phenytoin induced Stevens-Johnson syndrome is associated with HLA-B*1502 allele in Thai population. Epilepsia 2008, 49, 2087–2091. [Google Scholar] [CrossRef] [PubMed]
- Man, C.B.L.; Kwan, P.; Baum, L.; Yu, E.; Lau, K.M.; Cheng, A.S.H.; Ng, M.H.L. Association between HLA-B*1502 Allele and Antiepileptic Drug-Induced Cutaneous Reactions in Han Chinese. Epilepsia 2007, 48, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-I.; Chung, W.-H.; Liou, L.-B.; Chu, C.-C.; Lin, M.; Huang, H.-P.; Lin, Y.-L.; Lan, J.-L.; Yang, L.-C.; Hong, H.-S.; et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc. Natl. Acad. Sci. USA 2005, 102, 4134–4139. [Google Scholar] [CrossRef] [PubMed]
- Lonjou, C.; Borot, N.; Sekula, P.; Ledger, N.; Thomas, L.; Halevy, S.; Naldi, L.; Bouwes-Bavinck, J.-N.; Sidoroff, A.; de Toma, C.; et al. A European study of HLA-B in Stevens-Johnson syndrome and toxic epidermal necrolysis related to five high-risk drugs. Pharmacogenet. Genom. 2008, 18, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Kaniwa, N.; Saito, Y.; Aihara, M.; Matsunaga, K.; Tohkin, M.; Kurose, K.; Sawada, J.-I.; Furuya, H.; Takahashi, Y.; Muramatsu, M.; et al. HLA-B locus in Japanese patients with anti-epileptics and allopurinol-related Stevens-Johnson syndrome and toxic epidermal necrolysis. Pharmacogenomics 2008, 9, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Tassaneeyakul, W.; Jantararoungtong, T.; Chen, P.; Lin, P.-Y.; Tiamkao, S.; Khunarkornsiri, U.; Chucherd, P.; Konyoung, P.; Vannaprasaht, S.; Choonhakarn, C.; et al. Strong association between HLA-B*5801 and allopurinol-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in a Thai population. Pharmacogenet. Genom. 2009, 19, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Cornejo Castro, E.M.; Carr, D.F.; Jorgensen, A.L.; Alfirevic, A.; Pirmohamed, M. HLA-allelotype associations with nevirapine-induced hypersensitivity reactions and hepatotoxicity. Pharmacogenet. Genom. 2015, 25, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Carr, D.F.; Chaponda, M.; Jorgensen, A.L.; Castro, E.C.; van Oosterhout, J.J.; Khoo, S.H.; Lalloo, D.G.; Heyderman, R.S.; Alfirevic, A.; Pirmohamed, M. Association of Human Leukocyte Antigen Alleles and Nevirapine Hypersensitivity in a Malawian HIV-Infected Population. Clin. Infect. Dis. 2013, 56, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Yunis, J.J.; Corzo, D.; Salazar, M.; Lieberman, J.A.; Howard, A.; Yunis, E.J. HLA associations in clozapine-induced agranulocytosis. Blood 1995, 86, 1177–1183. [Google Scholar] [PubMed]
- Goldstein, J.I.; Jarskog, L.F.; Hilliard, C.; Alfirevic, A.; Duncan, L.; Fourches, D.; Huang, H.; Lek, M.; Neale, B.M.; Ripke, S.; et al. Clozapine-induced agranulocytosis is associated with rare HLA-DQB1 and HLA-B alleles. Nat. Commun. 2014, 5, 4757. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.K.; Donaldson, P.T.; Bhatnagar, P.; Shen, Y.; Pe’er, I.; Floratos, A.; Daly, M.J.; Goldstein, D.B.; John, S.; Nelson, M.R.; et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009, 41, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Kindmark, A.; Jawaid, A.; Harbron, C.G.; Barratt, B.J.; Bengtsson, O.F.; Andersson, T.B.; Carlsson, S.; Cederbrant, K.E.; Gibson, N.J.; Armstrong, M.; et al. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenom. J. 2007, 8, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Hautekeete, M.L.; Horsmans, Y.; van Waeyenberge, C.; Demanet, C.; Henrion, J.; Verbist, L.; Brenard, R.; Sempoux, C.; Michielsen, P.P.; Yap, P.; et al. HLA association of amoxicillin-clavulanate-induced hepatitis. Gastroenterology 1999, 117, 1181–1186. [Google Scholar] [CrossRef]
- O’Donohue, J.; Oien, K.A.; Donaldson, P.; Underhill, J.; Clare, M.; MacSween, R.M.; Mills, P.R. Co-amoxiclav jaundice: Clinical and histological features and HLA class II association. Gut 2000, 47, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, P.T.; Daly, A.K.; Henderson, J.; Graham, J.; Pirmohamed, M.; Bernal, W.; Day, C.P.; Aithal, G.P. Human leucocyte antigen class II genotype in susceptibility and resistance to co-amoxiclav-induced liver injury. J. Hepatol. 2010, 53, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Molokhia, M.; Shen, Y.; Urban, T.J.; Aithal, G.P.; Andrade, R.J.; Day, C.P.; Cabello, F.R.; Donaldson, P.T.; Stephens, C.L.; et al. Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology 2011, 141, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.B.; Lewitzky, S.; Leroy, E.; Yang, F.; Zhao, X.; Klickstein, L.; Wright, T.M.; Meyer, J.; Paulding, C.A. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat. Genet. 2010, 42, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Takagi, H.; Yamamoto, M.; Matsumoto, T.; Nishiya, T.; Mori, K.; Shimizu, S.; Masumoto, H.; Okutani, Y. Ticlopidine-induced hepatotoxicity is associated with specific human leukocyte antigen genomic subtypes in Japanese patients: A preliminary case-control study. Pharmacogenom. J. 2007, 8, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Hetherington, S.; McGuirk, S.; Powell, G.; Cutrell, A.; Naderer, O.; Spreen, B.; Lafon, S.; Pearce, G.; Steel, H. Hypersensitivity reactions during therapy with the nucleoside reverse transcriptase inhibitor abacavir. Clin. Ther. 2001, 23, 1603–1614. [Google Scholar] [CrossRef]
- Mallal, S.; Phillips, E.; Carosi, G.; Molina, J.-M.; Workman, C.; Tomazic, J.; Jägel-Guedes, E.; Rugina, S.; Kozyrev, O.; Cid, J.F.; et al. HLA-B*5701 screening for hypersensitivity to abacavir. N. Engl. J. Med. 2008, 358, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Vilar, F.J.; Ward, C.C.; Alfirevic, A.; Park, B.K.; Pirmohamed, M. Cost-effectiveness analysis of HLA B*5701 genotyping in preventing abacavir hypersensitivity. Pharmacogenetics 2004, 14, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Schackman, B.R.; Scott, C.A.; Walensky, R.P.; Losina, E.; Freedberg, K.A.; Sax, P.E. The cost-effectiveness of HLA-B*5701 genetic screening to guide initial antiretroviral therapy for HIV. AIDS 2008, 22, 2025–2033. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Blankenburg, M.; Bogner, J.R.; Becker, W.; Gorriahn, D.; Mueller, M.C.; Jaeger, H.; Welte, R.; Baudewig, M.; Walli, R.; et al. Cost Impact of Prospective Hla-B*5701-Screening Prior to Abacavir/Lamivudine Fixed Dose Combination Use in Germany. Eur. J. Med. Res. 2010, 15, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Schoonen, W.M.; Thomas, S.L.; Somers, E.C.; Smeeth, L.; Kim, J.; Evans, S.; Hall, A.J. Do selected drugs increase the risk of lupus? A matched case-control study. Br. J. Clin. Pharmacol. 2010, 70, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Moser, K.L.; Kelly, J.A.; Lessard, C.J.; Harley, J.B. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. 2009, 10, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.R.; Ortmann, W.A.; Langefeld, C.D.; Jawaheer, D.; Selby, S.A.; Rodine, P.R.; Baechler, E.C.; Rohlf, K.E.; Shark, K.B.; Espe, K.J.; et al. Visualizing Human Leukocyte Antigen Class II Risk Haplotypes in Human Systemic Lupus Erythematosus. Am. J. Hum. Genet. 2002, 71, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Fernando, M.M.A.; Stevens, C.R.; Sabeti, P.C.; Walsh, E.C.; McWhinnie, A.J.M.; Shah, A.; Green, T.; Rioux, J.D.; Vyse, T.J. Identification of Two Independent Risk Factors for Lupus within the MHC in United Kingdom Families. PLoS Genet. 2007, 3, e192–e113. [Google Scholar] [CrossRef] [PubMed]
- International MHC and Autoimmunity Genetics Network; Rioux, J.D.; Goyette, P.; Vyse, T.J.; Hammarström, L.; Fernando, M.M.; Green, T.; de Jager, P.L.; Foisy, S.; Wang, J.; et al. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2009, 106, 18680–18685. [Google Scholar] [PubMed]
- Barcellos, L.F.; May, S.L.; Ramsay, P.P.; Quach, H.L.; Lane, J.A.; Nititham, J.; Noble, J.A.; Taylor, K.E.; Quach, D.L.; Chung, S.A.; et al. High-density SNP screening of the major histocompatibility complex in systemic lupus erythematosus demonstrates strong evidence for independent susceptibility regions. PLoS Genet. 2009, 5, e1000696. [Google Scholar] [CrossRef] [PubMed]
- De Maat, M.M.R.; ter Heine, R.; Mulder, J.W.; Meenhorst, P.L.; Mairuhu, A.T.A.; van Gorp, E.C.M.; Huitema, A.D.R.; Beijnen, J.H. Incidence and risk factors for nevirapine-associated rash. Eur. J. Clin. Pharmacol. 2003, 59, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.J.; Boxwell, D.E.; Kim, N.Y.; Drolet, B.A. Nevirapine-associated Stevens-Johnson syndrome. Lancet 1998, 351, 567–561. [Google Scholar] [CrossRef]
- Alvir, J.M.J.; Lieberman, J.A.; Safferman, A.Z.; Schwimmer, J.L.; Schaaf, J.A. Clozapine-Induced Agranulocytosis—Incidence and Risk Factors in the United States. N. Engl. J. Med. 1993, 329, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Phillips, E.J.; Mallal, S.A. HLA-B*5701 and flucloxacillin associated drug-induced liver disease. AIDS 2013, 27, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Alfirevic, A.; Pirmohamed, M. predictiveg enetic testing for drug-induced liver injury: Considerations of clinical utility. Clin. Pharmacol. Ther. 2009, 92, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Fontana, R.J.; Bonkovsky, H.L.; Watkins, P.B.; Davern, T.; Serrano, J.; Yang, H.; Rochon, J. Causes, Clinical Features, and Outcomes from a Prospective Study of Drug-Induced Liver Injury in the United States. Gastroenterology 2008, 135, 1924–1934. [Google Scholar] [CrossRef] [PubMed]
- Stephens, C.; López-Nevot, M.-Á.; Ruiz-Cabello, F.; Ulzurrun, E.; Soriano, G.; Romero-Gómez, M.; Moreno-Casares, A.; Lucena, M.I.; Andrade, R.J. HLA alleles influence the clinical signature of amoxicillin-clavulanate hepatotoxicity. PLoS ONE 2013, 8, e68111–e68117. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Saide, K.; Farrell, J.; Faulkner, L.; Tailor, A.; Ogese, M.; Daly, A.K.; Pirmohamed, M.; Park, B.K.; Naisbitt, D.J. Characterization of amoxicillin- and clavulanic acid-specific T-cells in patients with amoxicillin-clavulanate-induced liver injury. Hepatology 2015, 62, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Farid, N.A.; Kurihara, A.; Wrighton, S.A. Metabolism and disposition of the thienopyridine antiplatelet drugs ticlopidine, clopidogrel, and prasugrel in humans. J. Clin. Pharmacol. 2013, 50, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Ariyoshi, N.; Iga, Y.; Hirata, K.; Sato, Y.; Miura, G.; Ishii, I.; Nagamori, S.; Kitada, M. Enhanced susceptibility of HLA-mediated ticlopidine-induced idiosyncratic hepatotoxicity by CYP2B6 polymorphism in Japanese. Drug Metab. Pharmacokinet. 2010, 25, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M.; Larrey, D.; Olsson, R.; Lewis, J.H.; Keisu, M.; Auclert, L.; Sheth, S. Hepatic findings in long-term clinical trials of ximelagatran. Drug Saf. 2005, 28, 351–370. [Google Scholar] [CrossRef] [PubMed]
- Pentikäinen, P.J.; Neuvonen, P.J.; Jostell, K.G. Pharmacokinetics of Chlormethiazole in healthy-volunteers and patients with cirrhosis of the liver. Eur. J. Clin. Pharmacol. 1980, 17, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, G.; Gabor, M.; Reiff, K. Pharmacokinetics and bioavailability of carvedilol in patients with liver cirrhosis. Drugs 1988, 36, 148–154. [Google Scholar] [CrossRef]
- Morgan, D.J.; McLean, A.J. Therapeutic implications of impaired hepatic oxygen diffusion in chronic liver disease. Hepatology 1991, 14, 1280–1282. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.F.; Zgheib, N.K.; Matzke, G.R.; Chaves-Gnecco, D.; Rabinovitz, M.; Shaikh, O.S.; Branch, R.A. Liver disease selectively modulates cytochrome P450—mediated metabolism. Clin. Pharmacol. Ther. 2006, 80, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, J.M.; Sabia, H.D.; Figueiredo, J.; Zimmermann, H.; Reynolds, C.; Dilzer, S.C.; Lasseter, K.; Rordorf, C. Influence of hepatic impairment on everolimus pharmacokinetics: Implications for dose adjustment. Clin. Pharmacol. Ther. 2001, 70, 425–430. [Google Scholar] [CrossRef]
- Chalasani, N.; Gorski, J.C.; Patel, N.H.; Hall, S.D.; Galinsky, R.E. Hepatic and intestinal cytochrome P450 3A activity in cirrhosis: Effects of transjugular intrahepatic portosystemic shunts. Hepatology 2001, 34, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Turvy, C.G. Comparison of levels of several human microsomal cytochrome P-450 enzymes and epoxide hydrolase in normal and disease states using immunochemical analysis of surgical liver samples. J. Pharmacol. Exp. Ther. 1991, 256, 1189–1194. [Google Scholar] [PubMed]
- George, J.; Murray, M.; Byth, K.; Farrell, G.C. Differential alterations of cytochrome P450 proteins in livers from patients with severe chronic liver disease. Hepatology 1995, 21, 120–128. [Google Scholar] [PubMed]
- Shull, H.J.; Wilkinson, G.R.; Johnson, R.; Schenker, S. Normal Disposition of Oxazepam in Acute Viral-Hepatitis and Cirrhosis. Ann. Intern. Med. 1976, 84, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Klotz, U.; Antonin, K.H.; Brügel, H.; Bieck, P.R. Disposition of Diazepam and Its Major Metabolite Desmethyldiazepam in Patients with Liver-Disease. Clin. Pharmacol. Ther. 1977, 21, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Crotty, B.; Watson, K.; Desmond, P.V.; Mashford, M.L.; Wood, L.J.; Colman, J.; Dudley, F.J. Hepatic Extraction of Morphine Is Impaired in Cirrhosis. Eur. J. Clin. Pharmacol. 1989, 36, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Taburet, A.M.; Naveau, S.; Zorza, G.; Colin, J.N.; Delfraissy, J.F.; Chaput, J.C.; Singlas, E. Pharmacokinetics of Zidovudine in Patients with Liver-Cirrhosis. Clin. Pharmacol. Ther. 1990, 47, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; De Bony, F.; Garret, C.; Altman, C.; Boige, V.; Castelnau, C.; Laurent-Puig, P.; Trinchet, J.C.; Rolan, P.; Chen, C.; et al. Influence of cirrhosis on lamotrigine pharmacokinetics. Br. J. Clin. Pharmacol. 2002, 51, 410–414. [Google Scholar] [CrossRef]
- Fisher, C.D.; Lickteig, A.J.; Augustine, L.M.; Ranger-Moore, J.; Jackson, J.P.; Ferguson, S.S.; Cherrington, N.J. Hepatic Cytochrome P450 Enzyme Alterations in Humans with Progressive Stages of Nonalcoholic Fatty Liver Disease. Drug Metab. Dispos. 2009, 37, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Kolwankar, D.; Vuppalanchi, R.; Ethell, B.; Jones, D.R.; Wrighton, S.A.; Hall, S.D.; Chalasani, N. Association Between Nonalcoholic Hepatic Steatosis and Hepatic Cytochrome P-450 3A Activity. Clin. Gastroenterol. Hepatol. 2007, 5, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Orellana, M.; Rodrigo, R.; Varela, N.; Araya, J.; Poniachik, J.; Csendes, A.; Smok, G.; Videla, L.A. Relationship between in vivo chlorzoxazone hydroxylation, hepatic cytochrome P450 2E1 content and liver injury in obese non-alcoholic fatty liver disease patients. Hepatol. Res. 2006, 34, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.S.; Baker, R.D.; Liu, W.; Nowak, N.J.; Zhu, L. Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS ONE 2010, 5, e9570. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Gorski, J.C.; Asghar, M.S.; Asghar, A.; Foresman, B.; Hall, S.D.; Crabb, D.W. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology 2003, 37, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Emery, M.G.; Fisher, J.M.; Chien, J.Y.; Kharasch, E.D.; Dellinger, E.P.; Kowdley, K.V.; Thummel, K.E. CYP2E1 activity before and after weight loss in morbidly obese subjects with nonalcoholic fatty liver disease. Hepatology 2003, 38, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Baranova, A.; Ziegler, K.; Del Giacco, L.; Schlauch, K.; Born, T.L.; Elariny, H.; Gorreta, F.; VanMeter, A.; Younoszai, A.; et al. A genomic and proteomic study of the spectrum of nonalcoholic fatty liver disease. Hepatology 2005, 42, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, M.; Hossain, N.; Afendy, A.; Perry, K.; Goodman, Z.D.; Baranova, A.; Younossi, Z. Hepatic Gene Expression of Caucasian and African-American Patients with Obesity-Related Non-Alcoholic Fatty Liver Disease. Obes. Surg. 2010, 20, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, R.N.; Fisher, C.D.; Canet, M.J.; Scheffer, G.L.; Cherrington, N.J. Variations in ATP-binding cassette transporter regulation during the progression of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 2011, 39, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Okushin, K.; Tsutsumi, T.; Enooku, K.; Kado, A.; Fujinaga, H.; Moriya, K.; Yotsuyanagi, H.; Koike, K. P0972: Expressions of bile acid transporters are inversely correlated with NAFLD activity score in the liver of patients with non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, S710. [Google Scholar] [CrossRef]
- Canet, M.J.; Merrell, M.D.; Hardwick, R.N.; Bataille, A.M.; Campion, S.N.; Ferreira, D.W.; Xanthakos, S.A.; Manautou, J.E.; A-Kader, H.H.; Erickson, R.P.; et al. Altered regulation of hepatic efflux transporters disrupts acetaminophen disposition in pediatric nonalcoholic steatohepatitis. Drug Metabo. Dispos. 2015, 43, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.F.; Garrett, M.E.; Koch, A.A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship Between Methylome and Transcriptome in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Fisel, P.; Schaeffeler, E.; Schwab, M. DNA Methylation of ADME Genes. Clin. Pharmacol. Ther. 2016, 99, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Westlind, A.; Löfberg, L.; Tindberg, N.; Andersson, T.B.; Ingelman-Sundberg, M. Interindividual differences in hepatic expression of CYP3A4: Relationship to genetic polymorphism in the 5′-upstream regulatory region. Biochem. Biophys. Res. Commun. 1999, 259, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, V.; Kalow, W.; Tang, B.K.; Paterson, A.D.; Walker, S.E.; Endrenyi, L.; Kashuba, A.D. Evaluation of the genetic component of variability in CYP3A4 activity: A repeated drug administration method. Pharmacogenetics 2000, 10, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Kacevska, M.; Ivanov, M.; Wyss, A.; Kasela, S.; Milani, L.; Rane, A.; Ingelman-Sundberg, M. DNA methylation dynamics in the hepatic CYP3A4 gene promoter. Biochimie 2012, 94, 2338–2344. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.; Kals, M.; Kacevska, M.; Barragan, I.; Kasuga, K.; Rane, A.; Metspalu, A.; Milani, L.; Ingelman-Sundberg, M. Ontogeny, distribution and potential roles of 5-hydroxymethylcytosine in human liver function. Genome Biol. 2013, 14, R83. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.; Kals, M.; Lauschke, V.M.; Barragan, I.; Ewels, P.; Käller, M.; Axelsson, T.; Lehtiö, J.; Milani, L.; Ingelman-Sundberg, M. Single base resolution analysis of 5-hydroxymethylcytosine in 188 human genes: Implications for hepatic gene expression. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Lechón, M.J.; Tolosa, L.; Conde, I.; Donato, M.T. Competency of different cell models to predict human hepatotoxic drugs. Exp. Opin. Drug Metab. Toxicol. 2014, 10, 1553–1568. [Google Scholar] [CrossRef] [PubMed]
- Sison-Young, R.L.; Lauschke, V.M.; Johann, E.; Alexandre, E.; Anthérieu, S.; Aerts, H.; Gerets, H.H.J.; Labbe, G.; Hoët, D.; Dorau, M.; et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Arch. Toxicol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Treyer, A.; Müsch, A. Hepatocyte polarity. Compr. Physiol. 2013, 3, 243–287. [Google Scholar] [PubMed]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Rowe, C.; Gerrard, D.T.; Jenkins, R.; Berry, A.; Durkin, K.; Sundstrom, L.; Goldring, C.E.; Park, B.K.; Kitteringham, N.R.; Hanley, K.P.; et al. Proteome-wide analyses of human hepatocytes during differentiation and dedifferentiation. Hepatology 2013, 58, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Heslop, J.A.; Rowe, C.; Walsh, J.; Sison-Young, R.; Jenkins, R.; Kamalian, L.; Kia, R.; Hay, D.; Jones, R.P.; Malik, H.Z.; et al. Mechanistic evaluation of primary human hepatocyte culture using global proteomic analysis reveals a selective dedifferentiation profile. Arch. Toxicol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lauschke, V.M.; Vorrink, S.U.; Moro, S.M.; Reyazee, F.; Nordling, Å.; Hendriks, D.F.; Bell, C.C.; Sison-Young, R.; Park, B.K.; Goldring, C.E.; et al. Massive rearrangements of cellular miRNA signatures are key drivers of hepatocyte dedifferentiation. Hepatology 2016. [Google Scholar] [CrossRef]
- Lauschke, V.M.; Hendriks, D.F.; Bell, C.C.; Andersson, T.B.; Ingelman-Sundberg, M. Novel 3D culture systems for studies of liver function and assessments of hepatotoxicity of drugs and drug candidates. Chem. Res. Toxicol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tostões, R.M.; Leite, S.B.; Serra, M.; Jensen, J.; Björquist, P.; Carrondo, M.J.T.; Brito, C.; Alves, P.M. Human liver cell spheroids in extended perfusion bioreactor culture for repeated-dose drug testing. Hepatology 2012, 55, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Messner, S.; Agarkova, I.; Moritz, W.; Kelm, J.M. Multi-cell type human liver microtissues for hepatotoxicity testing. Arch. Toxicol. 2012, 87, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Hendriks, D.F.G.; Moro, S.M.L.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson-Puigvert, L.; Dankers, A.C.A.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.D.; Ballinger, K.R.; Khetani, S.R. Long-term exposure to abnormal glucose levels alters drug metabolism pathways and insulin sensitivity in primary human hepatocytes. Sci. Rep. 2016, 6, 28178. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.D.; Xiong, W.; Bunker, A.M.; Vaughn, C.P.; Furtado, L.V.; Roberts, W.L.; Fang, J.C.; Samowitz, W.S.; Heichman, K.A. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Baden, J.; Green, G.; Painter, J.; Curtin, K.; Markiewicz, J.; Jones, J.; Astacio, T.; Canning, S.; Quijano, J.; Guinto, W.; et al. Multicenter evaluation of an investigational prostate cancer methylation assay. J. Urol. 2009, 182, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
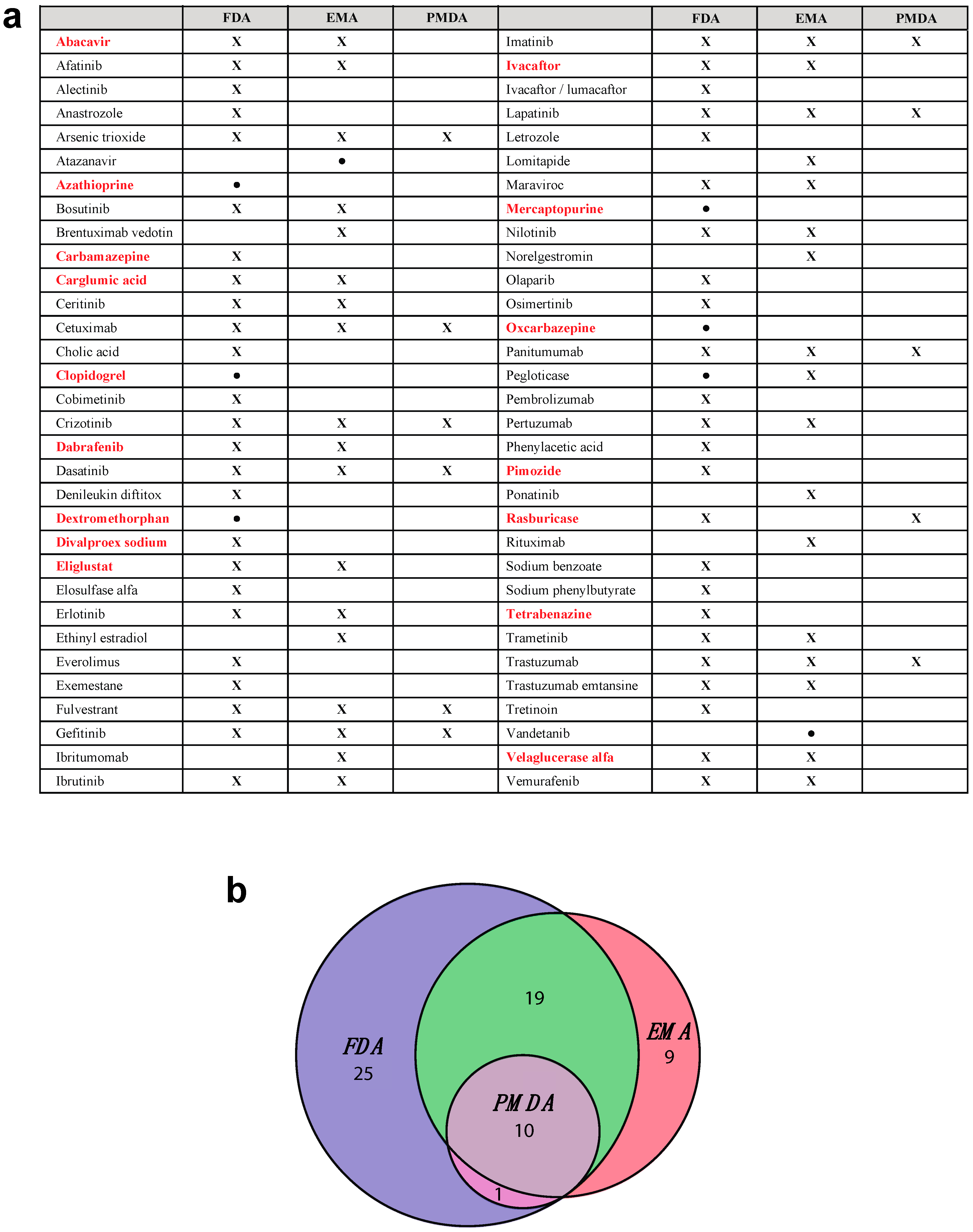
| Gene | Actionable Pairs | Medications |
|---|---|---|
| CYP2D6 | 20 | Amitriptyline, codeine, fluvoxamine, nortriptyline, tramadol, aripiprazole, atomoxetine, clomipramine, desipramine, doxepin, imipramine, protriptyline, trimipramine, vortioxetine, iloperidone, perphenazine, dextromethorphan, eliglustat, pimozide, tetrabenazine |
| DPYD | 2 | Capecitabine, fluorouracil |
| HLA-A | 1 | Carbamazepine |
| CACNA1S | 4 | Desflurane, isoflurane, sevoflurane, succinylcholine |
| RYR1 | 4 | Desflurane, isoflurane, sevoflurane, succinylcholine |
| UGT1A1 | 2 | Irinotecan, belinostat |
| HLA-B | 4 | Phenytoin, oxcarbazepine, abacavir, carbamazepine |
| TPMT | 3 | Thioguanine, azathioprine, mercaptopurine |
| CYP2C19 | 8 | Voriconazole, citalopram, dexlansoprazole, doxepin, esomeprazole, pantoprazole, carisoprodol, clopidogrel |
| CYP2C9 | 2 | Warfarin, celecoxib |
| VKORC1 | 1 | Warfarin |
| IFNL3 | 1 | Peginterferon α-2b |
| G6PD | 22 | Chloroquine, chlorpropamide, dapsone, glibenclamide, glimepiride, glipizide, mafenide, methylene blue, nalidixic acid, nitrofurantoin, norfloxacin, primaquine, probenecid, quinine, sodium nitrite, sulfadiazine, sulfasalazine, erythromycin, sulfisoxazole, dabrafenib, pegloticase, rasburicase |
| HPRT1 | 1 | Mycophenolic acid |
| ABL2 | 1 | Valproic acid |
| ASL | 1 | Valproic acid |
| ASS1 | 1 | Valproic acid |
| CPS1 | 1 | Valproic acid |
| NAGS | 1 | Valproic acid |
| OTC | 1 | Valproic acid |
| POLG | 2 | Valproic acid, divalproex sodium |
| CFTR | 1 | Ivacaftor |
| NAGS | 1 | Carglumic acid |
| GBA | 1 | Velaglucerase α |
| Drug | Gene | Activity Level (Exemplary Genotypes) | Pharmacological Consequence | Dosing Recommendation |
|---|---|---|---|---|
| Fluoropyrimidines | DPYD | Intermediate DPD activity (*1/*2A, *1/*13) | Decreased fluoropyrimidine catabolism and increased levels toxic metabolites | At least 50% initial dose reduction |
| DPD deficiency (*2A/*2A, *13/*13) | Select alternate drug | |||
| Mercaptopurine | TPMT | Intermediate TPMT activity (*1/*2, *1/*3A, *1/*3B, *1/*3C, *1/*4) | Increased levels of cytotoxic TGN metabolite | Reduction to 30%–70% of normal starting dose |
| TPMT deficiency (*3A/*3A, *2/*3A, *3C/*3A, *3C/*4, *3C/*2, *3A/*4) | Drastic dose reduction to <10% or consider alternative therapy | |||
| Codeine | CYP2D6 | Ultrarapid metabolizer (*1/*1xN, *1/*2xN) | Increased formation of morphine | Select alternate drug |
| Intermediate metabolizer (*5/*41, *4/*10) | Reduced formation of morphine | Dosage according to label. If no response, select alternate drug | ||
| Poor metabolizer (*4/*4, *4/*5, *5/*5, *4/*6) | Drastically reduced formation of morphine | Select alternate drug due to lack of efficacy | ||
| Irinotecan | UGT1A1 | Intermediate UGT1A1 activity (*1/*28, *1/*37) | Reduced glucuronidation of active metabolite SN-38 | Standard dose with rigorous clinical surveillance |
| Strongly reduced UGT1A1 activity (*28/*28, *37/*37) | Dose reduction of 30% for standard dose, no dose intensification | |||
| Clopidogrel | CYP2C19 | Ultrarapid metabolizer (*1/*17, *17/*17) | Increased formation of active metabolite, decreased platelet aggregation | Standard dose |
| Intermediate metabolizer (*1/*2, *1/*3, *2/*17) | Reduced formation of active metabolite, increased platelet aggregation | Select alternate drug | ||
| Poor metabolizer (*2/*2, *3/*3, *4/*4, *5/*5, *6/*6, *7/*7, *8/*8) | Select alternate drug | |||
| Omeprazole | CYP2C19 | Ultrarapid metabolizer (*1/*17, *17/*17) | Increased metabolic inactivation to 5-hydroxyomeprazole | Increase dose 2–3-fold for H. pylori eradication therapy |
| Intermediate metabolizer (*1/*2, *1/*3, *2/*17) | Decreased metabolic inactivation to 5-hydroxyomeprazole | Standard dose | ||
| Poor metabolizer (*2/*2, *3/*3, *4/*4, *5/*5, *6/*6, *7/*7, *8/*8) | Standard dose | |||
| Simvastatin | SLCO1B1 | Intermediate SLCO1B1 activity (*1a/*5, *1a/*15, *1a/*17, 1b/*5, *1b/*15, *1b/*17) | Decreased hepatic simvastatin uptake | High simvastatin doses (80 mg/day) not recommended, consider alternative statin |
| Strongly reduced SLCO1B1 activity (*5/*5, *15/*15, *17/*17) |
| Pathway | Drug | Reference |
|---|---|---|
| Mitochondrial permeability transition pore opening | Acetaminophen | Kon et al., 2004 [64] |
| Alpidem | Berson et al., 2001 [65] | |
| Diclofenac | Masubuchi et al., 2002 [66] | |
| Disulfiram | Balakirev et al., 2001 [67] | |
| Nimesulide | Mingatto et al., 2000 [68] | |
| Salicylic acid | Trost et al., 1996 [69] | |
| Troglitazone | Tirmenstein et al., 2002 and Lim et al., 2008 [70,71] | |
| Valproic acid | Trost et al., [69] | |
| Inhibition of mitochondrial respiratory chain | Acetaminophen | Meyers et al., 1988, Donnelly et al., 1994 and Lee et al., 2015 [72,73,74] |
| Amiodarone | Fromenty et al., 1990 [62] | |
| Buprenorphine | Berson et al., 2001 [75] | |
| Efavirenz | Blas-Garcia et al., 2010 [59] | |
| Methotrexate | Yamamoto et al., 1988 [76] | |
| Nefazodone | Dykens et al., 2008 [60] | |
| Nilutamide | Berson et al., 1994 [61] | |
| Perhexillin | Deschamps et al., 1994 [77] | |
| Tamoxifen | Cardoso et al., 2001 and Larosche et al., 2007 [78,79] | |
| Tetracycline | Pious and Hawley, 1972 [80] | |
| Ximelagatran | Neve et al., 2015 [63] | |
| Oxidative phosphorylation uncoupling | Amiodarone | Fromenty et al., 1990 [62] |
| Bupivacaine | Dabadie et al., 1997 [81] | |
| Buprenorphine | Berson et al., 2001 [75] | |
| Diclofenac | Ponsoda et al., 1995 and Syed et al., 2016 [82,83] | |
| Nimesulide | Mingatto et al., 2002 [84] | |
| Perhexillin | Deschamps et al., 1994 [77] | |
| Tacrine | Berson et al., 1996 [85] | |
| Tamoxifen | Cardoso et al., 2001 [78] | |
| Mitochondrial DNA depletion | Didanosine | Walker et al., 2004 [86] |
| Fialuridine | McKenzie et al., 1995 [87] | |
| Stavudine | Walker et al., 2004 [86] | |
| Tacrine | Mansouri et al., 2003 [88] | |
| Tamoxifen | Larosche et al., 2007 [79] | |
| Troglitazone | Rachek et al., 2009 [89] | |
| Zalcitabine | Walker et al., 2004 [86] | |
| Zidovudine | De la Asuncion et al., 1999 [90] | |
| Inhibition of β-oxidation and/or depletion of carnitine and Coenzyme A | Amineptine | Le Dinh et al., 1988 [91] |
| Amiodarone | Kennedy et al., 1996 [92] | |
| Buprenorphine | Berson et al., 2001 [75] | |
| Ibuprofen | Fréneaux et al., 1990 and Baldwin et al., 1998 [93,94] | |
| Panadiplon | Ulrich et al., 1998 [95] | |
| Perhexillin | Deschamps et al., 1994 and Kennedy et al., 1994 [77,92] | |
| Pirprofen | Genève et al., 1987 [96] | |
| Salicylic acid | Deschamps et al., 1991 [97] | |
| Tamoxifen | Larosche et al., 2007 [79] | |
| Tetracyclin | Fréneaux et al., 1988 [98] | |
| Troglitazone | Fulgencio et al., 1996 [99] | |
| Valproic acid | Aires et al., 2010 [100] |
| Drug | Class of Drug | HLA Allele | Adverse Reaction | Reference |
|---|---|---|---|---|
| Abacavir | Antiretroviral | B*57:01, DR7 and DQ3 | HSS | [111,112,113] |
| Hydralazine | Vasodilator | DR4 | SLE | [114] |
| Minocycline | Antibiotic | DQB1 alleles with tyrosine at position 30 | SLE | [115] |
| Carbamazepine | Anticonvulsant | B*15:02 and A*31:01 | HSS and SJS/TEN | [116,117,118,119,120] |
| Phenytoin | Anticonvulsant | B*15:02 | SJS/TEN | [120,121] |
| Allopurinol | Uricosuric | B*58:01 | SJS/TEN | [122,123,124,125] |
| Nevirapine | Antiretroviral | B*35:05 and C*04:01 | SJS/TEN | [126,127] |
| Clozapine | Antipsychotic | Multiple | Agranulocytosis | [128,129] |
| Flucloxacillin | Antibiotic | B*57:01 | DILI | [130] |
| Ximelagatran | Anticoagulant | DRB1*07:01 and DQA1*02:01 | DILI | [131] |
| Co-amoxiclav | Antibiotic | DRB1*15:01 and A*02:01 and B*18:01 | DILI | [132,133,134,135] |
| Lumiracoxib | NSAID | DRB*15:01 and DQA*01:02 | DILI | [136] |
| Ticlopidine | Anticoagulant | A*33:03 | DILI | [137] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauschke, V.M.; Ingelman-Sundberg, M. The Importance of Patient-Specific Factors for Hepatic Drug Response and Toxicity. Int. J. Mol. Sci. 2016, 17, 1714. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101714
Lauschke VM, Ingelman-Sundberg M. The Importance of Patient-Specific Factors for Hepatic Drug Response and Toxicity. International Journal of Molecular Sciences. 2016; 17(10):1714. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101714
Chicago/Turabian StyleLauschke, Volker M., and Magnus Ingelman-Sundberg. 2016. "The Importance of Patient-Specific Factors for Hepatic Drug Response and Toxicity" International Journal of Molecular Sciences 17, no. 10: 1714. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101714